
Abstract
Background
Rye (Secale cereale), one of the drought and cold-tolerant crops, is an important component of the Triticae Dumortier family of Gramineae plants. Basic helix-loop-helix (bHLH), an important family of transcription factors, has played pivotal roles in regulating numerous intriguing biological processes in plant development and abiotic stress responses. However, no systemic analysis of the bHLH transcription factor family has yet been reported in rye.
Results
In this study, 220 bHLH genes in S. cereale (ScbHLHs) were identified and named based on the chromosomal location. The evolutionary relationships, classifications, gene structures, motif compositions, chromosome localization, and gene replication events in these ScbHLH genes are systematically analyzed. These 220 ScbHLH members are divided into 21 subfamilies and one unclassified gene. Throughout evolution, the subfamilies 5, 9, and 18 may have experienced stronger expansion. The segmental duplications may have contributed significantly to the expansion of the bHLH family. To systematically analyze the evolutionary relationships of the bHLH family in different plants, we constructed six comparative genomic maps of homologous genes between rye and different representative monocotyledonous and dicotyledonous plants. Finally, the gene expression response characteristics of 22 ScbHLH genes in various biological processes and stress responses were analyzed. Some candidate genes, such as ScbHLH11, ScbHLH48, and ScbHLH172, related to tissue developments and environmental stresses were screened.
Conclusions
The results indicate that these ScbHLH genes exhibit characteristic expression in different tissues, grain development stages, and stress treatments. These findings provided a basis for a comprehensive understanding of the bHLH family in rye.
Similar content being viewed by others

Background
The basic helix-loop-helix (bHLH) transcription factor family is the second largest transcription factor family after MYB in plants, which is widely present in eukaryotes [1]. The bHLH transcription factor was first discovered in animals [2]. The molecular architecture of the Lc gene within the maize R locus, responsible for encoding proteins comprising 610 amino acids, was elucidated through cDNA sequencing. This intricate mechanism governs the biosynthesis of anthocyanins, representing the first reported instance of a plant-based bHLH gene [3]. The domain of the bHLH protein consists of two parts with different functions, namely the basic region and the helix-loop-helix region (HLH) [4, 5]. Among them, the basic domain is located in the N-terminal of bHLH proteins, with about 15 ~ 20 amino acids. This region is diversified in general, except for some amino acid residue sites. These conserved amino acid residues, namely E9 and R13, play a crucial role in conferring the binding capacity of bHLH proteins to specific cis-elements such as E-box (CANNTG) and G-box (CACGTG) elements [6, 7]. Moreover, the HLH region, comprising approximately 40 amino acids, is situated at the C-terminal of the bHLH proteins. The HLH region consists of two alpha helixes and a connecting region (loop) of variable length. Five amino acid residues in the HLH region (Leu23/64, Leu/Iso54, Val61) and two amino acid residues in the ring region (Lys/Arg47) are highly conserved in the plant bHLH genes [8,9,10,11,12]. Most bHLH proteins perform their biological functions by forming dimers. For example, Brownlie et al. [13] reported that homodimers and heterodimers of bHLH proteins can compete for a common DNA target site (E-box), where Leu23/54 in the HLH region is an essential amino acid residue for dimer formation. Ciarapica et al. [14] suggested that the dimer function of plant bHLH proteins were determined by the formation of molecular scaffolds, affinity interfaces between key amino acids involved in dimer molecular recognition in conserved HLH domains, and their interactions with chaperone proteins.
In animals, bHLH proteins are classified into six major groups (from A to F), according to their DNA-binding specificities, sequence homology, and structural features, which are in turn divided into smaller subfamilies. Each group contains subfamilies of bHLH proteins that have highly conserved DNA-binding and protein-protein interaction domains [15, 16]. At present, the classification of bHLH transcription factors in different plants is still uncertain. Pires and Dolan [17] used transcription factor sequences of the bHLH proteins of nine terrestrial plant and algal species to classify their evolutionary relationships, dividing these members into 26 subfamilies. Among them, 23 subfamilies contained the bHLH proteins of both plants, which were lycopods and angiosperms. Twenty subfamilies were identified as descendants of the common ancestor that gave rise to both mosses and vascular plants. On the other hand, six subfamilies within the vascular plants’ group do not include any mosses. These six subfamilies have continued to evolve and diversify since their separation from the ancestral group that included both mosses and vascular plants. In model plants, phylogenetic analysis of 147 bHLH genes divided these members into 21 subfamilies in Arabidopsis thaliana [11]. The 167 OsbHLH TFs have been divided into 22 subfamilies in rice [7]. The bHLH genes have been identified in many plants, including Brassica rapa subsp. pekinensis [18], Solanum lycopersicum [19], Phaseolus vulgaris [20], Malus domestica [21], Arachis hypogaea [22], Brachypodium distachyon [23], Zea mays [24], Triticum aestivum [25], Phyllostachys edulis [26], Ziziphus jujuba [27], Capsicum annuum [28], Panax ginseng [29], Ananas comosus [30], Fagopyrum tataricum [31], Sorghum bicolor [32], Setaria italica [33], Dactylis glomerata [34], Brassica oleracea var. botrytis [35], and Jatropha curcas [36]. These bHLH genes are mainly involved in the defense response of plants to abiotic stresses such as high temperature, cold, drought, and elevated salinity. Most of these stresses are unique to terrestrial plants. Therefore, genome-wide analysis of the bHLH family will provide a useful aid to comprehend the adaptation and evolution of different plants in their environments.
The bHLH gene family is known to exist in organisms for a very long time. As early as 415 million years ago, bHLH transcription factors were present in the ancestors of angiosperms [37, 38]. Pires et al. [17] reported that these transcription factors evolved to include a series of highly conserved short amino acid prescriptions that were present in plants that existed 400 million years ago. However, the evolution of ancestral bHLH members into different subfamilies and the gene expansion induced by structural variation in different subfamilies is still unclear, which is speculated to be related to repetitive events, including segment and tandem duplications [31, 32]. The bHLH family of plants with monocotyledon amplifies members in a monophyletic manner, and this relationship may be established after its separation from the bHLH members of dicotyledon, several types of ancestors did not converge or evolve in parallel. Nevertheless, the origin of the affinity observed in the bHLH members of Triticeae Dumortier may be attributed to evolutionary clustering within the Gramineae family [39]. The interplay and evolutionary relationship between these members and other biological bHLH proteins remain shrouded in ambiguity. Therefore, examining the homologous bHLH genes in many Triticeae Dumortier plants is significant to analyze the relatives of members of different subfamilies and sort out the evolution and derivation of these bHLH proteins.
bHLH transcription factors play important roles in various metabolic and developmental processes in plants, such as light response, hormone signaling, fruit and seed development, and plant morphological development. For example, SPATULA was the first bHLH gene found in A. thaliana to regulate the development of carpel margin and inner pollen tissue, and also redundantly regulate the growth of stem tip meristem, leaves, stamens, and roots [40, 41]. The LAX gene, a bHLH member, can regulate the formation of the axillary bud primordium in rice [42]. AtRGE1 (RETARDED GROWTH OF EMBRY01) is specifically expressed in the endosperm during embryonic development and controls seed growth in A. thaliana [43]. SPEECHLESS and two homologous genes are required for the initiation, proliferation, and terminal differentiation of cells in the stomatal lineage, ultimately leading to the formation of functional stomata on the leaf surface [44]. TabHLH123 interacts with TaMOR, an essential regulator of crown root initiation, to regulate stem base and root development of wheat, and participate in plant height building and auxin metabolism [45]. OsbHLH98 exerts negative regulation on the leaf angle by modulating the quantity and dimensions of paraxial parenchyma cells at the leaf junction. Its involvement extends to plant morphogenesis and photosynthesis, presenting a multifaceted role in plant biology [46]. bHLH is also involved in response to various hormones. JAZ protein, bHLH (TTG8, GL3), and R2R3-MYB transcription factors (MYB75 and Glabra1) jointly formed WD-repeat/bHLH/MYB transcription complex, which regulated anthocyanin accumulation by inhibiting jasmonic acid content [47]. CESTA and its homologs induce the expression of the gibberellin 2-oxidase gene GA2ox7 in A. thaliana are enhanced by brassinosteroids (BRs) [48]. SlPRE2 regulates fruit development and plant response to gibberellin in tomatoes. When SlPRE2 is silenced through genetic modification, the resulting plants exhibit reduced grain size, as well as thinner peels, smaller placentas, and smaller seeds [49]. In addition, the C-terminal domain (SD) of BIGPETALp promotes petal growth by interacting with ARF8 to participate in the auxin transport pathway and influence cell expansion in Arabidopsis [50].
More and more studies have revealed the importance of bHLH transcription factors in regulating the plant response to abiotic stresses such as drought, salinity, and high temperature. For example, SlbHLH96 has been shown to act as a positive regulator of drought tolerance in tomatoes. Overexpression of SlbHLH96 leads to enhanced drought tolerance by activating the expression of genes encoding antioxidants, ABA signaling molecules, and stress-related proteins, which ultimately leads to better stress-adaptive responses in the plant [51]. Overexpression FtbHLH3 of Tartary buckwheat can improve the tolerance to drought stress by activating the antioxidant system and up-regulating the expression of various metabolic pathway genes in A. thaliana [52]. Overexpression of PebHLH35 of Populus euphratica resulted in longer initial roots, more leaves, and increased tolerance to drought stress in A. thaliana by regulating stomatal density, stomatal opening, photosynthesis, and growth [53]. In sorghum, SbbHLH85 significantly increases the number and length of root hairs through participation in ABA and auxin signaling pathways and leads to sodium ion absorption and improved resistance to salt stress [54]. CabHLH035 regulates Na+ homeostasis and proline biosynthesis to enhance salt tolerance. It accomplishes this by binding to the CaSOS1 and CaP5CS gene promoters and actively stimulating their expression in capsicum [55]. In wheat, TabHLH1 reduces leaf water loss rate and increases proline and soluble sugar content, through osmotic regulation of substance accumulation and intracellular ROS homeostasis mediated by the ABA signaling pathway, thus contributing to drought and salt stress tolerance [56]. In a high-temperature environment, SPEECHLESS and PIF4 (PHYTOCHROME-INTERACTING FACTOR 4) jointly mediated porosity development processes, modulating adaptation to stress through negative feedback [57]. Although progress has been made in the function and identification of bHLH transcription factors in several plant species, the specific physiological roles and regulatory mechanisms of many bHLH proteins in rye have been poorly reported.
Rye (S. cereale), an ancient and important cereal crop, is a monocotyledonous plant mainly cultivated in Europe, Asia, and Latin America [58]. Rye is the only crop rich in gluten protein next to wheat, which is widely used by the food industry around the world for making bread, biscuits, flour, drinks, beer, and other products [59]. Compared with wheat, rye possesses a very balanced distribution of amino acids, so it is considered to be an important dietary source [60, 61]. In addition, S. cereale is a diploid plant belonging to the Secale genus of the Triticeae Dumortier, an important related species of barley and wheat, which is critical for the study of cross-breeding, evolutionary relationship and genetic improvement of Gramineous crops [62]. In this study, we systematically studied the ScbHLH family by studying the recently published whole genome sequence of S. cereale [63]. We identified 220 bHLH genes in S. cereale, and their gene structures, motif compositions, repetition events, chromosome distributions, and phylogenetic relationships were observed. These analyses can provide insights into the evolutionary history and functional diversity of the bHLH gene family in rye, as well as the potential roles of specific bHLH genes in different biological processes. And, the expression patterns of ScbHLH members under different stress and hormone induction were further analyzed. Our results provide valuable clues to the functional identification and evolutionary relationship of the bHLH gene in S. cereale.
Results
Identification of bHLH genes in S. cereale
Two BLAST methods were used to identify all possible bHLH genes in the rye genome (Table S1). The resulting genes were subsequently assigned names ranging from ScbHLH1 to ScbHLH220 based on their respective positions on the rye chromosomes. A comprehensive analysis was conducted to determine their fundamental characteristics, including gene coding sequence (CDS), protein molecular weight (MW), isoelectric point (PI), and subcellular localization.
Of the 220 ScbHLH proteins, ScbHLH195 was the smallest with 85 amino acids. The largest was ScbHLH82 with 887 amino acids. The molecular weight of the proteins ranged from 9.61 kDa (ScbHLH195) to 96.60 kDa (ScbHLH108). The pI ranged from 4.67 (ScbHLH31) to 11.95 (ScbHLH154), with a mean of 6.75. All of the ScbHLH proteins contained the HLH domain, which is necessary for their function as transcription factors. Additionally, 15 of the ScbHLH proteins contained the bHLH-MYC domain, which may contribute to additional functions or regulatory mechanisms specific to these genes. In the predicted subcellular localization results, 187 ScbHLHs were located in the nucleus, 19 in the chloroplast, 9 in the cytoplasmic, three (ScbHLH16, ScbHLH43, ScbHLH168) in the mitochondria, one (ScbHLH184) in the cytoskeleton, one (ScbHLH220) in the extracellular compartment, one (ScbHLH85) in the peroxisome (Table S1). The ratio of ScbHLH genes to total genes in the S. cereale genome was approximately 0.50%, which was lower than that in Arabidopsis (0.59%) [11] and maize (0.53%) [24], but higher than that in wheat (0.46%) [25], barley (0.35%) [64], rice (0.44%) [7], S. italica (0.48%) [33], tomato (0.46%) [19], and buckwheat (0.49%) [31]. This information provides insights into the evolution and diversification of the bHLH gene family in different plant species.
Multiple sequence alignment, phylogenetic analysis, and classification of ScbHLH genes
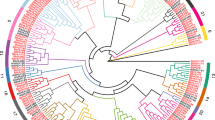
To determine the subfamily classification of bHLH proteins in rye, we constructed phylogenetic trees of S. cereale (220 ScbHLHs) and A. thaliana (54 AtbHLHs) using the neighbor-joining method (Fig. 1, Table S1). Referring to the classification method proposed by Toledo-Ortiz et al. [11] and Pires and Dolan [17], 274 bHLH proteins are divided into 21 main topological clades (subfamilies 1 ~ 21) and one unclassified protein (ScbHLH71). The 21 main subfamilies contained 219 ScbHLH protein members, which is consistent with the classification of bHLH proteins in Arabidopsis [11], indicating that these subfamilies of bHLH proteins have not been lost in S. cereale during long-term evolution. The cluster tree shows that ScbHLH71 protein forms a unique topological structure, which is inconsistent with the classification of Arabidopsis, it may indicate a new feature of the diversity evolution of S. cereale (Fig. 1, Table S1). We have noticed bHLH proteins in wheat [25], barley [64], and other Triticeae Dumortier members, may have generated new evolutionary features that are different from Arabidopsis [11], tomato [19], common bean [20], and Moso bamboo [26], but more evidence is needed. Among these subfamilies, subfamily 5 contains the largest number of members (23 ScbHLH proteins), while subfamily 13 has the fewest (three ScbHLH proteins). The topology tree shows a close genetic relationship between some ScbHLH proteins and many AtbHLH proteins (bootstrap support ≥ 70), such as ScbHLH130, ScbHLH216, and ScbHLH98, indicating that these homologous proteins may have similar gene structures and physiological functions.

Unrooted phylogenetic tree showing relationships among bHLH proteins of S. cereale and Arabidopsis. The phylogenetic tree was derived using the neighbor-joining method in MEGA7.0. The tree shows the 21 phylogenetic subfamilies, which are denoted in red font. bHLH proteins from Arabidopsis are denoted by the prefix ‘At’
To discern conserved amino acid residues across various subfamilies, a subset of AtbHLHs and ScbHLHs were randomly selected from 21 distinct subfamilies for comprehensive multi-sequence comparisons (Fig. S1, Table S1). Based on previous studies [11], the basic regions in ScbHLH proteins may be diverse, such as subfamily 7,9,12,13,14, and 20. The amino acid residues in the basic region can regulate the specific binding of bHLHs to DNA [6, 7], which may indicate the complex physiological functions of ScbHLH proteins. As shown in Fig. S1, the bHLH protein domain span of S. cereale is about 50 ~ 60 amino acids, which is different from rice [7] and Arabidopsis [11].
Conserved motifs, gene structures, and cis-acting elements analysis of ScbHLH genes
By comparing the DNA structures of these ScbHLH members in the rye genome, the number and arrangement characteristics of exons and introns of these genes were further analyzed (Fig. 2, Table S1). The results showed that 220 ScbHLH genes had varying number of exons ranging from 1 to 13 (Table S1). Fifteen (6.82%) ScbHLH genes contain one exon and are distributed in three different subfamilies (8, 14, and 19). The other genes have two or more exons, with the largest proportion of ScbHLH genes (n = 49) having three exons. The member of subfamily 18 (ScbHLH200) had the maximum number of exons (13). We note that members of subfamily 11 have similar complex genetic structures, with 9, 11, or 12 exons. Subfamilies 8 and 10 contained one or two exons, except for individual genes (ScbHLH53). Subfamilies 9 and 18 have the highest number of exon types, with six different exon types of genes. In general, the gene structures of members of the same subfamily exhibit certain similarities, although their exon and intron distributions may be inconsistent.

Phylogenetic relationships, gene structure analysis, and motif distributions of S. cereale bHLH proteins. (A) Phylogenetic tree was constructed using the neighbor-joining method with 1000 replicates for each node. (B) Exons and introns are indicated by yellow rectangles and grey lines, respectively. (C) Amino acid motifs in the ScbHLH proteins (1–10) are represented by colored boxes. The black lines indicate relative protein lengths
Ten conserved motifs were found by the MEME online program in these ScbHLH proteins, and the characteristic amino acid regions of ScbHLH members were arranged (Fig. 2C). The motifs 1 and 2 are widely distributed in most bHLH proteins, except in ScbHLH107, ScbHLH156, ScbHLH40, and ScbHLH71. In most members, the positions of these two motifs are very close. In general, ScbHLH proteins within the same subfamily have similar motif arrangements. For example, subfamilies 2, 3, 4, 8, and 11 contained motifs 1, 2, and 6; subfamily 3 contained motifs 2, 5, and 6; subfamily 5 contained motifs 1, 2, 3, and 5; subfamilies 16 and 17 contained motifs 4, 1, and 2. Almost all members of subfamily 7 contained motifs 7, 10, 1, 2, and 6. In addition, some motifs exist only in specific subfamilies. Motif 3 was specific to subfamilies 5, and 8; motif 9 was specific to subfamilies 3; motif 5 was specific to subfamilies 3, 4, 5, 7, and 8. Further analysis indicates that some motifs are always located at specific locations in the structures of these ScbHLHs. In subfamilies 1, 2, 3, 6, 10, 10, 12, 13, 14, and 15, motifs 1 and 2 are always distributed at the beginning of the pattern. Motif 7 is almost always positioned at the beginning of subfamilies 7 and 8; motif 4 is almost always distributed at the beginning of subfamilies 16, 17, and 18. In subfamily 3, motif 9 is almost always distributed at the end of the pattern. From the arrangement of these motifs, the motif structure of the same subfamily is similar, indicating that the protein structure of the same subgroup may be conservative, which supports the classification of phylogenetic trees.
To analyze the complex physiological processes that these genes may involve, we identified cis-acting elements in the promoter region (upstream 2000 bp) of 220 ScbHLH genes. A total of 126 cis-regulating elements have been identified (Table S3), which involve 48 different physiological functions. These cis-regulatory elements could be divided into eight categories: site binding-related, development-related, environmental stress-related, hormone responsive-related, light responsive-related, promoter-related, wound responsive-related, and other elements. Among the promoter elements of the ScbHLH genes, light-response elements accounted for the largest proportion, including 36 cis-regulatory factors. Some elements may participate in complex physiological functions, such as the P-box in the ScbHLH100 promoter, which is a gibberellin-responsive and part of a light-responsive element. Five promoter-related elements (CAAT-box, TATA-box, A-box, Box II-like sequence, and 3-AF3 binding site) in the promoter region were identified in all ScbHLH genes. There were 12 hormone-responsive elements in the 213 ScbHLH genes of the S. cereale, which covered most plant hormones, including abscisic acid-responsive (ABRE, AAGAA-motif), auxin-responsive (AuxRR-core, AuxRE, and TGA-box), gibberellin-responsive (P-box, GARE-motif, and TATC-box), salicylic acid-responsive (TCA-element), and jasmonic acid-responsive elements (TGACG-motif, CGTCA-motif). In addition, cis-regulatory elements related to drought, low-temperature, salt stresses, anaerobic conditions, other defenses, and stress responses were found in ScbHLH genes. Nearly 80.45% of ScbHLH genes contained abscisic acid-responsiveness and Methyl jasmonate (MeJA) responsive elements; whereas only approximately 49.55% of bHLH genes contained gibberellin-responsive elements. Twelve cis-acting elements are involved in the expression of regulatory processes of different tissues (endosperm, meristem, leaf, root, and seed) during development in S. cereale. Therefore, the ScbHLH genes can not only participate in the development process of multiple tissues but also respond to various abiotic and wound stressed.
Chromosomal spread and gene duplication of ScbHLH genes
A total of 214 ScbHLH genes are unevenly distributed on chromosomes 1R to 7R (Fig. 3, Table S1). To locate all ScbHLH members, the unassigned chromosome (ChrUn) based on the annotated sequence was also considered. Moreover, six ScbHLH members (ScbHLH215 to ScbHLH220) were located on ChrUn. Each ScbHLH gene corresponds to the physical location on different chromosomes in the rye. Among them, chromosome 5R contains the most ScbHLH members (47 bHLH genes, ~ 21.36%), followed by 6R (n = 33, ~ 15.00%). 1R contained the lowest (n = 20, ~ 9.09%). The chromosomes 7R, 4R, 3R, and 2R contained 32 (~ 14.55%), 31 (~ 14.09%), 30 (~ 13.64%), and 21 (~ 9.55%) ScbHLH genes, respectively. The presence of two or more gene members from the same family within the 200 kb chromosome region is defined as the presence of tandem duplications (tandemly arrayed genes) [32, 33]. Some tandem duplications were observed in the bHLH family in S. cereale (Fig. 3, Table S4). Seventeen tandem duplications involving 27 ScbHLH genes were observed on chromosomes 3R, 4R, 5R, 6R, and 7R. All ScbHLH genes involved in tandem duplications belong to the same subfamily. Members of subfamily 5 have more tandem duplications, which we speculate may be one of the reasons why this subfamily has the largest number of members. For example, ScbHLH150 and ScbHLH151 are tandemly arrayed genes that aggregate in subfamily 9. There were six pairs of segmental duplications (duplications of large chromosomal regions) in ScbHLH genes (Fig. 4, Table S5). Chromosomes 4R and 5R had the most ScbHLH members (n = 3) and 1R and 3R had the least (n = 1). For all identified ScbHLH genes, subfamily 19 had two segmental duplications (four ScbHLH genes), while groups 12, 18, 20, and 21 had only one ScbHLH gene. These gene replication events formed the expansion of many ScbHLH members, which may play an important role in the evolution and the occurrence of new functions in environmental adaptation for the bHLH gene family in rye.

Schematic representation of the chromosomal distribution of the S. cereale bHLH genes. Vertical bars represent the chromosomes of S. cereale. The chromosome number is indicated to the left of each chromosome. The scale on the left represents chromosome length

Schematic representation of the chromosomal distribution and interchromosomal relationships of S. cereale bHLH genes. Colored lines indicate all synteny blocks in the S. cereale genome, and the red lines indicate duplicated bHLH gene pairs. The chromosome number is indicated at the bottom of each chromosome
Synteny analysis of ScbHLH genes
To elucidate the evolutionary relationships between these bHLH proteins in several plants, the synteny maps based on homologous genes from rye and six representative plants were constructed. Five monocotyledonous plants included three Triticeae Dumortier plants (Triticum aestivum, Aegilops tauschii, Hordeum vulgare), one model plant (Oryza sativa), and one C4 plant (Zea mays). In addition, a dicotyledonous model plant (A. thaliana) was also included in the comparison (Fig. 5, Table S6). 148 ScbHLH proteins exhibit homologous relationships with those in A. thaliana (7 genes), O. sativa (88), Z. mays (88), A. tauschii (112), T. aestivum (138), and H. vulgare (100) (Table S6). The number of collinear gene pairs between rye and other representative species (A. thaliana, O. sativa, Z. mays, A. tauschii, T. aestivum, and H. vulgare) was 9, 116, 147, 133, 380, and 117, respectively. In the orthologous pairs of the Triticeae Dumortier, rye with A. tauschii and H. vulgare involved a relatively high proportion of bHLH genes, which was 85.47% and 84.21%, respectively. Ten ScbHLHs paired with at least two genes in five monocots, notably including S. cereale and other members of the Triticeae Dumortier plant family. These genes included ScbHLH28, ScbHLH62, ScbHLH82, ScbHLH86, ScbHLH108, ScbHLH123, ScbHLH155, ScbHLH159, ScbHLH205, and ScbHLH212, indicated that they may have an undeniable role in the evolution of these monocotyledonous plants.

Synteny analyses of the bHLH genes between S. cereale and six representative species (T. aestivum, A. tauschii, H. vulgare, O. sativa, Z. mays, and A. thaliana). Gray lines on the background indicate the collinear blocks in S. cereale and other plant genomes; red lines highlight the syntenic S. cereale bHLH gene pairs
Moreover, a considerable number of collinear gene pairs (consisting of fifty ScbHLH genes) were discovered between S. cereale and Triticeae Dumortier plants, with genes that were not found in A. thaliana, O. sativa, and Z. mays. These included ScbHLH5 with AET1Gv20229700 / ARI1A01G110900 / HORVU1Hr1G020370 and ScbHLH65 with AET1Gv20762600 / ARI3A01G346700 / HORVU1Hr1G066250, indicating that these ScbHLH genes may exhibit stronger expansion within the Triticeae Dumortier plants, which may be different from dicotyledonous plants such as Arabidopsis, and other monocotyledonous plants. Furthermore, in these dicotyledonous and monocotyledonous plants, many ScbHLH genes have been found to have at least one pair of homologous pairs (especially between S.cereale and H.vulgare). For example, ScbHLH11, ScbHLH65, ScbHLH86, ScbHLH120, ScbHLH154, and ScbHLH198, indicated that these homologous genes may have been preserved in long-term evolution, and may exist before the differentiation of ancestral plants. To better distinguish the targeted or balanced selection of these 220 ScbHLH genes, these members were subjected to Tajima-D neutrality testing (Table S7). The D value is 13.82 (significant deviation from 0), which indicates that the ScbHLH gene family may participate in the evolution of neutral selection.
Evolutionary analysis of ScbHLH and bHLH genes of several different species
To analyze the genetic relationship of the bHLH proteins between rye and these six representative plants (A. thaliana, O. sativa, Z. mays, A. tauschii, T. aestivum, and H. vulgare), an unrooted Neighbor-joining tree was constructed. Based on the MEME online service software, 10 conserved motifs were identified in the sequence of 589 bHLH proteins from these plants (Fig. 6, Table S2). The detailed conservative motif domains are shown in Tables S1 and S2. ScbHLH proteins tend to aggregate with the bHLH members of A. tauschii, T. aestivum, and H. vulgare. Except for a few ScbHLH proteins, such as ScbHLH71, ScbHLH85, and ScbHLH107, all other ScbHLH proteins contained motifs 1 and 2. Moreover, some motifs only exist in specific topological structures, such as motifs 7, 8, and 10. Overall, these bHLH genes of Triticeae Dumortier plants and S. cereale on the same topological branches had similar motif arrangements. In these plants, some specific bHLH protein subfamilies often contain similar motifs, which suggests their evolutionary relationship. Motifs 1, 2, 5, and 3 form a conserved structure and tend to aggregate within subfamily 5, while motifs 7, 10, 1, 2, 5, and 6 tend to aggregate within subfamily 7.

Phylogenetic relationship and motif composition of the bHLH proteins from S. cereale with six different plant species (T. aestivum, A. tauschii, H. vulgare, O. sativa subsp. Indica, Z. mays, and A. thaliana). Outer panel: an unrooted phylogenetic tree constructed using Geneious R11 with the neighbor-joining method. Inner panel: distribution of conserved motifs in bHLH proteins. The differently colored boxes represent different motifs and their positions in each bHLH protein sequence. The sequence information for each motif is provided in Table S2
Expression patterns of ScbHLHs in several plant organs
To evaluate the physiological functions of 22 bHLH genes in S. cereale, we used qRT-PCR to detect the expression levels of 22 ScbHLH members. As far as possible, the selected members of different subfamilies exhibit significant differences in amino acid structures and distant clustering relationships. The accumulation of transcripts of these ScbHLH genes in five organs (leaf, stem, root, flower, and grain) was detected (Fig. 7A). Many ScbHLH genes are preferentially expressed in certain tissues of S. cereale. Eight genes (ScbHLH11, ScbHLH78, ScbHLH86, ScbHLH120, ScbHLH138, ScbHLH198, ScbHLH211, and ScbHLH214) displayed the highest expression in the root; the expression of six genes (ScbHLH23, ScbHLH48, ScbHLH138, ScbHLH203, ScbHLH210, and ScbHLH211) was highest in the stem, whereas the expression of ScbHLH98 and ScbHLH138 was highest in the leaf, and the expression of seven genes (ScbHLH5, ScbHLH71, ScbHLH110, ScbHLH120, ScbHLH154, ScbHLH172, and ScbHLH210) was highest in the flower, and ScbHLH27, ScbHLH65, ScbHLH162, and ScbHLH198 were highly expressed in the grain.

Expression patterns of 22 S. cereale bHLH genes in several plant organs. (A) Expression patterns of 22 S. cereale bHLH genes in the roots, stems, leaves, flowers and grains were examined via qRT-PCR. Error bars were obtained from three measurements. Lowercase letters above the bars indicate significant differences (α = 0.05, The least significant difference test [LSD]) among the treatments. (B) Expression patterns of 22 S. cereale bHLH genes were examined during different grain development stages: 7 DPA (early filling stage), 14 DPA (mid filling stage), 21 DPA (early ripening stage), 28 DPA (mid ripening stage), and 35 DPA (full ripening stage). The statistical method is consistent with Fig. 7A
Some ScbHLHs may regulate the grain development of S. cereale, thus affecting its nutritional composition and development rate. To determine the genes that may regulate the development of rye grain, the expression of 22 ScbHLH genes from five grain-filling stages after flowering were evaluated. The expression levels of most ScbHLH genes were different in grain during the five grain development stages. In the grain of S. cereale, the expression of most genes decreased with grain development. The expression levels of four genes (ScbHLH11, ScbHLH23, ScbHLH138, and ScbHLH210) were highest at 14 DPA(days post-anthesis), whereas the expression levels of three genes (ScbHLH71, ScbHLH86, and ScbHLH198) were highest at 28 DPA (Fig. 7B). The expression levels of most ScbHLH genes, except for ScbHLH11, and ScbHLH172, were lowest at the full-ripening stage.
In addition, the expression patterns of some ScbHLH members were observed to exhibit some possible coordinated expression in several plant organs (Figs. S2 and S3). Most bHLH members exhibit a significant positive correlation. For example, nine genes ScbHLH5, ScbHLH23, ScbHLH71, ScbHLH110, ScbHLH120, ScbHLH127, ScbHLH154, ScbHLH172, and ScbHLH210 were significantly positively correlated in several plant organs, and ScbHLH11, ScbHLH23, ScbHLH27, ScbHLH48, ScbHLH65, ScbHLH78, ScbHLH98, ScbHLH110, and ScbHLH154 were significantly positively correlated the five development in grain. And, some pairs of ScbHLH genes (ScbHLH98 and ScbHLH86 / ScbHLH127) showed a significant negative correlation.
Expression patterns of ScbHLH genes in response to different treatments
To further observe if the transcription patterns of these rye bHLH members may change under different abiotic stress, the rye seedlings were examined under six abiotic stresses, including ultraviolet radiation (UV), flooding, PEG (10%), NaCl (5%), heat (40˚C), and cold (4˚C). Based on qRT-PCR, the expression patterns of 22 ScbHLH genes responding to different abiotic stress in roots, leaves, and stems were analyzed (Fig. S4). Many ScbHLH members are significantly up-regulated or inhibited under different stresses. Most bHLH genes are sensitive to environmental changes, and their expression levels show significant differences under short-term stress, indicating that they may be involved in these stress regulations. For example, the expression of ScbHLH11, ScbHLH27, and ScbHLH86 significantly increased in roots, stems, and leaves in 1 h during heat stress. In addition, with the increase in treatment time, the expression level of ScbHLH showed changes in different organs, depending on the specific treatment. The ScbHLH86, ScbHLH211, and ScbHLH214 were significantly up-regulated and then down-regulated under cold stress. ScbHLH48 expression was gradually significantly up-regulated in the roots, whereas it was significantly down-regulated in the stems. Interestingly, many ScbHLH genes exhibit opposite expression patterns in these abiotic stresses. For example, the transcript levels of ScbHLH5 were up-regulated in cold, but down-regulated in ultraviolet radiation treatment in leaves. The expression of many ScbHLH genes showed similar patterns during these stress treatments. For example, ScbHLH86 and ScbHLH154 expression was significantly up-regulated first and then down-regulated in the leaves by many different treatments. Other genes exhibit characteristics in specific tissues and exposure times. For example, ScbHLH110 responded quickly to flooding treatment in roots and stems, while ScbHLH198 showed a significant response to cold treatment in leaves. Negative correlations were observed between most ScbHLH genes (Fig. S5). However, some ScbHLHs are significantly positively correlated, such as ScbHLH23, ScbHLH71, ScbHLH138, ScbHLH154, as well as ScbHLH5, ScbHLH110, and ScbHLH210 (P < 0.05).
The bHLH family of rye is speculated to be involved in abiotic stresses and hormone responses. Therefore, we considered detecting the expression patterns of these members in the grain-filling period under different hormone treatments. Three representative hormones were considered for application, namely gibberellin, auxin, and abscisic acid. The expression levels of these members were used to construct a correlation network (Fig. S6). Overall, the expression of some genes may be synergistic. The expression of these genes, ScbHLH5, ScbHLH98, ScbHLH154, and ScbHLH162, were positively co-expressed under gibberellin induction, and ScbHLH23, ScbHLH48, ScbHLH172, and ScbHLH211, were positively co-expressed under auxin induction. In addition, the expression patterns of some genes may be contradictory, for example, the expression pattern of ScbHLH154 is significantly positively correlated with ScbHLH48, ScbHLH138, ScbHLH172, and ScbHLH210, while it is negatively correlated with ScbHLH65, ScbHLH110, and ScbHLH162 under short-term treatment with abscisic acid. This reflects the complexity of the functions in different subfamily members, which is speculated to be related to the diversity of promoter elements and amino acid structure. However, more evidence is needed to illustrate it.
Discussion
ScbHLH gene structures and evolutionary analyses
In this study, a total of 220 ScbHLH genes were discerned, each encoding proteins that displayed noteworthy structural variations, thus elucidating the profound intricacies in the S. cereale. (Figures 1 and 2, Table S1). The N-terminal base region of the bHLH transcription factor was considered to have a conserved amino acid region that determines DNA binding ability. In general, transcription factors with similar gene structures bind to the specific cis-acting elements [10, 11]. A total of 163 ScbHLHs (74.1%) were classified as E-box binding proteins, 121 ScbHLHs (55.0%) were classified as G-box binding proteins, and 42 ScbHLHs (19.1%) were classified as non-G-box binding proteins. Due to the lack of Glu-13 or Arg-16 in the basic region in the alkaline region, the remaining 18 bHLH proteins (8.2%) are considered to have lost their ability to bind to DNA (Fig. S1, Table S1). This ratio may be similar in monocotyledonous plants, especially in the Triticeae Dumortier plants [25, 32, 33, 65]. Although most ScbHLH proteins have G-box binding ability, their relative abundance is lower than those of S. italica (n = 99, 56.9%), and O. sativa (n = 95, 56.9%) [12, 33]. Based on the conserved amino acid structures of bHLH proteins, they can function by forming homologous and heterologous complexes [66]. However, the specific molecular mechanisms have not yet been fully understood. Previous studies have reported that the HLH domain has the function of forming dimers, and the Leu-23 and Leu-52 amino acid residues located in the helixes regions are essential for forming dimers [12].
According to the phylogenetic tree constructed by Li et al. [7], similar to Arabidopsis [11], rice [7], millet [33], wheat [25], and barley [64], these 21 subfamily members have been preserved. The diversity of bHLH proteins may already exist in early terrestrial plants, possibly earlier than before the isolation of monocotyledonous and dicotyledonous plants. In addition, an unclassified gene (UC, ScbHLH71) is independently supported in the topological structures of bHLH proteins, which is speculated to be further differentiation of the bHLH genes in S. cereale, which has also been observed in some monocotyledonous plants such as sorghum [32] and barley [64]. Although some proteins may have more intense evolution or expansion, the distance within different families is convergent, that is, the bHLH proteins may have a closer genetic distance in the same family of plants, whether C3 or C4 plants. These bHLH proteins in Gramineae plants may come from one or several ancestral proteins, which is supported by the distant genetic relationship between angiosperms and animal bHLH proteins [10]. Based on the topology tree analysis, it was observed that the most numerous rye subfamilies were: 5 (n = 23, 10.45%), 9 (n = 21, 9.55%), 19 (n = 20, 9.09%) and 18 (n = 19, 8.64%). This pattern differs from Arabidopsis but aligns with findings in sorghum [32] and barley [64]. The similarity in member count suggests that these specific subfamilies of bHLH genes have experienced extensive expansion during the long evolutionary process of monocotyledonous plants. The number and proportion of members in subfamilies 1, 5, and 9 were significantly higher than those in Arabidopsis [11], and similar to S. italica [33]. Whether this differentiation is beneficial for Gramineae or Triticeae Dumortier plants has not yet been determined.
Most ScbHLH genes (n = 205, 93.18%) contain more than two exons. Some subfamilies have observed the complexity of the number of introns, mainly reflected in subfamilies 9, 11, 18, and 19, which may lead to the expansion of the bHLH family and the generation of new functions in S. cereale. In addition, previous studies have shown that genes with few or no introns are usually less expressed in plants [67]. The non-intron genes mainly belong to subfamily 8, which is consistent with sorghum [32] and millet [33]. Tandem duplications can rapidly expand and contract in response to environmental changes, maintaining an equal number of functionally related genes, without increasing genetic complexity during evolution [68]. In addition, segmental duplications are commonly present in some animal and plant genomes, which helps to form genetic diversity [69]. Therefore, tandem and segmental duplications contribute to the expansion of this gene family and play an important role in genome evolution. As expected, all amplified events are within this subfamily, including tandem and segment duplications, similar to Arabidopsis [11], rice [7], and buckwheat [31], indicating that repeated events did not cause gene expansion between different subfamilies. However, tandem duplications of the ScbHLH genes may have a higher contribution to the amplification of the bHLH family in S. cereale, which is inconsistent with S. bicolor [32] and S. italica [33]. These gene replication events formed the expansion of many ScbHLH members, which may play an important role in the evolution and the occurrence of new functions in environmental adaptation for the bHLH gene family in rye.
Expression patterns and function prediction of ScbHLHs
In this investigation, the expression patterns of 22 ScbHLH genes exhibiting significant distinctions in phylogenetic trees were analyzed across various organs and developmental stages of grain (Fig. 7). Almost all ScbHLH genes were found to exhibit significant differential expression (more than 2-fold differences). ScbHLH86 is classified to subfamily 18, which has the highest expression in stems, roots, flowers, and grains. Similar to the expression pattern of the homologous gene CIB2 (AT5G48560) in Arabidopsis, which participates in roots and stems development and activates FT protein regulation of flowering and grain-filling processes by forming various combinations of heterodimers with CIB4, CIB5, and CIB1. ScbHLH120, classified in subfamily 16, is highly expressed in the flowers and roots in S. cereale, and its homologous gene HBP1 (Os08g0506700) interacts with POH1 to directly regulate the expression of the key factor Hd1 during rice flowering [70]. In addition, the expression pattern of ScbHLH120 was observed to be similar to the homologous gene (AT4G30410) in Arabidopsis [71]. ScbHLH78 showed the highest expression in roots and leaves, which is consistent with the expression pattern of the same family gene AT2G2270. AT2G22770, belonging to the subfamily 5, may play a key role in the roots and leaves of Arabidopsis [72]. The expression of AT1G06170, a member of the subfamily 4, is higher in grains than in roots, stems, leaves, and flowers [73], similar to the expression pattern of ScbHLH162. The expression of most ScbHLH genes was significantly positively correlated in S. cereale, indicating that their combination may have synergistic effects in five plant organs (Fig. S2). These findings provide direction for the study of their gene function. As a Triticeae Dumortier plant, the grain development process is very important for S. cereale, which is divided into five representative stages. Furthermore, the ScbHLH genes that may regulate the development of rye grains have been identified. This study investigated the expression levels of 22 bHLH members in grains during the filling period. The results showed that most of these genes were expressed in the early and middle stages of grain filling. ScbHLH172 is stably expressed at almost all stages. In addition, ScbHLH71 and ScbHLH210 have the highest expression in the early and middle stages of grain filling. We attempt to further validate the role of these genes in the growth and grain development of S. cereale.
The growth and developmental processes in plants are intricately influenced by both external environmental factors and hormonal cues. Rye, as a cold and drought-tolerant crop, exhibits the ability to adapt to stress conditions and respond to hormone stimulation through elaborate endogenous networks and transcriptional signals, akin to its counterparts wheat and barley [74, 75]. However, a comprehensive understanding of its response to complex abiotic stresses remains elusive. In our study, we conducted a systematic analysis of the expression patterns of 22 ScbHLHs in rye seedlings subjected to various stresses (Fig. S4), encompassing six distinct abiotic stresses. Under drought stress conditions, we observed a significant upregulation in the expression levels of 18 ScbHLH genes in the roots, 21 genes in the leaves, and 14 genes in the stems, with variations depending on the duration of treatment. These molecular responses likely contribute to the adaptive mechanisms of S. cereale in arid environments, aligning with its characterization as a drought-tolerant crop. Different bHLH subfamilies may play complex physiological roles in the environmental adaptation process of rye. In Arabidopsis, AT1G63650 (AtbHLH18), belonging to subfamily 7, displays preferential expression within Arabidopsis root epidermal cells and is speculated to play a role in stress adaptation through the regulation of root development [76]. ScbHLH5, member of subfamily 7, displayed remarkable responses at the roots in multiple abiotic stresses. Within the subfamily 8, ZBF1 (At1g32640) encodes an MYC-bHLH-related transcriptional activator that plays a vital role in drought stress and ABA response [77, 78]. Similarly, the expression of ScbHLH11 was significantly upregulated in almost all abiotic stresses, which may enhance the adaptability of rye to the environment in a similar manner. The expression of some members, such as ScbHLH23, ScbHLH86, and ScbHLH214, significantly up-regulated under flooding stress, which may contribute to stomatal expansion and increased respiration in leaves. The correlation network indicates that these bHLH transcription factors are involved in a complex cross-regulatory network regarding stresses and hormone induction. For example, ScbHLH5 and ScbHLH65 exhibit a significant positive correlation under different hormone induction, indicating that they have synergistic regulatory effects under various endogenous metabolism.
Conclusion
In summary, we provided a systematic genome-wide analysis of the bHLH gene family in S. cereale. These 220 members were divided into 21 subfamilies and an unclassified gene, and their evolutionary relationships, gene structures, conserved motifs, gene replications, and expression patterns were further analyzed. Both tandem duplications and segmental duplications contribute to the expansion of the bHLH family in S. cereale. Tandem duplications may play a crucial role in the expansion of the bHLH genes in the evolution of rye. In addition, the expression patterns of ScbHLH in different tissues and grain-filling stages were observed, as well as their responses to different stresses and hormones, were detected. Some candidate genes related to tissue development and environmental stresses have been screened, such as ScbHLH11, ScbHLH48, and ScbHLH172. These data will provide a useful aid for future research on the function of the bHLH gene family in rye.
Materials and methods
Gene identification
We downloaded the latest high-quality reference genome of S. cereale from the NCBI (National Center for Biotechnology Information) GenBank website, the access number is JADQCU000000000 [79]. Firstly, all of the bHLH proteins of Arabidopsis (54 AtbHLHs) [11] were used to search for candidate bHLH proteins from the rye genome via the BLASTp program [80]. The candidate genes were searched by BLASTP using a score value of ≥ 100 and an e-value ≤ e − 10. Secondly, the Hidden Markov Model (HMM) file of the bHLH domain (PF00010) is downloaded from the PFAM protein family database (http://pfam.xfam.org/). Based on the HMM model in the HMMER3.0 online software, the bHLH protein sequence in S. cereale was identified with a decision value of 0.01 (http://plants.ensembl.org/hmmer/index.html) [81]. Based on PFAM and SMART in thread sequencing, conserved motifs were found in the bHLH proteins in rye (http://smart.embl-heidelberg.de/) [82, 83]. Accordingly, a total of 220 ScbHLH proteins were identified in the rye. Then, in the NCBI protein database, these ScbHLH proteins were used as the initial query for re-verification (https://blast.ncbi.nlm.nih.gov/Blast.cgi? PROGRAM = blastp&PAGE_TYPE = BlastSearch&LINK_LOC = blast home). Finally, the ExPasy online program was used to identify the basic features of the bHLH gene in S. cereale, including sequence length, protein molecular weight, isoelectric points, and subcellular localization (http://web.expasy.org/protparam/).
bHLH gene structures and conserved motif analysis
Based on the default parameters of the ClusterW program, 220 rye bHLH proteins were subjected to multiple sequence alignment projects to further analyze their characteristic domains [84]. MEGA 7.0 and GeneDoc 2.7(http://genedoc.software.informer.com/2.7/) were used for the manual regulation of conserved domains in the amino acid sequences of these bHLH proteins. Gene Structure Display Server (GSDS; http://gsds.cbi.pku.edu.cn) online software was used to analyze the exon-intron Substructure of these ScbHLH genes [85]. Then, the MEME online program (http://meme.nbcr.net/meme/intro. HTML) was used to analyze the conserved motifs and gene structural differences in these bHLH proteins [86]. The optimization parameters for conservative motifs were as follows: the maximum number was 10, and the optimal width of residues was 6 to 200 [32]. In addition, The PlantCARE online software was used to predict cis-acting elements in the upstream 2000 bp promoter region of 220 bHLH genes (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/?tdsourcetag=s_pcqq_aiomsg) [87].
Chromosomal distribution and gene duplication
Firstly, based on the physical location of these genes in the annotation file, all ScbHLH genes have been designated as chromosomal details. And, Circos software was used to analyze these ScbHLH genes for chromosomal location information [88]. Multiple collinear scanning toolkits (MCScanX) were used to analyze gene replication events based on default parameters [81, 89]. The homology of the bHLH genes between S. cereale and six other plants (T. aestivum, A. tauschii, H. vulgare, O. sativa subsp. Indica, Z. mays, and A. thaliana) was analyzed by using the project of dual synteny plotter in TBtools software (v1.120) [90].
Phylogenetic analysis and classification of the ScbHLH family
According to the classification of AtbHLH proteins, 220 bHLH proteins in S. cereale are divided into 21 main subfamilies. In MEGA 7.0, the Jukes-Cantor model is used to construct NJ (neighbor-joining method) trees. The bootstrap value of the constructed phylogenetic tree was set to 1000, and assigned with Geneious R11 with BLOSUM62 cost matrix. In addition, this study also constructed a multi-species phylogenetic tree containing bHLH protein sequences from rye and six plant species (T. aestivum, A. tauschii, H. vulgare, O. sativa subsp. Indica, Z. mays, and A. thaliana), which were obtained from the UniProt website [32, 33].
Plant materials, growth conditions, and abiotic stresses in S. cereale
The study employed S. cereale cv. Weining, is a well-established traditional variety native to Guizhou Province in southwest China. Since 2022, ‘Weining’ has been cultivated within the greenhouse facilities at Chengdu University Farm. The seedling tray was put in the greenhouse. Rye cultivation entailed potting the plants in a blend of soil and vermiculite (in a 1:1 ratio), within a controlled growth chamber. The chamber was regulated to maintain a diurnal cycle of 16 h at a temperature of 25℃, followed by 8 h at a temperature of 20℃ during the night, while sustaining a relative humidity level of 75%. Plant nutrient solution (10 g of urea, 6 g of potassium dihydrogen phosphate, 2 g of calcium sulfate, and 1 g of magnesium sulfate dissolved in 20 l) was sprayed once a month, with 300mL sprayed into potted plants each time. Generally, pure water irrigation was carried out every 5 days. At the early ripening stage of rye, we carefully collected samples from the roots, stems, leaves, flowers, and grains. Due to the inconsistent filling period of all spikelets in rye, individual spikelets were peeled off with tweezers. Spikelets in the flowering stage (prior to filling stage), including the florets with outer lemma, inner lemma, stamens and pistils were called flowers. For grain analysis, we focused on five key developmental stages: 7 days (early filling stage), 14 days (mid-filling stage), 21 days (early ripening stage), 28 days (mid-ripening stage), and 35 days (full ripening stage). Each organ sample was obtained from five individual plants grown under identical growth conditions. To preserve their physiological state, the samples were swiftly placed in liquid nitrogen, pre-cooled to maintain their integrity, and subsequently stored at -80℃ until further use. Total RNA was rapidly extracted from these samples for subsequent qRT-PCR analysis, with each sample being subjected to at least three technical replicates. Furthermore, we also investigated the expression patterns of these ScbHLH genes in rye seedlings subjected to six different abiotic stresses. Rye seeds were sown in seedling trays, and after 28 days of germination, the seedling trays were treated with a 50 mL solution, which effectively submerged the seedling roots. The six distinct abiotic treatments administered to S. cereale plant seedlings included ultraviolet radiation (UV, 70 µW/cm2, 220 V, 30 W), flooding (affecting the whole plant), salt (5% NaCl), drought (10% PEG6000), high temperature (40℃), and cold (4℃). For each stress treatment, we established five parallel sets. To analyze the expression patterns, we examined the leaves, roots, and stems of rye seedlings at 0, 1, 4, and 12 h following stress induction. Lastly, considering the presence of diverse hormone response elements within the promoter region of these genes, we conducted three distinct hormone treatments during the flowering stage. These treatments included gibberellin (GA3, 100 µM), auxin (indole-3-acetic acid, IAA, 100 µM), and abscisic acid (50 µM).
Total RNA extraction, cDNA reverse transcription, and qRT-PCR analysis
Total RNA was extracted from fresh tissues using a plant RNA extraction kit (TianGen RNA Easy Fast Plant Tissue Kit, DP452). The reverse transcription kit was selected from TianGen FastKing One-Step RT-qPCR Kit (SYBR, FP313-01). Primer 5.0 software was used to design qRT-PCR primers for all genes (Table S8). Actin was used as the internal control. The standard expression for SYBR Premix ExTaqII (Takara Bio) was repeated on the CFX96 real-time system (Bio-Rad) for 3 replicates. The total reaction system of qRT PCR was 20 µL, containing 1µL cDNA (100ng/µL− 1), 10µL SYBR Green Realtime PCR Master Mix (Takara Bio), 0.5µL forward and reverse primers, and 8µL RNase-free water, respectively. All quantitative primers for genes were analyzed for their practicality through melting curves. The expression of these bHLH genes was analyzed using the 2− (ΔΔCt) method [91].
Statistical analysis
The least significant difference test (LSD) was conducted using significance levels of 0.05 in JMP6.0 software. Origin 2016 (OriginLab Corporation, Northampton, Massachusetts, USA) was used for the histogram drawing. Based on the Pearson correlation program, we defined the correlation coefficient of ScbHLH genes using Sigmaplot 12.0 software. The Pearson correlation matrix of the bHLH genes in rye was constructed using R2.11, and network analysis (CNA) was performed using Cytoscape 2.7.0 software. The correlation coefficient was defined at P < 0.05.
Data availability
The entire Secale cereale genome sequence information was obtained from the NCBI (National Center for Biotechnology Information) GenBank website, the access number is JADQCU000000000. Rye materials (Weining) used in the experiment were supplied by Prof. Kuiying Li of Anshun University. The datasets supporting the conclusions of this study are included in the article and its additional files.
Abbreviations
- bHLH :
-
Basic helix-loop-helix
- ScbHLH :
-
Secale cereale bHLH
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- AtbHLH :
-
Arabidopsis thaliana bHLH
- HMM:
-
Hidden Markov Model
- pI:
-
Isoelectric point
- CDS:
-
Gene coding sequence
- DPA:
-
Days post-anthesis
References
Sun X, Wang Y, Sui N. Transcriptional regulation of bHLH during plant response to stress. Biochem Biophys Res Commun. 2018;503(2):397–401. https://doi.org/10.1016/j.bbrc.2018.07.123.
Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56(5):777–83. https://doi.org/10.1016/0092-8674(89)90682-x.
Ludwig SR, Habera LF, Dellaporta SL, Wessler SR. Lc, a member of the maize R gene family responsible for tissue-specific anthocyanin production, encodes a protein similar to transcriptional activators and contains the myc-homology region. Proc Natl Acad Sci U S A. 1989;86(18):7092–6. https://doi.org/10.1073/pnas.86.18.7092.
Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20(2):429–40. https://doi.org/10.1128/MCB.20.2.429-440.2000.
Yin J, Chang X, Kasuga T, Bui M, Reid MS, Jiang CZ. A basic helix-loop-helix transcription factor, PhFBH4, regulates flower senescence by modulating ethylene biosynthesis pathway in petunia. Hortic Res. 2015;2:15059. https://doi.org/10.1038/hortres.2015.59. Published 2015 Dec 16.
Jones S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004;5(6):226. https://doi.org/10.1186/gb-2004-5-6-226.
Li X, Duan X, Jiang H, et al. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 2006;141(4):1167–84. https://doi.org/10.1104/pp.106.080580.
Ellenberger T, Fass D, Arnaud M, Harrison SC. Crystal structure of transcription factor E47: E-box recognition by a basic region helix-loop-helix dimer. Genes Dev. 1994;8(8):970–80. https://doi.org/10.1101/gad.8.8.970.
Shimizu T, Toumoto A, Ihara K, et al. Crystal structure of PHO4 bHLH domain-DNA complex: flanking base recognition. EMBO J. 1997;16(15):4689–97. https://doi.org/10.1093/emboj/16.15.4689.
Atchley WR, Terhalle W, Dress A. Positional dependence, cliques, and predictive motifs in the bHLH protein domain. J Mol Evol. 1999;48(5):501–16. https://doi.org/10.1007/pl00006494.
Toledo-Ortiz G, Huq E, Quail PH. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell. 2003;15(8):1749–70. https://doi.org/10.1105/tpc.013839.
Carretero-Paulet L, Galstyan A, Roig-Villanova I, Martínez-García JF, Bilbao-Castro JR, Robertson DL. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, Poplar, rice, moss, and algae. Plant Physiol. 2010;153(3):1398–412. https://doi.org/10.1104/pp.110.153593.
Brownlie P, Ceska T, Lamers M, et al. The crystal structure of an intact human Max-DNA complex: new insights into mechanisms of transcriptional control. Structure. 1997;5(4):509–20. https://doi.org/10.1016/s0969-2126(97)00207-4.
Ciarapica R, Rosati J, Cesareni G, Nasi S. Molecular recognition in helix-loop-helix and helix-loop-helix-leucine zipper domains. Design of repertoires and selection of high affinity ligands for natural proteins. J Biol Chem. 2003;278(14):12182–90. https://doi.org/10.1074/jbc.M211991200.
Atchley WR, Fitch WM. A natural classification of the basic helix-loop-helix class of transcription factors. Proc Natl Acad Sci U S A. 1997;94(10):5172–6. https://doi.org/10.1073/pnas.94.10.5172.
Simionato E, Ledent V, Richards G, et al. Origin and diversification of the basic helix-loop-helix gene family in metazoans: insights from comparative genomics. BMC Evol Biol. 2007;7:33. https://doi.org/10.1186/1471-2148-7-33. Published 2007 Mar 2.
Pires N, Dolan L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol Biol Evol. 2010;27(4):862–74. https://doi.org/10.1093/molbev/msp288.
Song XM, Huang ZN, Duan WK, et al. Genome-wide analysis of the bHLH transcription factor family in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol Genet Genomics. 2014;289(1):77–91. https://doi.org/10.1007/s00438-013-0791-3.
Sun H, Fan HJ, Ling HQ. Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genomics. 2015;16(1):9. Published 2015 Jan 22. https://doi.org/10.1186/s12864-014-1209-2.
Kavas M, Baloğlu MC, Atabay ES, Ziplar UT, Daşgan HY, Ünver T. Genome-wide characterization and expression analysis of common bean bHLH transcription factors in response to excess salt concentration. Mol Genet Genomics. 2016;291(1):129–43. https://doi.org/10.1007/s00438-015-1095-6.
Mao K, Dong Q, Li C, Liu C, Ma F. Genome wide identification and characterization of Apple bHLH Transcription Factors and Expression Analysis in response to Drought and Salt stress. Front Plant Sci. 2017;8:480. https://doi.org/10.3389/fpls.2017.00480. Published 2017 Apr 11.
Gao C, Sun J, Wang C, et al. Genome-wide analysis of basic/helix-loop-helix gene family in peanut and assessment of its roles in pod development. PLoS ONE. 2017;12(7):e0181843. https://doi.org/10.1371/journal.pone.0181843. Published 2017 Jul 27.
Niu X, Guan Y, Chen S, Li H. Genome-wide analysis of basic helix-loop-helix (bHLH) transcription factors in Brachypodium distachyon. BMC Genomics. 2017;18(1):619. Published 2017 Aug 15. https://doi.org/10.1186/s12864-017-4044-4.
Zhang T, Lv W, Zhang H et al. Genome-wide analysis of the basic Helix-Loop-Helix (bHLH) transcription factor family in maize. BMC Plant Biol. 2018;18(1):235. Published 2018 Oct 16. https://doi.org/10.1186/s12870-018-1441-z.
Wei K, Chen H. Comparative functional genomics analysis of bHLH gene family in rice, maize and wheat. BMC Plant Biol. 2018;18(1):309. https://doi.org/10.1186/s12870-018-1529-5. Published 2018 Nov 29.
Cheng X, Xiong R, Liu H, et al. Basic helix-loop-helix gene family: genome wide identification, phylogeny, and expression in Moso bamboo. Plant Physiol Biochem. 2018;132:104–19. https://doi.org/10.1016/j.plaphy.2018.08.036.
Li H, Gao W, Xue C, et al. Genome-wide analysis of the bHLH gene family in Chinese jujube (Ziziphus jujuba Mill.) And wild jujube. BMC Genomics. 2019;20(1):568. https://doi.org/10.1186/s12864-019-5936-2. Published 2019 Jul 10.
Zhang Z, Chen J, Liang C, Liu F, Hou X, Zou X. Genome-wide identification and characterization of the bHLH transcription factor family in Pepper (Capsicum annuum L). Front Genet. 2020;11:570156. https://doi.org/10.3389/fgene.2020.570156. Published 2020 Sep 25.
Zhu L, Zhao M, Chen M, et al. The bHLH gene family and its response to saline stress in Jilin ginseng, Panax ginseng C.A. Meyer. Mol Genet Genomics. 2020;295(4):877–90. https://doi.org/10.1007/s00438-020-01658-w.
Aslam M, Jakada BH, Fakher B, et al. Genome-wide study of pineapple (Ananas comosus L.) bHLH transcription factors indicates that cryptochrome-interacting bHLH2 (AcCIB2) participates in flowering time regulation and abiotic stress response. BMC Genomics. 2020;21(1):735. https://doi.org/10.1186/s12864-020-07152-2. Published 2020 Oct 22.
Sun W, Jin X, Ma Z, Chen H, Liu M. Basic helix-loop-helix (bHLH) gene family in Tartary buckwheat (Fagopyrum tataricum): genome-wide identification, phylogeny, evolutionary expansion and expression analyses. Int J Biol Macromol. 2020;155:1478–90. https://doi.org/10.1016/j.ijbiomac.2019.11.126.
Fan Y, Yang H, Lai D, et al. Genome-wide identification and expression analysis of the bHLH transcription factor family and its response to abiotic stress in sorghum [Sorghum bicolor (L.) Moench]. BMC Genomics. 2021;22(1):415. https://doi.org/10.1186/s12864-021-07652-9. Published 2021 Jun 5.
Fan Y, Lai D, Yang H, et al. Genome-wide identification and expression analysis of the bHLH transcription factor family and its response to abiotic stress in foxtail millet (Setaria italica L). BMC Genomics. 2021;22(1):778. https://doi.org/10.1186/s12864-021-08095-y. Published 2021 Oct 30.
Lu X, Zhang H, Hu J, et al. Genome-wide identification and characterization of bHLH family genes from orchardgrass and the functional characterization of DgbHLH46 and DgbHLH128 in drought and salt tolerance. Funct Integr Genomics. 2022;22(6):1331–44. https://doi.org/10.1007/s10142-022-00890-4.
Jiang H, Liu L, Shan X, et al. Genome-wide identification and expression analysis of the bHLH gene family in cauliflower (Brassica oleracea L). Physiol Mol Biol Plants. 2022;28(9):1737–51. https://doi.org/10.1007/s12298-022-01238-9.
Zhang L, Chen W, Liu R, Shi B, Shu Y, Zhang H. Genome-wide characterization and expression analysis of bHLH gene family in physic nut (Jatropha curcas L). PeerJ. 2022;10:e13786. https://doi.org/10.7717/peerj.13786. Published 2022 Aug 9.
Bennici A. Origin and early evolution of land plants: problems and considerations. Commun Integr Biol. 2008;1(2):212–8. https://doi.org/10.4161/cib.1.2.6987.
Morris JL, Puttick MN, Clark JW, et al. The timescale of early land plant evolution. Proc Natl Acad Sci U S A. 2018;115(10):E2274–83. https://doi.org/10.1073/pnas.1719588115.
Sokoloff DD, Fomichev CI, Rudall PJ, Macfarlane TD, Remizowa MV. Evolutionary history of the grass gynoecium. J Exp Bot. 2022;73(14):4637–61. https://doi.org/10.1093/jxb/erac182.
Alvarez J, Smyth DR. CRABS CLAW and SPATULA, two Arabidopsis genes that control carpel development in parallel with AGAMOUS. Development. 1999;126(11):2377–86. https://doi.org/10.1242/dev.126.11.2377.
Heisler MG, Atkinson A, Bylstra YH, Walsh R, Smyth DR. SPATULA, a gene that controls development of carpel margin tissues in Arabidopsis, encodes a bHLH protein. Development. 2001;128(7):1089–98. https://doi.org/10.1242/dev.128.7.1089.
Komatsu K, Maekawa M, Ujiie S, et al. LAX and SPA: major regulators of shoot branching in rice. Proc Natl Acad Sci U S A. 2003;100(20):11765–70. https://doi.org/10.1073/pnas.1932414100.
Kondou Y, Nakazawa M, Kawashima M, et al. RETARDED GROWTH OF EMBRYO1, a new basic helix-loop-helix protein, expresses in endosperm to control embryo growth. Plant Physiol. 2008;147(4):1924–35. https://doi.org/10.1104/pp.108.118364.
MacAlister CA, Ohashi-Ito K, Bergmann DC. Transcription factor control of asymmetric cell divisions that establish the stomatal lineage. Nature. 2007;445(7127):537–40. https://doi.org/10.1038/nature05491.
Wang J, Li C, Mao X, et al. The wheat basic helix-loop-helix (bHLH) protein TabHLH123 positively regulates the formation of crown root and associates with plant height and 1000-grain weight in wheat under various conditions [published online ahead of print, 2023 Feb 7]. J Exp Bot. 2023. erad051.
Guo J, Li W, Shang L, et al. OsbHLH98 regulates leaf angle in rice through transcriptional repression of OsBUL1. New Phytol. 2021;230(5):1953–66. https://doi.org/10.1111/nph.17303.
Qi T, Song S, Ren Q, et al. The Jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell. 2011;23(5):1795–814. https://doi.org/10.1105/tpc.111.083261.
Albertos P, Wlk T, Griffiths J, et al. Brassinosteroid-regulated bHLH transcription factor CESTA induces the gibberellin 2-oxidase GA2ox7. Plant Physiol. 2022;188(4):2012–25. https://doi.org/10.1093/plphys/kiac008.
Zhu Z, Liang H, Chen G, et al. The bHLH transcription factor SlPRE2 regulates tomato fruit development and modulates plant response to gibberellin. Plant Cell Rep. 2019;38(9):1053–64. https://doi.org/10.1007/s00299-019-02425-x.
Varaud E, Brioudes F, Szécsi J, Leroux J, Brown S, Perrot-Rechenmann C, Bendahmane M. AUXIN RESPONSE FACTOR8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell. 2011;23(3):973–83. https://doi.org/10.1105/tpc.110.081653. Epub 2011 Mar 18. PMID: 21421811; PMCID: PMC3082276.
Liang Y, Ma F, Li B, et al. A bHLH transcription factor, SlbHLH96, promotes drought tolerance in tomato. Hortic Res. 2022;9:uhac198. https://doi.org/10.1093/hr/uhac198. Published 2022 Sep 6.
Yao PF, Li CL, Zhao XR, Li MF, Zhao HX, Guo JY, Cai Y, Chen H, Wu Q. Overexpression of a Tartary Buckwheat Gene, FtbHLH3, enhances Drought/Oxidative stress tolerance in transgenic Arabidopsis. Front Plant Sci. 2017;8:625. https://doi.org/10.3389/fpls.2017.00625. PMID: 28487715; PMCID: PMC5403918.
Dong Y, Wang C, Han X, et al. A novel bHLH transcription factor PebHLH35 from Populus Euphratica confers drought tolerance through regulating stomatal development, photosynthesis and growth in Arabidopsis. Biochem Biophys Res Commun. 2014;450(1):453–8. https://doi.org/10.1016/j.bbrc.2014.05.139.
Song Y, Li S, Sui Y, et al. SbbHLH85, a bHLH member, modulates resilience to salt stress by regulating root hair growth in sorghum. Theor Appl Genet. 2022;135(1):201–16. https://doi.org/10.1007/s00122-021-03960-6.
Zhang H, Guo J, Chen X, et al. Pepper bHLH transcription factor CabHLH035 contributes to salt tolerance by modulating ion homeostasis and proline biosynthesis. Hortic Res. 2022;9:uhac203. https://doi.org/10.1093/hr/uhac203. Published 2022 Sep 6.
Yang T, Yao S, Hao L, Zhao Y, Lu W, Xiao K. Wheat bHLH-type transcription factor gene TabHLH1 is crucial in mediating osmotic stresses tolerance through modulating largely the ABA-associated pathway. Plant Cell Rep. 2016;35(11):2309–23. https://doi.org/10.1007/s00299-016-2036-5.
Lau OS, Song Z, Zhou Z, Davies KA, Chang J, Yang X, Wang S, Lucyshyn D, Tay IHZ, Wigge PA, Bergmann DC. Direct Control of SPEECHLESS by PIF4 in the high-temperature response of Stomatal Development. Curr Biol. 2018;28(8):1273–1280e3. Epub 2018 Apr 5. PMID: 29628371; PMCID: PMC5931714.
Kaur P, Singh Sandhu K, Singh Purewal S, Kaur M, Kumar Singh S, Rye. A wonder crop with industrially important macromolecules and health benefits. Food Res Int. 2021;150(Pt A):110769. https://doi.org/10.1016/j.foodres.2021.110769.
Stępniewska S, Cacak-Pietrzak G, Szafrańska A, Ostrowska-Ligęza E, Dziki D. Assessment of the Starch-Amylolytic Complex of Rye Flours by Traditional methods and Modern one. Mater (Basel). 2021;14(24):7603. https://doi.org/10.3390/ma14247603. Published 2021 Dec 10.
Johansson DP, Gutiérrez JLV, Landberg R, Alminger M, Langton M. Impact of food processing on rye product properties and their in vitro digestion. Eur J Nutr. 2018;57(4):1651–66. https://doi.org/10.1007/s00394-017-1450-y. Epub 2017 Apr 17. PMID: 28417207; PMCID: PMC5959992.
Torbica A, Belović M, Popović L, Čakarević J, Jovičić M, Pavličević J. Comparative study of nutritional and technological quality aspects of minor cereals. J Food Sci Technol. 2021;58(1):311–22. https://doi.org/10.1007/s13197-020-04544-w.
Li K, Jiang W, Hui Y, et al. Gapless indica rice genome reveals synergistic contributions of active transposable elements and segmental duplications to rice genome evolution. Mol Plant. 2021;14(10):1745–56. https://doi.org/10.1016/j.molp.2021.06.017.
Li G, Wang L, Yang J, et al. A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat Genet. 2021;53(4):574–84. https://doi.org/10.1038/s41588-021-00808-z.
Ke Q, Tao W, Li T, et al. Genome-wide identification, evolution and expression analysis of basic Helix-loop-helix (bHLH) Gene Family in Barley (Hordeum vulgare L). Curr Genomics. 2020;21(8):621–44. https://doi.org/10.2174/1389202921999201102165537.
Ma XF, Xia X, Liu S, Baenziger PS, Özkan H. Editorial: Genomics-enabled Triticeae Improvement. Front Plant Sci. 2022;13:871816. https://doi.org/10.3389/fpls.2022.871816. PMID: 35371118; PMCID: PMC8971988.
Nair SK, Burley SK. Recognizing DNA in the library. Nature. 2000;404(6779):715–8. https://doi.org/10.1038/35008182.
Henriksson M, Lüscher B. Proteins of the Myc network: essential regulators of cell growth and differentiation. Adv Cancer Res. 1996;68:109–82. https://doi.org/10.1016/s0065-230x(08)60353-x.
Graham GJ. Tandem genes and clustered genes. J Theor Biol. 1995;175(1):71–87. https://doi.org/10.1006/jtbi.1995.0122.
Bretani G, Rossini L, Ferrandi C, et al. Segmental duplications are hot spots of copy number variants affecting barley gene content. Plant J. 2020;103(3):1073–88. https://doi.org/10.1111/tpj.14784.
Yin Y, Yan Z, Guan J, et al. Two interacting Basic Helix-Loop-Helix transcription factors Control Flowering Time in Rice [published online ahead of print, 2023 Feb 9]. Plant Physiol. 2023;kiad077. https://doi.org/10.1093/plphys/kiad077.
Lu R, Zhang J, Wu YW, et al. bHLH transcription factors LP1 and LP2 regulate longitudinal cell elongation. Plant Physiol. 2021;187(4):2577–91. https://doi.org/10.1093/plphys/kiab387.
Taylor-Teeples M, Lin L, de Lucas M, et al. An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature. 2015;517(7536):571–5. https://doi.org/10.1038/nature14099.
Zhu E, You C, Wang S, et al. The DYT1-interacting proteins bHLH010, bHLH089 and bHLH091 are redundantly required for Arabidopsis anther development and transcriptome. Plant J. 2015;83(6):976–90. https://doi.org/10.1111/tpj.12942.
Kosová K, Vítámvás P, Prášil IT. Wheat and barley dehydrins under cold, drought, and salinity - what can LEA-II proteins tell us about plant stress response? Front Plant Sci. 2014;5:343. Published 2014 Jul 9. https://doi.org/10.3389/fpls.2014.00343.
Hura T. Wheat and barley: acclimatization to Abiotic and biotic stress. Int J Mol Sci. 2020;21(19):7423. https://doi.org/10.3390/ijms21197423. Published 2020 Oct 8.
Bruex A, Kainkaryam RM, Wieckowski Y, et al. A gene regulatory network for root epidermis cell differentiation in Arabidopsis. PLoS Genet. 2012;8(1):e1002446. https://doi.org/10.1371/journal.pgen.1002446.
Wilkinson SW, Hannan Parker A, Muench A, et al. Long-lasting memory of jasmonic acid-dependent immunity requires DNA demethylation and ARGONAUTE1. Nat Plants. 2023;9(1):81–95. https://doi.org/10.1038/s41477-022-01313-9.
He K, Du J, Han X, et al. PHOSPHATE STARVATION RESPONSE1 (PHR1) interacts with JASMONATE ZIM-DOMAIN (JAZ) and MYC2 to modulate phosphate deficiency-induced jasmonate signaling in Arabidopsis [published online ahead of print, 2023 Feb 28]. Plant Cell. 2023. koad057.
Li G, Wang L, Yang J, He H, Jin H, Li X, Ren T, Ren Z, Li F, Han X, Zhao X, Dong L, Li Y, Song Z, Yan Z, Zheng N, Shi C, Wang Z, Yang S, Xiong Z, Zhang M, Sun G, Zheng X, Gou M, Ji C, Du J, Zheng H, Doležel J, Deng XW, Stein N, Yang Q, Zhang K, Wang D. A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat Genet. 2021;53(4):574–84. https://doi.org/10.1038/s41588-021-00808-z. Epub 2021 Mar 18. PMID: 33737755; PMCID: PMC8035075.
Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. https://doi.org/10.1093/nar/25.17.3389.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;W29–W37. https://doi.org/10.1093/nar/gkr367. 39(Web Server issue).
Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The pfam protein families database. Nucleic Acids Res. 2000;28(1):263–6. https://doi.org/10.1093/nar/28.1.263.
Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46(D1):D493–6. https://doi.org/10.1093/nar/gkx922.
Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;Chap. 2:. https://doi.org/10.1002/0471250953.bi0203s00.
Guo AY, Zhu QH, Chen X, Luo JC. [GSDS: a gene structure display server]. Yi Chuan. 2007;29(8):1023–6.
Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;W202–8. https://doi.org/10.1093/nar/gkp335. 37(Web Server issue).
Lescot M, Déhais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, Rouzé P, Rombauts S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30(1):325–7. https://doi.org/10.1093/nar/30.1.325. PMID: 11752327; PMCID: PMC99092.
Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. https://doi.org/10.1101/gr.092759.109.
Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4:10. https://doi.org/10.1186/1471-2229-4-10. PMID: 15171794; PMCID: PMC446195.
Chen C, Chen H, Zhang Y, et al. TBtools: an integrative Toolkit developed for interactive analyses of big Biological Data. Mol Plant. 2020;13(8):1194–202. https://doi.org/10.1016/j.molp.2020.06.009.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–8. https://doi.org/10.1006/meth.2001.1262.
Acknowledgements
We thank all our colleagues for providing useful discussions and technical assistance. We are very grateful to the editor and reviewers for critically evaluating the manuscript and providing constructive comments for its improvement.
Funding
This research was supported by the Talent Initiation Funding Project of Chengdu University (No. 2081923015), the Incubation and Cultivation Program for Innovative Training of College Students (No. CDUCX2023403), the Sichuan Science and Technology Program (No. 2022YFS0435) and the Chengdu Science and Technology Program (No. 2022-YF05-00447-SN).
Author information
Authors and Affiliations
Contributions
XC planned and designed the research and analyzed the data. XC wrote the manuscript. CY, and Jh-L studied gene expression using qRT-PCR. Jt-L identified the S. cereale bHLH gene family and analyzed its gene structures. JF studied chromosome distribution, gene duplication, and syntenic analysis of the S. cerealerye bHLH genes. XC and HD analyzed the evolutionary relationship between bHLH genes in several different species. XC and XG supervised the study. QY and TK revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies involving human participants or animals performed by the authors. These methods were carried out by relevant guidelines and regulations. S. cereale cv. Weining, a traditional variety in Guizhou Province in southwest China. Based on international standards, all the experimental research and field studies on plants, including the collection of plant material were approved by Chengdu University. This material was stored in the Sichuan Provincial Crop Germplasm Bank of Chengdu University, numbered Sc032.
Consent for publication
Not applicable.
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Table S1.
List of the 220 S. cereale bHLH genes identified in this study
Supplementary Material 2: Table S2.
Analysis and distribution of conserved motifs of bHLH proteins in seven species
Supplementary Material 3: Table S3.
Cis-regulatory elements in the promoter region of ScbHLH genes
Supplementary Material 4: Table S4.
The tandem duplication clusters of ScbHLH genes
Supplementary Material 5: Table S5.
The six pairs of segmental duplicates in S. cereale bHLH genes
Supplementary Material 6: Table S6.
One-to-one orthologous relationships between S. cereale with other plants
Supplementary Material 7: Table S7.
Results of Tajima’s D neutrality test
Supplementary Material 8: Table S8.
Primer sequences for qRT-PCR
Supplementary Material 9:
Figure S1–S6 in this study
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, X., Yao, C., Liu, J. et al. Basic helix-loop-helix (bHLH) gene family in rye (Secale cereale L.): genome-wide identification, phylogeny, evolutionary expansion and expression analyses. BMC Genomics 25, 67 (2024). https://doi.org/10.1186/s12864-023-09911-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09911-3