
Abstract
Developmental delay (DD), or intellectual disability (ID) is a very large group of early onset disorders that affects 1–2% of children worldwide, which have diverse genetic causes that should be identified. Genetic studies can elucidate the pathogenesis underlying DD/ID. In this study, whole-exome sequencing (WES) was performed on 225 Chinese DD/ID children (208 cases were sequenced as proband-parent trio) who were classified into seven phenotype subgroups. The phenotype and genomic data of patients with DD/ID were further retrospectively analyzed. There were 96/225 (42.67%; 95% confidence interval [CI] 36.15–49.18%) patients were found to have causative single nucleotide variants (SNVs) and small insertions/deletions (Indels) associated with DD/ID based on WES data. The diagnostic yields among the seven subgroups ranged from 31.25 to 71.43%. Three specific clinical features, hearing loss, visual loss, and facial dysmorphism, can significantly increase the diagnostic yield of WES in patients with DD/ID (P = 0.005, P = 0.005, and P = 0.039, respectively). Of note, hearing loss (odds ratio [OR] = 1.86%; 95% CI = 1.00-3.46, P = 0.046) or abnormal brainstem auditory evoked potential (BAEP) (OR = 1.91, 95% CI = 1.02–3.50, P = 0.042) was independently associated with causative genetic variants in DD/ID children. Our findings enrich the variation spectrums of SNVs/Indels associated with DD/ID, highlight the value genetic testing for DD/ID children, stress the importance of BAEP screen in DD/ID children, and help to facilitate early diagnose, clinical management and reproductive decisions, improve therapeutic response to medical treatment.
Similar content being viewed by others


Introduction
Developmental delay or intellectual disability (DD/ID) is one of the most common neurodevelopmental disabilities with high clinical heterogeneity [1, 2]. It is often classified as either isolated or syndromic DD/ID, and syndromic DD/ID patients present with additional clinical manifestations, such as congenital anomalies, dysmorphic features, epilepsy or unusual behavior [3, 4]. DD/ID affects 1–2% of children worldwide and pose heavy medical, psychological, financial, and social burden [5, 6].
DD/ID might be caused by environmental factors, such as gestational substance abuse, birth complications, infections, and traumas [7, 8]. DD/ID can also be caused by genetic factors, more than 700 genes have been identified to date [9,10,11,12,13]. As genomic technologies progress, new DD/ID genes can be identified rapidly. Whole-exome sequencing (WES) mainly focuses the detection of single nucleotide variants (SNVs) and small insertions/deletions (Indels), which has been proven to result in a high overall diagnostic yield of 30–40% in patients with DD/ID [12, 14,15,16,17]. In 2021, the American College of Medical Genetics and Genomics (ACMG) strongly recommended WES as a first- or second-tier tool for diagnosis of DD/ID to reduce “diagnostic odyssey” [12]. Furthermore, the results of WES may lead to earlier diagnosis, improve therapeutic response, facilitate clinical management, and impact reproductive decisions [12, 15]. Therefore, the objective of this study was to determine the diagnostic yield of DD/ID by WES, to better characterize the genetic landscape of DD/ID and to determine whether WES results can impact medical management.
Furthermore, there is a paucity of information about associations of clinical manifestations with identified causative variants for DD/ID. According to previous studies, the diagnostic rates of WES for isolated and syndromic DD/ID were equivalent [1, 4, 16, 17],, while a meta-analysis reported that the diagnostic yield was 54% for syndromic DD but 31% for isolated DD [18]. Furthermore, several studies have shown that specific clinical features, such as craniofacial anomalies and abnormal head circumference, can increase the diagnostic yield of WES in patients with DD/ID, but none of these impacts are statistically significant [4, 16]. However, Michelle VS et al. reported that the diagnostic yield of WES was significantly greater in DD patients with dysmorphic features than in patients without dysmorphic features [14]. Therefore, another objective of this study was to determine whether specific clinical features can increase the genetic diagnostic yield of DD/ID, and to highlight the importance of routine physiological and biochemical tests in genomic screens.
Methods
Study participant
As illustrated in Fig. 1, this study recruited 225 DD/ID children after obtaining signed informed consents from their parents or legal guardians, between March 2018 and December 2021 in Seventh Medical Center of PLA General Hospital. The detailed clinical data (e.g. age, gender, perinatal history, birth history, neurodevelopmental history, family history) and clinical examinations data (such as physiological testing, biochemical testing) of all patients were reviewed. The exclusion criteria were as follows: (1) parents/guardians refused to sign informed consents; (2) children had nervous system infections or traumas; (3) maternal substance abuse or infections; (4) birth complications; (5) incomplete medical records; (6) positive karyotype test result. Subsequently, WES was performed and analyzed by bioinformatics. The clinical examinations and genetic diagnostic tests were recommended by physicians based on the clinical judgment, but the final decision was made by the parents/guardians. Diagnostic results (including physiological, metabolic and genetic results) were reported to the parents/guardians. Relevant recommendations (e.g., medical management changes, dietary changes, physiotherapy/psychosocial support, follow-up assessment and reproductive planning) were proposed by physicians, but autonomous decisions were made by the parents/guardians. During the study process, the parents/guardians signed waivers of informed consent and could withdraw from the study at any time. This study was approved by the Ethics Committee of PLA General Hospital (No. S2016-120-02). Work was performed in accordance with the Declaration of Helsinki.

Flow chart of the study desig
WES analysis
Genomic DNA was isolated from peripheral blood of the probands and/or their biological parents. WES was performed and analyzed by (Kaiumph Medical Diagnostic Lo. Ltd, (Beijing, China) [19], Angen Gene Medicine Tech (Beijing, China) [20] or Running Gene Inc. (Beijing, China) [21] using their own bioinformatics pipelines as previously described. The laboratory-specific WES methodologic parameters were shown in Supplementary Table 1. Briefly, reads were cleaned to pass quality controls and were aligned to the reference human genome (GRCH37/hg19, genome.ucsc.edu) by BWA-MEM. SNVs and Indels were detected by GATK, and annotated by ANNOVAR (annovar.openbioinformatics.org/en/latest/). Variants were filtered using public databases (including dbSNP142, 1000 Genomes, and ESP6500, ExAc, and in-house Chinese Exome Database) [22,23,24], and/or published papers in WOSCC and PubMed database. Deleterious SNVs were predicted by ReVe (varcards.biols.ac.cn/); VEST3 (wiki.chasmsoftware.org/index.php/Main_Page); REVEL (sites.google.com/site/revelgenomics); and CADD (cadd.gs.washington.edu). Variants were classified using the recommended terminology “pathogenic (P)”, “likely pathogenic (LP)”, “uncertain significance (VUS)”, “likely benign (LB)”, and “benign (B)” according to the recommendation of ACMG [25]. P/LP variations were selected as causative variations for DD/ID in this study. The diagnostic yield of DD/ID was calculated as the total number of DD/ID children with P/LP variants divided by the total number of DD/ID children.
Statistical analysis
Statistical analyses were performed using SPSS version 28.0. Descriptive statistics was performed to describe demographic and basic clinical features. Results were presented as numbers, median, percentage, and 95% confidence interval (CI). The Wilcoxon rank-sum test was used for age-group comparison. Categorical variables were presented as numbers, and a chi-square test was used for between group comparisons. The variance inflation factor (VIF) was used for the multicollinearity test and variable selection. Clinical variables found to be associated with P/LP variants in a univariate analysis (p < 0.2) were further included in a multivariate logistic regression analysis, and results were presented as odds ratios (ORs) and 95% CI. P < 0.05 was considered statistically significant.
Results
Demographic and basic clinical features of enrolled patients
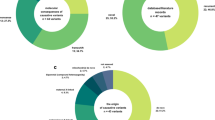
We reviewed and analyzed the WES results of 225 patients diagnosed with DD/ID, of which 208 were trio-WES data (trio: proband and biological parents), one was duo-WES data (duo: proband and one biological parent), and another 16 was singleton-WES data (singleton: proband only, no parents were available). The study group had a median age of 2.58 years and was 64.44% male. Detailed demographic and basic clinical features are shown in Table 1. Overall, 56.21%, 55.84%, 79.31%, and 88.89% patients were found to have an abnormal brain magnetic resonance imaging (MRI), electroencephalogram (EEG), abnormal brainstem auditory evoked potential (BAEP), and visual evoked potential (VEP) results, respectively. Affected children displayed multiple clinical manifestations with 64.89% of patients displaying ID, 52.44% with speech delay, 46.22% with motor delay, 31.56% with hearing loss, 15.11% visual loss, 19.56% with seizures, and 12.44% facial dysmorphism. Based on clinical phenotypes, children were classified into two main groups: an isolated DD/ID group (n = 80, 35.56%) and a syndromic DD/ID group (n = 145, 64.44%). Furthermore, children in the latter group were further divided into six subgroups: (1) DD/ID with hearing loss (n = 71, 48.97%); (2) DD/ID with malformations (n = 54, 37.24%); (3) DD/ID with epilepsy (n = 44, 30.34%); (4) DD/ID with visual loss (n = 34, 23.45%); (5) DD/ID with behavioural troubles (n = 16, 11.03%); and (6) DD/ID with metabolic disorders (n = 7, 4.83%). Since an affected child may have multiple clinical manifestations, the same child may be classified in different subgroups. Figure 2a shows the detailed information on patient classification among six subgroups of syndromic DD/ID.

Diagnostic rate of WES. (a) Patient classification among six subgroups of syndromic DD/ID. (b) Patient with diagnostic SNV/Indels in these six subgroups of syndromic DD/ID. The six colored irregular graphics in (a) and (b) represent the six subgroups of syndromic DD/ID: brown for DD/ID with hearing loss, pink for DD/ID with malformations, blue for DD/ID with epilepsy, orange for DD/ID with visual loss, yellow for DD/ID with behavioural troubles and purple for DD/ID with metabolic disorders. Overlap between the different irregular graphics shows the overlap of patients among these subgroups. (c) Hearing loss, visual loss, facial dysmorphism, and metabolic disorders were analyzed in the logistic regression model. (d) Abnormal BAEP, visual loss, facial dysmorphism, and metabolic disorders were analyzed in the logistic regression model. DD, developmental disorder; ID, intellectual disability; BAEP, brainstem auditory evoked potential; 95% CI, 95% confidence interval; OR, odds ratio
Diagnostic rate of WES
As shown in Table 1, an overall diagnostic yield of 42.67% (96/225, 95% CI: 36.15-49.18%) was achieved, and there was no significant difference in different age groups (P = 0.536), gender groups (P = 0.548), or family history (P = 0.442). In the isolated DD/ID group, the yield was 40.00% (32/80, 95% CI: 29.03-50.97%), close to that of syndromic DD/ID group (64/145, 95% CI: 35.96-52.32%), with no significant difference (P = 0.548). And in the six phenotype subgroups of syndromic DD/ID group, there were no significant difference from one another (P = 0.221), although the diagnostic yield ranged from 31.25 to 71.43%. Figure 2b shows the details of children with diagnostic SNV/Indels in these six subgroups of syndromic DD/ID.
Furthermore, the diagnostic yield varied in different clinical features (Table 1), hearing loss, visual loss and facial dysmorphism can raise the diagnostic yield, and these three effects were all statistically significant (P = 0.005, P = 0.005, and P = 0.039, respectively). As for clinical examinations, none of these had statistical significance, except for abnormal BAEP (P = 0.030), which indicated that BAEP evaluation can help identify causative genetic variants in DD/ID children. Of note, children with hearing loss were mostly identified by BAEP signals (69/71), and VIF between abnormal BAEP and hearing loss was 24.61, thus, abnormal BAEP and hearing loss were separately included in a multivariate logistic regression analysis.
In the logistic regression model, hearing loss/abnormal BAEP, visual loss, facial dysmorphism, and metabolic disorders were analyzed (Fig. 2c, d). Hearing loss (OR = 1.86, 95%CI: 1.01–3.46, P = 0.046) or abnormal BAEP (OR = 1.91, 95%CI: 1.02–3.56, P = 0.042) was independently associated with causative genetic variations (Fig. 2c, d).
These findings confirmed our hypothesis that systematic clinical phenotyping of DD/ID is important for increasing the diagnostic yield of WES, and we should emphasize the great value of physiological, biochemical and genetic tests in the diagnosis of DD/ID.
Inheritance patterns among diagnostic SNVs/Indels
In the cohort, 108 diagnostic SNVs/Indels were found in 96 patients with variants spanning 81 genes. The detailed diagnostic SNVs/Indels are shown in Supplementary Table 2. The inheritance patterns in 100 conditions (96 cases, two cases with two conditions caused by P/LP variants in different genes, one case with three conditions caused by P/LP variants in different genes) were identified, including 61.00% were autosomal dominant and de novo, 11.00% were X-linked and inherited, 9.00% were autosomal dominant and inherited, 8.00% were autosomal recessive, 7.00% were X-linked and de novo, 3.00% were indeterminately autosomal dominant or recessive, and 1.00% were autosomal dominant of unknown origin due to lack of parental samples (Fig. 3a).

Diagnostic SNVs/Indels were identified in our cohort. (a) Inheritance patterns among diagnostic SNVs/Indels. (b) Mutation types among diagnostic SNVs/Indels. (c) Heatmap of identified causative genes with diagnostic SNVs/Indels among phenotype groups. Genes appeared in ≥ 2 patients are displayed. The color of each cell represents the number of patients diagnosed by the specific gene (row) in the relevant phenotype group (column). (d) Distribution of diagnostic genes in different clinical features. The three circles represent the three clinical features: yellow for hearing loss, purple for visual loss, and pink for facial dysmorphism. Overlap between the different circles shows the overlap of genes among these clinical features. DD, developmental disorder; ID, intellectual disability; SNVs, single nucleotide variations; Indels, small insertions/deletions
Mutation types among the 108 diagnostic SNVs/Indels
Among the 108 diagnostic SNVs/Indels, 53 (49.07%) were missense variants, 34 (31.48%) were frameshift variants, 13 (12.04%) were stop-gained variants, 7 (6.48%) were predicted splice variants, and 1 (0.93%) was inframe deletion (Fig. 3b). Half of the 108 mutant alleles were truncating (nonsense, splicing, or frameshift) and others were nontruncating (missense or in-frame deletions).
Genes with diagnostic variants within multiple patients
Mutations were identified in 81 different genes, 14 (17.28%) of which were identified in two or more patients (Supplementary Table 2). Mutations in two genes (ASXL3 and UBE3A) were each found in four patients, mutations in MECP2 were found in three patients, and mutations in 11 genes (MMACHC, VPS13B, ARID1B, BRPF1, CASK, FOXP1, HNRNPH2, IQSEC2, KCNQ2, KIDINS220, and KMT2D) were each found in two patients (Supplementary Table 2). Furthermore, among these 14 genes, seven genes influenced the isolated DD/ID group, HNRNPH2 and IQSEC2 were only detected in this group (Fig. 3c). Meanwhile, FOXP1 was only detected in DD/ID with malformation group, and ASXL3 was most frequently involved in DD/ID with hearing loss group (Fig. 3c).
Figure 3d shows the diagnostic genes identified in patients with hearing loss, visual loss and/or facial dysmorphism. Forty-one genes were involved, and six genes: MUC6, KMT2D, CHD7, BCL11B, SMC1A, and ASXL3, were detected in all three clinical manifestations (Fig. 3d).
Multiple gene findings in one patient
Although cases with multiple genetic diagnoses are rare, in this study, two cases revealed two genetic diagnoses, and one case had three distinct genetic diagnoses (Supplementary Table 3). Both (or all three) gene findings can explain the major or all nonoverlapping/overlapping clinical features. For example, one patient with DD and hypotonia had a de novo pathogenic variant in three genes: PRDM16, SETD2, and KRT9. One patient with epilepsy and ID had a de novo missense pathogenic variant in EIF4G1 and HSPB1.
Impact of WES on medical management
Supplementary Table 4 showed that WES results changed medical management and impacted family planning for DD/ID children (45/96), including reproductive decision changes (n = 22), initiation of disease monitoring/systemic investigation (n = 5), discontinuation medication (n = 6), addition of medication (n = 30), physiotherapy/psychosocial support (n = 12), and ending a diagnostic “odyssey” (n = 6). For example, a compound heterozygous pathogenic variant in MMACHC was detected in a child (Patient 210) with DD, seizures, methylmalonic acidaemia and homocysteinaemia that was treatable with vitamin B12, betaine, folate, and levetiracetam. Hypermethioninemia due to MAT1A mutation was diagnosed in a child (Patient 156) with DD, who was subsequently treated with a methionine-restricted diet in combination with rehabilitation treatment after diagnosis and experienced significant growth improvement. A child with DD (Patient 119) who harboured anSTXBP1 variant had been empirically started on phenobarbital to stop the seizures but showed no response; after molecular diagnosis, levetiracetam was added to the treatment protocol, which was successful, and the patient was seizure free for years. In summary, the data emphasize the significant implications of genetic diagnosis established by WES for patients and their families.
Discussion
In this study, WES was performed for 225 Chinese children with DD/ID, an overall diagnostic yield of 42.67% was achieved. 108 diagnostic SNVs/Indels in 81 genes were found in the cohort, most of which were de novo and protein altering. Hearing loss, visual loss, and facial dysmorphism had significant effect on diagnostic yield. Of note, hearing loss or abnormal BAEP was more likely to have causative genetic mutations.
In heterogeneous populations, the diagnostic yields of DD/ID with WES vary widely depending on the sample size of the study [26,27,28]. In a study of 38 patients with ID and microcephaly, a positive diagnosis was revealed by WES in 29% (11/38) [27]. In a cohort of 232 children with DD/ID, WES identified P/LP SNVs in 39% of the patients (91/232) [28]. In our paediatric DD/ID cohort, the diagnostic yield of WES was relatively high at 42.67%, which was partly due to the following reasons: (1) There was a high rate of trio sequencing in our cohort (208/225, 92.44%), which numerous studies have proven that trio sequencing can increase the diagnostic yield [29]. (2) Subjects in our cohort may exhibit selection bias. Our paediatric clinic is one of the top paediatric clinics in China, and the most severely affected children are referred to the top clinics for diagnosis and management. Thus, the children seen in our clinic had a relatively high rate of dysmorphism and/or multiple organ system abnormalities. Given that specific clinical features can increase the diagnostic yield of WES [4, 16], the comparatively severe clinical features of the children in our study may have increased the diagnostic yield of WES; furthermore, these children had undergone extensive prior routine physiological and biochemical testing, and WES was conducted due to a high suspicion that the child had a genetic disorder [15].
In this study, we found that the diagnostic yields for isolated and syndromic DD/ID were equivalent, which was consistent with previous reports [1, 4, 16, 17]. Dong XR et al. found that although there was no significant difference between isolated and syndromic DD, there were significant differences among the four subgroups of syndromic DD. The diagnostic rate of DD in the behavioural troubles subgroup was significantly lower than that in the other three subgroups (i.e., DD with malformation, DD with epilepsy, and DD with metabolic disorder), and these three subgroups were not significantly different from one another [1]. Conversely, in our study, we found that there were no significant differences among the six subgroups of syndromic DD/ID, which can be partially attributed to the smaller sample sizes of some subgroups, such as the DD/ID with behavioural troubles group (n = 16), and the DD/ID with metabolic disorder group (n = 7), which may have resulted in underpowered statistical tests. It is worth noting that although the numbers of patients in our study with some specific clinical features were still modest, our study demonstrated a diagnostic yield of at least 30% for these clinical features (Table 1), which supported the powerful and valuable effects of WES in identifying the genetic aetiology of DD/ID. Furthermore, we found that hearing loss, visual loss, and facial dysmorphism significantly increased the diagnostic yield of WES in patients with DD/ID; notably, hearing loss and abnormal BAEP were independently associated with causative genetic variations, which further confirmed that specific clinical features can significantly increase genetic diagnostic yields in DD/ID and emphasized the importance of routine physiological tests in genetic aetiology analysis in DD/ID patients.
Hearing loss is one of the common specific impairments that were modeled as sequelae of specific health disorders of children [12, 30, 31], and it is also a common clinical feature in DD/ID patients [31,32,33]. And early detection of hearing loss is vital to language development [34, 35]. But few studies have tested the relationship between hearing loss and the diagnostic rate of DD/ID. Hearing evaluation through subjective tests is difficult in young and uncooperative children, BAEP is reliable and effective tool in this setting [35,36,37]. Lau WL et al. had found the prevalence of hearing deficit in children with Down syndrome in Hong Kong was 36% (18/55) measured by BAEP [35]. BAEP is also used to assess neuronal maturation [37, 38]. In our study, 69 children (97.18%) were identified having hearing loss by BAEP and most children had a mild bilateral lesion (Supplementary Table 5). Furthermore, abnormal BAEP was independently associated with causative genetic variations, which suggested BAEP screen should be encouraged in DD/ID children.
There were 39 genes were identified in DD/ID children with abnormal BAEP in the cohort (Supplementary Table 6), most genes were associated with neurodevelopmental disorders such as DD, ID, epilepsy, and ear/hearing anomalies. And their biological processes are mainly related with nervous system development, positive regulation of cellular biosynthetic process, generation of neurons, brain development, neurogenesis, single-multicellular organism process, sensory perception of sound, inner ear morphogenesis (Supplementary Table 6). These findings suggested a need for detailed research on these genes in future.
Importantly, positive genetic results can not only end a diagnostic “odyssey”, but also beneficially influence medical care and reproductive decision [39,40,41], which were also observed in our studies (Supplementary Table 4). A random-effects meta-analysis showed that genetic results changed clinical management (range: 2–49%, n = 6 studies) and impacted reproductive planning (range: 42–100%, n = 4 studies) for patients with neurodevelopmental disorders (ID/DD, and/or ASD) [18]. These data strongly indicate the extremely beneficial of WES in early diagnose and personalized treatment of DD/ID, as well as in genetic counseling for DD/ID patients and families.
Although WES should be considered in the early stage of the diagnosis process, physicians should not ignore the importance of routine physiological and biochemical tests, since these examinations often substantiate the genetic testing results. In our study, specific clinical features (such as hearing loss, visual loss, and facial dysmorphism) significantly increased the diagnostic yield of WES in patients with DD/ID. Moreover, in our study, we found that hearing loss and abnormal BAEP were more likely to have causative genetic mutations. Given that early diagnosis of hearing loss and hearing rehabilitation promote language, academic and social development [42,43,44], hearing impairment/BAEP tests should be conducted as part of newborn screening, as well as in evaluations of DD/ID children. Early intervention and treatment based on physiological, metabolic and genetic findings can lead to better prognoses, even preventing the development of DD/ID. For example, in a study of 149 Chinese patient with cobalamin C deficiency harbouring the MMACHC c.609G > A homologous mutation, 1.3% (2/149) were prenatally diagnosed with metabolic and genetic tests, treated after birth and showed normal development; 10.1% (15/149) were diagnosed by newborn screening(10 children were treated at 15 days of age and showed normal development, while the other five children were treated after onset and all developed severe DD because of poor treatment compliance); and 88.6% (132/149) were diagnosed after onset and received personalized treatment, with various neurological sequelae (including DD, seizure, etc.) observed although most patients improved [45]. Consistent with these results, in our study, we identified a genetic aetiology in two DD/ID children (Patient 210, Patient 156) by metabolic and WES tests, and early intervention/treatment was applied; one patient showed normal development and the other patient showed significant improvement. Taken together, these data strongly indicate that physiological, metabolic, and genetic screening and early personalized treatment are pivotal for preventing DD/ID.
With the advancement of genomic technology, genetic findings in research concerning individual health are an ethical challenge and concern [15]: (1) Since children cannot legally sign informed consent on their own behalf, the parents/guardians ultimately made the final decision in this cohort, which was consistent with other studies [46]. Ross LF et al. reported that hindering children’s involvement increased the risk that medical professionals and parents would lose the children’s trust if they believed that they had no right to express their feelings and suggested that minors should be able to make informed decisions regarding their genomic evaluation [47]. (2) In this study, laboratories classified variants as P, LP, and/or VUS. After reviewing the data, we returned all genomic results to the parents/guardians, including “incidental” or “secondary” findings, which were unrelated to the reason for ordering WES but may have future medical implications [15]. Notably, we focused more on medically “actionable” findings (e.g., the availability of relevant targeted therapies or relevant risk reduction interventions) and avoided overinterpreting the clinical significance of VUSs. (3) We were bound to provide recommendations, but we respected the parents’/guardians’ beliefs, feelings, religion, and cultural traditions; autonomous decisions (e.g. therapy, reproductive planning, and follow-up assessment.) should be made by parents/guardians, although genetic results may have the potential to improve a patient’s health through effective medical intervention, or to impact family planning [48,49,50]. (4) Although we encouraged more family members to participate in WES testing to explore genetic aetiology [25, 48], we did not disclose the WES results to other family members without permission.
This the study has several limitations: (1) WES will not reliably detect large deletion/insertion, translocation/transversion, mitochondrial DNA, epigenic variants, or nonexonic regulatory regions, which could be caused DD/ID [4, 15, 51]. (2) There was potential selection bias for this single-center study. Some children with DD/ID did not been recruited in this cohort for various reasons (e.g. parents/guardians refused to perform WES or refused to sign informed consents, incomplete medical records, etc.). (3) WES was performed through three different WES laboratories that use their own bioinformatics pipelines [15, 16]. However, these three laboratories used the same variant-level classification according to the recommendations of the ACMG; there were no significant differences in the diagnostic yields among them (Supplementary Table 7); and all genetic results were reviewed by the ordering physicians. (4) The clinical phenotype of a child may be the result of interactions of different genes and/or environmental factors [15]. (5) Although clinical management can be guided by genetic results, cases with significantly improved effectiveness were still relatively rare (n = 12) compared with the number of DD/ID children (n = 225) (Supplementary Table 4). There are large gaps in the current knowledge on personalized genomic treatment in DD/ID patients, which underscores the importance of collaboration between genetic researchers and clinical physicians (including paediatricians, paediatric rehabilitation specialists, and paediatric neurologists) to accelerate basic and clinical research.
Conclusion
In conclusion, our study identified genetic etiology in 42.67% of patients with DD/ID in Beijing, China, which supported that the powerful and valuable effects of WES in identifying the genetic etiology of DD/ID. Given that abnormal BAEP is independently associated with causative genetic variations, there is a need for the development of BAEP screen in DD/ID children. Despite present limitations, WES still serves as a critical tool in pediatric neurology practices.
Data availability
These sequence data have been submitted to the NCBI Sequence Read Archive (SRA) under accession number: PRJNA1067565 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1067565).
Abbreviations
- DD:
-
Developmental Delay
- ID:
-
Intellectual Disability
- WES:
-
Whole-exome Sequencing
- SNVs:
-
Single Nucleotide Variants
- BAEP:
-
Brainstem Auditory Evoked Potential
- ACMG:
-
American College of Medical Genetics and Genomics
- DNA:
-
Deoxyribonucleic Acid
- P:
-
Pathogenic
- LP:
-
Likely Pathogenic
- VUS:
-
Uncertain Significance
- LB:
-
Likely Benign
- B:
-
Benign
- CI:
-
Confidence Interval
- VIF:
-
Variance inflation factor
- ORs:
-
Odds Ratios
- MRI:
-
Magnetic Resonance Imaging
- EEG:
-
Electroencephalogram
- VEP:
-
Visual Evoked Potential
References
Dong X, Liu B, Yang L, Wang H, Wu B, Liu R, Chen H, Chen X, Yu S, Chen B, et al. Clinical exome sequencing as the first-tier test for diagnosing developmental disorders covering both CNV and SNV: a Chinese cohort. J Med Genet. 2020;57(8):558–66.
Chen Y, Tang X, Liu L, Huang Q, Lin L, Liu G, Xiao N. Comprehensive genome sequencing analyses identify novel gene mutations and copy number variations associated with infant developmental delay or intellectual disability (DD/ID). Genes Dis. 2022;9(5):1166–9.
Carapito R, Ivanova EL, Morlon A, Meng L, Molitor A, Erdmann E, Kieffer B, Pichot A, Naegely L, Kolmer A, et al. ZMIZ1 variants cause a syndromic neurodevelopmental disorder. Am J Hum Genet. 2019;104(2):319–30.
AlMutiri R, Malta M, Shevell MI, Srour M. Evaluation of individuals with Non-syndromic Global Developmental Delay and Intellectual Disability. Child (Basel) 2023, 10(3).
Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, Cochran JN, Brothers KB, East KM, Gray DE, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9(1):43.
Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. 2011;32(2):419–36.
Froukh TJ. Next generation sequencing and genome-wide genotyping identify the Genetic Causes of Intellectual Disability in ten Consanguineous families from Jordan. Tohoku J Exp Med. 2017;243(4):297–309.
Miclea D, Osan S, Bucerzan S, Stefan D, Popp R, Mager M, Puiu M, Zimbru C, Chirita-Emandi A, Alkhzouz C. Copy number variation analysis in 189 Romanian patients with global developmental delay/intellectual disability. Ital J Pediatr. 2022;48(1):207.
Li Y, Anderson LA, Ginns EI, Devlin JJ. Cost effectiveness of Karyotyping, Chromosomal Microarray Analysis, and targeted next-generation sequencing of patients with unexplained Global Developmental Delay or Intellectual Disability. Mol Diagn Ther. 2018;22(1):129–38.
Gurkan H, Atli EI, Atli E, Bozatli L, Altay MA, Yalcintepe S, Ozen Y, Eker D, Akurut C, Demir S, et al. Chromosomal Microarray Analysis in Turkish patients with unexplained Developmental Delay and Intellectual Developmental disorders. Noro Psikiyatr Ars. 2020;57(3):177–91.
Wan RP, Liu ZG, Huang XF, Kwan P, Li YP, Qu XC, Ye XG, Chen FY, Zhang DW, He MF, et al. YWHAZ variation causes intellectual disability and global developmental delay with brain malformation. Hum Mol Genet. 2023;32(3):462–72.
Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, Massingham LJ, Miller D, Yu TW, Hisama FM, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(11):2029–37.
Xiang J, Ding Y, Yang F, Gao A, Zhang W, Tang H, Mao J, He Q, Zhang Q, Wang T. Genetic analysis of Children with Unexplained Developmental Delay and/or intellectual disability by whole-exome sequencing. Front Genet. 2021;12:738561.
van Slobbe M, van Haeringen A, Vissers L, Bijlsma EK, Rutten JW, Suerink M, Nibbeling EAR, Ruivenkamp CAL, Koene S. Reanalysis of whole-exome sequencing (WES) data of children with neurodevelopmental disorders in a standard patient care context. Eur J Pediatr 2023.
Srivastava S, Cohen JS, Vernon H, Baranano K, McClellan R, Jamal L, Naidu S, Fatemi A. Clinical whole exome sequencing in child neurology practice. Ann Neurol. 2014;76(4):473–83.
Baldridge D, Heeley J, Vineyard M, Manwaring L, Toler TL, Fassi E, Fiala E, Brown S, Goss CW, Willing M, et al. The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet Med. 2017;19(9):1040–8.
Gieldon L, Mackenroth L, Kahlert AK, Lemke JR, Porrmann J, Schallner J, von der Hagen M, Markus S, Weidensee S, Novotna B et al. Diagnostic value of partial exome sequencing in developmental disorders. PLoS ONE 2018, 13(8).
Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, Firth HV, Frazier T, Hansen RL, Prock L, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21(11):2413–21.
Wang J, Wang Y, Wang L, Chen WY, Sheng M. The diagnostic yield of intellectual disability: combined whole genome low-coverage sequencing and medical exome sequencing. BMC Med Genomics. 2020;13(1):70.
Gu Y, Xiang B, Zhu L, Ma X, Chen X, Cai T. Three intellectual disability-associated de novo mutations in MECP2 identified by trio-WES analysis. BMC Med Genet. 2020;21(1):99.
Wang X, Shen X, Fang F, Ding CH, Zhang H, Cao ZH, An DY. Phenotype-driven virtual panel is an effective method to analyze WES Data of Neurological Disease. Front Pharmacol. 2018;9:1529.
Cai T, Yang L, Cai W, Guo S, Yu P, Li J, Hu X, Yan M, Shao Q, Jin Y, et al. Dysplastic spondylolysis is caused by mutations in the diastrophic dysplasia sulfate transporter gene. Proc Natl Acad Sci U S A. 2015;112(26):8064–9.
Guo S, Yang L, Liu H, Chen W, Li J, Yu P, Sun ZS, Chen X, Du J, Cai T. Identification of novel compound mutations in PLA2G6-Associated Neurodegeneration patient with characteristic MRI imaging. Mol Neurobiol. 2017;54(6):4636–43.
Wang T, Wang J, Ma Y, Zhou H, Ding D, Li C, Du X, Jiang YH, Wang Y, Long S, et al. High genetic burden in 163 Chinese children with status epilepticus. Seizure. 2021;84:40–6.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–9.
Rump P, Jazayeri O, van Dijk-Bos KK, Johansson LF, van Essen AJ, Verheij JB, Veenstra-Knol HE, Redeker EJ, Mannens MM, Swertz MA, et al. Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly. Bmc Med Genomics. 2016;9:7.
Anazi S, Maddirevula S, Faqeih E, Alsedairy H, Alzahrani F, Shamseldin HE, Patel N, Hashem M, Ibrahim N, Abdulwahab F, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2017;22(4):615–24.
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704.
Zablotsky B, Black LI, Maenner MJ, Schieve LA, Danielson ML, Bitsko RH, Blumberg SJ, Kogan MD, Boyle CA. Prevalence and Trends of Developmental Disabilities among children in the United States: 2009–2017. Pediatrics 2019, 144(4).
Olusanya BO, Wright SM, Nair MKC, Boo NY, Halpern R, Kuper H, Abubakar AA, Almasri NA, Arabloo J, Arora NK et al. Global Burden of Childhood Epilepsy, Intellectual disability, and sensory impairments. Pediatrics 2020, 146(1).
Weiss K, Lazar HP, Kurolap A, Martinez AF, Paperna T, Cohen L, Smeland MF, Whalen S, Heide S, Keren B, et al. The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis. Genet Med. 2020;22(2):389–97.
Richard EM, Bakhtiari S, Marsh APL, Kaiyrzhanov R, Wagner M, Shetty S, Pagnozzi A, Nordlie SM, Guida BS, Cornejo P, et al. Bi-allelic variants in SPATA5L1 lead to intellectual disability, spastic-dystonic cerebral palsy, epilepsy, and hearing loss. Am J Hum Genet. 2021;108(10):2006–16.
Shott SR, Joseph A, Heithaus D. Hearing loss in children with Down syndrome. Int J Pediatr Otorhinolaryngol. 2001;61(3):199–205.
Lau WL, Ko CH, Cheng WW. Prevalence and parental awareness of hearing loss in children with Down syndrome. Chin Med J (Engl). 2015;128(8):1091–5.
Kazan HM, Samelli AG, Neves-Lobo IF, Magliaro FC, Limongi SC, Matas CG. Electrophysiological characterization of hearing in individuals with Down syndrome. Codas. 2016;28(6):717–23.
Wang X, Carroll X, Wang H, Zhang P, Selvaraj JN, Leeper-Woodford S. Prediction of delayed neurodevelopment in infants using Brainstem Auditory Evoked potentials and the Bayley II scales. Front Pediatr. 2020;8:485.
Kassis I, Bero D, Hafner H, Chistyakov A, Meir M. Brainstem auditory pathway maturation in term neonates with congenital cytomegalovirus infection: a cohort study. Eur J Pediatr. 2023;182(1):95–100.
Nambot S, Thevenon J, Kuentz P, Duffourd Y, Tisserant E, Bruel AL, Mosca-Boidron AL, Masurel-Paulet A, Lehalle D, Jean-Marcais N, et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet Med. 2018;20(6):645–54.
Mahfouz NA, Kizhakkedath P, Ibrahim A, El Naofal M, Ramaswamy S, Harilal D, Qutub Y, Uddin M, Taylor A, Alloub Z, et al. Utility of clinical exome sequencing in a complex Emirati pediatric cohort. Comput Struct Biotechnol J. 2020;18:1020–7.
Evers C, Staufner C, Granzow M, Paramasivam N, Hinderhofer K, Kaufmann L, Fischer C, Thiel C, Opladen T, Kotzaeridou U, et al. Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Mol Genet Metab. 2017;121(4):297–307.
Priner R, Brand D. The complexity of hearing Aid Fitting: children with Congenital Hearing Loss and middle ear dysfunction. Child (Basel) 2023, 10(10).
Yoshinaga-Itano C, Sedey AL, Mason CA, Wiggin M, Chung W, Pediatrics. 2020, 146(Suppl 3):S270–7.
Tomblin JB, Oleson JJ, Ambrose SE, Walker E, Moeller MP. The influence of hearing aids on the speech and language development of children with hearing loss. JAMA Otolaryngol Head Neck Surg. 2014;140(5):403–9.
He R, Mo R, Shen M, Kang L, Song J, Liu Y, Chen Z, Zhang H, Yao H, Liu Y, et al. Variable phenotypes and outcomes associated with the MMACHC c.609G > A homologous mutation: long term follow-up in a large cohort of cases. Orphanet J Rare Dis. 2020;15(1):200.
Vickers RR, Gibson JS. A review of the Genomic Analysis of Children Presenting with Developmental Delay/Intellectual Disability and Associated Dysmorphic Features. Cureus. 2019;11(1):e3873.
Ross LF, Saal HM, David KL, Anderson RR, American College of Medical G. Genomics: technical report: ethical and policy issues in genetic testing and screening of children. Genet Med. 2013;15(3):234–45.
Jamal L, Schupmann W, Berkman BE. An ethical framework for genetic counseling in the genomic era. J Genet Couns. 2020;29(5):718–27.
Fine B. The Evolution of Nondirectiveness in Genetic Counseling and Implications of the Human Genome Project,[w:] DM Bartels, BS LeRoy, AL Caplan (red.), Prescribing our Future. Ethical Challenges in Genetic Counseling. In.: New York: Aldine de Gruyter; 1993.
Stern AM. A quiet revolution: the birth of the genetic counselor at Sarah Lawrence College, 1969. J Genet Couns. 2009;18(1):1–11.
Corominas J, Smeekens SP, Nelen MR, Yntema HG, Kamsteeg EJ, Pfundt R, Gilissen C. Clinical exome sequencing-mistakes and caveats. Hum Mutat. 2022;43(8):1041–55.
Funding
This study was funded by the National Natural Science Foundation of China (82160620); Natural Science Foundation of Guangxi Province (2023GXNSFAA026036); National College Students Innovation and Entrepreneurship Training Program (S202310601164).
Author information
Authors and Affiliations
Contributions
X.M. and X.Z. were involved in the conception and design of the study. H.M., L.Z. and X.Y. performed WES analysis and interpretation, wrote the original draft. H.M., L.Z., X.Y., and S.Z. performed public databases analysis. M.A., S.Z., M.G. performed WES analysis and drafted the figures. L.Z., X.Y. and X.M. collected and analyzed clinical data for the study. H.M., X.M., X.Z. reviewed clinical data. H.M., S.Z., M.G., and X.D. performed sequencing data analysis and prepared the tables. X.Z. substantively revised the draft. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was performed in line with the principles of the Declaration of Helsinki. Informed consents had been obtained from the children’s parents and/or legal guardians. The study did not contain information or images that could lead to identification of a study participant. Approval was granted by the Ethics Committee of PLA General Hospital (No. S2016-120-02).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Acknowledgments.
We thank the patients and their families for agreeing to participate in this study.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, H., Zhu, L., Yang, X. et al. Genetic and phenotypic analysis of 225 Chinese children with developmental delay and/or intellectual disability using whole-exome sequencing. BMC Genomics 25, 391 (2024). https://doi.org/10.1186/s12864-024-10279-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10279-1