
Abstract
Background
Plant-parasitic nematodes (PPNs) that cause most damage include root-knot nematodes (RKNs) which are a major impediment to crop production. Root-knot nematodes, like other parasites, secrete proteins which are required for parasite proliferation and survival within the host during the infection process.
Results
Here, we used various computational tools to predict and identify classically and non-classically secreted proteins encoded in the Meloidogyne javanica genome. Furthermore, functional annotation analysis was performed using various integrated bioinformatic tools to determine the biological significance of the predicted secretome. In total, 7,458 proteins were identified as secreted ones. A large percentage of this secretome is comprised of small proteins of ≤ 300 aa sequence length. Functional analyses showed that M. javanica secretome comprises cell wall degrading enzymes for facilitating nematode invasion, and migration by disintegrating the complex plant cell wall components. In addition, peptidases and peptidase inhibitors are an important category of M. javanica secretome involved in compatible host-nematode interactions.
Conclusion
This study identifies the putative secretome encoded in the M. javanica genome. Future experimental validation analyses can greatly benefit from this global analysis of M. javanica secretome. Equally, our analyses will advance knowledge of the interaction between plants and nematodes.
Similar content being viewed by others


Background
Soil-borne plant parasites, members of the genus Meloidogyne (root-knot nematodes, RKNs) are some of the most yield-limiting plant-parasitic nematodes (PPNs). On a global scale, PPNs cause huge economic losses in the agricultural sector with annual losses estimated at $US157 billion [1, 2]. To date, approximately 100 RKN species are documented. The four dominant globally distributed RKN species are M. arenaria, M. javanica, M. incognita, and M. hapla. These highly polyphagous nematodes are capable of infecting nearly all higher vascular plants including economically important crops such as potato, tomato, maize, rice, tobacco, sweet potato, pineapple, and woody perennial plants [1, 3].
Initial infection of any parasitic organism is challenged by the host defense system. However, these parasites have devised ways to dampen the host immune system and mechanisms for their survival [4]. Parasites produce a diverse range of excretory-secretory products (ESPs, also known as secretome) during the infection process to interact with the host proteins. The secretome constitutes functionally distinct molecules including digestive and detoxification enzymes, toxins, proteases, serpins, as well as small RNAs that can imitate host miRNAs [4, 5]. The ESPs are the main actors that mediate successful interactions at the host-parasite interface [6]. The ESPs are essential for parasite survival and are involved in various cellular activities including host invasion, nutrient acquisition, parasite development, evasion, and modulation of the host immune system [5].
As effective management strategies against RKN species diminish and become obsolete, there is a need for the development of new environmentally benign approaches [7, 8]. Identifying so far unexplored nematode ESPs and understanding their mechanism in host cells unfolds the possibility of developing novel control strategies against nematode pests.
In the genus Meloidogyne, many secretory proteins have been identified and studies geared towards deciphering their mechanism in plant parasitism are developing at an increasing pace [9]. Candidate secretory proteins have been identified from proteomes, genomes, and/or transcriptome analyses. For instance, mass spectrometry analysis has been used directly to identify 486 M. incognita secretory proteins [10]. Previously, [11] and [12] mined 37 and 18 putative effectors, respectively from M. incognita cDNA libraries of esophageal glands.
Global transcriptomic analysis of pre-parasitic and parasitic phases of M. graminicola identified putative secretory proteins [13]. Moreover, dual RNA sequencing analysis, a tool that can simultaneously profile both host and pathogen transcriptional changes during the pathogenicity, led to the discovery of M. incognita secretory proteins in a stage-specific manner [14, 15]. Secretome analysis using computational tools identified 1,886 secretory proteins encoded in M. incognita genome [16]. In addition, phylogenomics in conjunction with RNA-seq analysis allowed the discovery of 68 phytoparasite-specific proteins strongly expressed during the endophytic stages of M. incognita [17].
In view of the above, it is evident that previous studies have largely focused on identification of secretory proteins from M. incognita using in silico tools, proteomics, and genome-wide approaches. This could be due to the availability and accessibility of M. incognita genome sequences earlier than other RKN species [18], its highly polyphagous nature, and its classification as the most damaging crop pest worldwide [2]. Now, with the accessibility of genome sequences from other important Meloidogyne spp. including M. arenaria, M. javanica, M. hapla, M. graminicola, M. floridensis, M. luci, M. exigua, and M. enterolobii [19,20,21,22,23,24] genome-wide analysis, and/or comparative genomics are plausible to reveal the common mechanism that drive RKN infections and potentially guide towards identification of new control strategies specifically directed against Meloidogyne spp.
The availability of M. javanica genome sequences allowed us to explore its secretome systematically and comprehensively using bioinformatics approaches. Indeed, computational tools have been used widely in the discovery of putative ESPs in PPNs and animal parasitic nematodes (APNs) [16, 25,26,27]. Here, we identified classically and non-classically secreted proteins, followed by functional annotation and in-depth insights into the predicted M. javanica secretome. The present study not only adds to the secretory proteins repertoire from M. javanica but also provides a resourceful biological database that lays a framework for future functional characterization of these putative M. javanica secretory proteins. In addition, this research provides a foundation for understanding the interaction between RKNs and their host plants.
Results
In silico identification of putative secretory proteins from the M. javanica genome
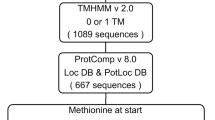
Nematode secretions released by the infective juveniles are crucial for a successful infection process and participate in various stages of nematode parasitism [28]. Here, we applied various bioinformatics tools to predict M. javanica secreted proteins. Of the 97,208 proteins encoded in the M. javanica genome, 5,258 and 2,369 were identified as classically and non-classically secreted proteins, using SignalP and SecretomeP softwares. The 7,627 secreted proteins identified were without transmembrane domains according to TMHMM and Phobius analysis. TargetP analysis identified 11 proteins as mitochondrial-target proteins, and these were discarded.
Further, PS-SCAN was used to scan endoplasmic reticulum-based proteins while PreGPI detected secretory proteins but linked to the membrane by a GPI anchor. In total, 26 proteins with endoplasmic reticulum retention signal and 132 proteins predicted to be GPI anchored proteins were removed from the data set. Thus, the bioinformatic pipeline predicted a total of 7,458 (7.7%) of the entire proteome as the secretome, and these were used for further analysis (Fig. 1, Additional file 2: Table S1). Overall, 1,269 (17%) predicted secreted proteins (Additional file 2: Table S2) matched to the NCBI dbEST database (https://www.ncbi.nlm.nih.gov/nuccore/?term=Meloidogyne+javanica). This implies that these proteins are expressed during the nematode infection cycle and may be involved in nematode parasitism.

Overview of bioinformatic pipeline to predict putative secretory proteins from the genome of root-knot nematode, Meloidogyne javanica (See the materials and methods section for detailed criteria for secreted proteins prediction)
The size distribution analysis of the predicted secreted proteins showed that 50.1% (3,739) of the proteins were between 101 and 300 aa, followed by proteins with a sequence length of below 100 aa accounted for 32.4% (2,413 proteins). The remaining secretome is made up of 906 (12.1%) proteins with sequence lengths between 301 and 500 aa, 344 (4.6%) proteins between 501 and 1000 aa, and 56 (0.2%) proteins with more than 1000 aa (Additional file 1: Figure S1). The large proportion of the predicted secretome constituted small-sized proteins that can be easily released to the extracellular medium of the parasite where they mediate interactions with their host.
We searched for the Mel-DOG promoter motif (Meloidogyne DOrsal Gland; TGCMCTT) identified in the promoter of Meloidogyne incognita dorsal gland encoding effectors [29] within 2 kbupstream of the coding region of genes of the predicted M. javanica secretome. The core motif was detected in the 2 kb upstream of the coding region of 1,608 secretory proteins out of the 7,256 promoter regions retrieved from WormBase database (Additional file 2: Table S3), adding to the criteria for identifying RKN effectors.
Functional annotation of M. javanica secretory proteins
Gene ontology (GO) analysis
Out of the 7,458 secreted proteins, 24.1% (1,797) were assigned to 3,513 GO terms with 1,044 GO terms from biological process category, 1,216 GO terms from molecular function and 1,253 GO terms from cellular component category. A summary of the GO term annotation is shown in Fig. 2, Additional file 2: Table S4.

Gene ontology distribution of secretory proteins from Meloidogyne javanica
GO terms such as cellular (GO:0009987; 43.6%), and metabolic process (GO:0008152; 47.2%), biological regulation (GO:0065007; 26%), regulation of biological process (GO:0050789; 23.8%), developmental process (GO:0032502; 13.9%) and response to stimulus (GO:0050896; 9.5%) were the most represented GO terms under biological process category (Fig. 2, Additional file 2: Table S4).
The top represented GO terms at the molecular function category level were catalytic activity (GO:0003824; 35.7%) binding (GO:0005488; 39.3%), and molecular function regulator (GO:0098772; 5.8%) (Fig. 2). The most dominant sub-categories in catalytic activity terms were hydrolase activity (GO:0016787; 20.1%), catalytic activity action on a protein (GO:0140096; 14.4%), and transferase activity (GO:0016740; 8%). In binding term, ion (GO:0043167; 29.4%), heterocyclic compound (GO:1,901,363; 20.4%), organic cyclic compound (GO:0097159;20.4%), protein (GO:0005515; 5%), drug binding (GO:0008144; 5.6%), small molecule (GO:0036094; 6.1%) and carbohydrate derivative binding (GO:0097367; 5.8%) were the most predominant sub-categories (Additional file 1: Figure S3). Lastly, enzyme regulator activity (GO:0030234; 4.9%) was the abundant sub-category in molecular function regulator terms.
At the cellular component category, membrane (GO:0016020; 50.9%), membrane part (48.9%) cell (GO:0005623; 31.6%), cell part (GO:0044464; 31.6%) organelle (GO:0043226; 24.2%), and extracellular region (GO:0005576; 16.5%) were found to be highly represented GO terms within this category (Fig. 2).
Gene ontology enrichment analysis
GO enrichment analysis was performed using the WEGO genomics tool to detect significantly enriched GO terms in the predicted secretome compared to the total proteome at a P-value of 0.05. In the cellular component category, cell surface, extracellular region, extracellular region part, extracellular space, extracellular matrix, transporter complex, and receptor complex were found to be significantly enriched terms (Fig. 3, Additional file 1: Figure S3). Carbohydrate binding, molecular function regulator, catalytic, activity acting on proteins, and enzyme regulator activity were over-represented in the molecular function category. Finally, under biological process category, anatomical structure development, response to biotic stress stimulus, regulation of metabolic process, regulation of molecular function, response to other organisms, biological and cell adhesion, secondary metabolic process, and pigment metabolic process showed significant enrichment in the predicted secretome in comparison to the distribution of the same GO terms in the total proteome (Fig. 3, Additional file 1: Figure S3).

Gene Ontology enrichment analysis of Meloidogyne javanica secretome in comparison to the total proteome. This figure only displays secretome GO terms that are significantly enriched at (P ˂0.05)
Identification of peptidases and peptidase inhibitors in M. javanica secretome
Peptidases are hydrolytic enzymes found in parasitic nematodes. These enzymes are important virulence factors that influence nematode-host interactions and facilitate penetration of host tissues, sustenance through host protein digestion, parasite growth and survival, and immune system evasion [30]. Here, 276 (3.7%) secreted proteins were identified as peptidase enzymes representing the five major catalytic classes belonging to 43 families presented in Fig. 4, Additional file 2: Table S5. These include 65 cysteines (C), 104 serine (S), 70 metallo (M), 17 aspartic (A), and 10 threonines (T) peptidases distributed into 43 families. The most abundant peptidases were C1A, S9, S10, S1A, and M12A. Moreover, 248 (3.3%) proteins were annotated as peptidase inhibitors (I) belonging to 18 families in the predicted M. javanica secretome. The most represented families include I2 (Kunitz inhibitors, 50 proteins) I63 (90 proteins), I35 (19 proteins), and I43 (25 proteins) among others (Fig. 4, Additional file 2: Table S4).

Distribution of peptidases and peptidase inhibitors identified in Meloidogyne javanica secretome
Analysis of cell wall degrading enzymes (CWDEs)
Cell wall degrading enzymes are a major category of PPNs secretome that mediate compatible interactions [31]. Here, 146 secretory proteins were mapped into 31 families representing five main functional classes of CWDEs including glycoside hydrolases (GH, 90 proteins), glycosyltransferases (GT, 19 proteins), pectate lyases (PL, 36 proteins), and carbohydrate-binding modules (CBM, 1 protein) shown in Additional file 1: Figure S4, Additional file 2: Table S5. The most represented Carbohydrate-Active Enzymes (CAZymes) sub-families include GH5-2 (27 proteins), GH30 (9 proteins), GH28 (6 proteins), GH18 (10 proteins) PL3-3 (21 proteins), and PL3 (13 proteins) (Additional file 1: Figure S4 and Additional file 2: Table S5).
Protein domain analysis
Using InterProScan domain search, 1915 (25.7%) of the predicted secreted proteins were mapped into 412 known protein domains and families. The most abundant InterProScan domains summarized in Table 1, Additional file 2: Table S7 include protein kinase domain, peptidase C1A papain C-terminal (cysteine peptidase family C1, sub-family C1A (papain family, clan CA), C-type lectin-like domain, Transthyretin-like, CAP (cysteine-rich secretory proteins) domain, Pancreatic trypsin inhibitor Kunitz domain, Modulator of levamisole receptor-1, Peptidase S10 serine carboxypeptidase, Pectate lyase PlyH/PlyE-like, Glycoside hydrolase, family 5, and DnaJ domain.
Metabolic pathway analysis
Out of the 7,458 predicted secretory proteins, 672 (9.0%) were mapped into 470 KEGG pathways presented in Additional file 2: Table S8. The most abundant KEGG pathways displayed in Table 2 include lysosome, protein processing in endoplasmic reticulum, autophagy, apoptosis pathways of neurodegeneration-multiple diseases, pathways in cancer, antigen processing, and representation, and calcium signaling pathway. In addition, KEGG functional hierarchies revealed molecular entities linked to membrane trafficking, exosome, peptidases and peptidase inhibitors, chaperones and folding catalyst, transcription factors, protein kinases, chromosome, and associated proteins and spliceosome were found to be highly represented (Additional file 2: Table S9) which are essential for the parasite’s existence inside the host [16].
Identification of Meloidogyne javanica species-specific secretome and discovery of potential targets for functional analysis
The 7,458 predicted secreted proteins of M. javanica were searched for homologues in other closely related Meloidogyne species. From this comparative analysis, the tropical Meloidogyne spp. shared 323 orthologous groups with 1,737 proteins which can be explored further to uncover typical mechanisms RKNs use to undermine host immunity and promote disease processes. A set of 599 clusters with 1,684 proteins was specific to M. javanica secretome (Fig. 5, Additional file 1: Figure S5).

A Venn diagram depicting the orthologous protein clusters shared in the secretome of four root-knot nematode species
A further BLAST analysis of the 1,684 proteins against the NCBI non-redundant proteins database showed that the majority of these proteins had homologues to M. graminicola and M. enterolobii suggesting that RKN-secreted proteins are highly conserved within the genus. Among these, a total of 382 proteins were identified as M. javanica species-specific secreted proteins since there were no homologous proteins in the NCBI non-redundant protein database (Additional file 2: Table S10). According to Argot functional prediction analysis [32], 63 protein sequences out of 382 proteins were annotated with 123 different GO terms (Additional file 2: Table S11). The REVIGO tool was used to obtain of non-redundant GO terms which were classified into functional groups [33]. Major biological processes associated M. javanica species-specific secreted proteins include spliceosomal complex assembly, carbohydrate metabolic process, cell death, defense response, protein ubiquitation, DNA modification, regulation of DNA-templated transcription, microtubule-based process, negative regulation of Wnt signaling pathway, homophilic cell adhesion via plasma membrane adhesion molecules and positive regulation of siRNA production. The major molecular function categories include transcription cis-regulatory region binding, DNA binding, DNA-binding transcription factor activity, actin binding, aspartic-type endopeptidase activity, hydrolase activity, hydrolyzing O-glycosyl compounds, hydrolase activity, acting on glycosyl bonds, lipid binding, N-methyltransferase activity, protein-containing complex binding, and collagen binding. The major cellular component represented include extracellular region, intracellular anatomical structure, spliceosomal complex, nuclear pore, nucleus, cytoskeleton, external enscapulating structure and other organism part (Additional file 2: Table S12).
Of the 382 secretory proteins, 36 secretory proteins were supported at the transcriptional level (EST mapping, Additional file 2: Table S10). In addition, 50 predicted secreted proteins had the Mel-DOG promoter motif (Additional file 2: Table S10) which is associated with effector proteins expressed within the nematode dorsal oesophageal glands [29]. Taken together, these secreted protein sets constitute the best candidates for functional studies, as revealed by our analysis of their likely involvement in M. javanica infection process. The host target repertoire of this effectome can be uncovered by further functional research, which will unvarel the virulence mechanism and host range susceptibility of M. javanica species.
We subjected the 382 protein sequences to a fold recognition analysis using the Phyre2 algorithm to obtain additional functional information for this set of specific secreted proteins. The Phyre2 tool determines the structure and function of novel protein sequences using advanced homology detection algorithms [34, 35]. In this case, if the predicted structure of the query protein is derived with high confidence, the template protein function’s can be tentatively attributed to the query protein. Table 3 shows the proteins that attained confidence scores of 50% which was the minimum cut off value. The protein M. Javanica_Scaff8743g053112 had the highest structural similarity to repeat five residue (rfr) protein whose function is yet to be known.
We performed BLAST analysis to identify M.javanica secreted proteins with homologues in C. elegans. M. javanica proteins homologous to C.elegans RNAi lethal phenotype genes can be potential targets for functional analysis using RNA interference (RNAi) tool and inform designing of control strategies. Meloidogyne javanica protein sequences with homologues in host proteomes were excluded from this analysis considering that they are less likely to be exploited as nematode control targets due to off-targets effects. In total, 5,289 secretory proteins were detected as RKN-specific shown in Additional file 1: Figures S6 were blasted against C. elegans’ lethal phenotype genes. On the premise that homologous genes retain similar functions in other organisms and share other crucial features [36]. In total 227 secretory proteins were similar to C. elegans RNAi lethal phenotype genes including transthyretin-like protein family, peptidases, and peptidase inhibitors (Additional file 2: Table S13).
Discussion
Meloidogyne javanica and M. incognita are both tropical Meloidogyne species with comparable secretomes. Despite being closely related, the two RKN species exhibit differences in their host ranges [37]. It is now understood that phytonematode genes encoding effector proteins are key determinants of host specificity [38, 39]. Consequently, variations in pathogen effector repertoires could potentially influence pathogen host range changes, including host range expansion, including host range expansion, which leads to reproductive isolation and subsequent pathogen speciation [40]. It is therefore important to identify secreted proteins of an individual parasite as these are essential for molecular dialogue between the host and the parasite as well as the parasite’s survival within host tissues [5, 6].
The ever-increasing number of sequenced genomes of PPNs provides an unparalleled opportunity to delve into their secretome, which is critical for their ability to survive within host cells. To this end, we applied various computational approaches to predict and annotate the secretome encoded in the M. javanica genome (Fig. 1). This work represents the first in silico prediction of M. javanica secretome which accounts for 7.7% of the total proteome. The size of M. javanica secretome is comparable to those reported in other PPNs in comparison to their proteome size [16, 27, 41] indicating that ˂10% of the total proteome is secreted.
We examine the composition of RKNs secretomes by comparing the functional annotation of M. javanica secretome, reported in this study with the secretome of M. incognita predicted by [16]. Parallels can be drawn between M. javanica and M. incognita secretome, given that Meloidogyne species have the same lifestyle, as obligate biotrophs [42]. One observation of GO enrichment analysis is that RKN-secreted proteins are mostly present on the cell surface, extracellular region, space, and matrix cell compartments (Fig. 3) that are probably involved in virulence and mediate parasite-host interactions [43, 44].
Molecular entities linked to hydrolase activity, lyase activity, and carbohydrate-binding activity under MF were enriched in the secretome compared to the proteome in the GO enrichment analysis (Fig. 3). These molecular activities are associated with the functions of CWDEs. RKN predicted proteins were mapped into various CWDEs families, namely glycoside hydrolase, glycosyltransferase, polysaccharide lyases, and carbohydrate-binding modules. CWDEs provide an essential catalog of RKN secretomes, which not only facilitate nematode’s penetration and migration through host tissue, but they also help nematodes utilize plant nutrients as a source of food and develop nematode feeding sites [31, 45, 46]. The composition of plant cell walls is important in the interaction between plants and nematodes [47]. As a result, RKNs harbour various families of CWDEs in their secretome. This reflects the strategic ways employed by these parasites to break down and modify the multifaceted plant cell wall structure to invade and exploit host nutrients necessary for a successful infection.
Pectate lyases and glycoside hydrolase 5 represent the most abundant families found in this study and are also among the differentially expressed effector-encoding genes implicated in cell wall modification during M. javanica infection process [48]. The functional characterization of M. graminicola pectate lyase (Mg-PEL1) revealed that it is involved in virulence and facilitates RKN parasitism [49].
Protein domain analysis revealed that M. javanica secretome harbors secretory proteins with distinct protein domains which are largely shared with the closely related M. incognita secretome. The protein kinase domain (IPR000719, also supported by our KEGG analysis) was, for example, one of the most represented domains according to InterProscan analyses (Table 1, Additional file 2: Table S7). Signaling transduction pathways such as protein kinases are critical in facilitating microbes’ transition and adaptation inside a host [50]. Several C-type (IPR001304) proteins were among the top represented domains (Table 1, Additional file 2: Table S7), which have previously been shown to aid nematode parasitism by regulating plant defense responses [51, 52].
Additionally, secreted proteins containing transthyretin-like (IPR001534) and thioredoxin (IPR013766) domains (Table 1, Additional file 2: Table S7) facilitate compatible host-nematode interactions by protecting the nematode against toxic reactive oxygen species molecules generated when the host perceives a parasitic nematode [53, 54]. Altogether these findings eminently show that the various domains represented by RKNs secretome play diverse roles in shaping plant-nematode interactions. Functional annotation analysis indicates that RKN-secreted proteins associated with lysosome, protein processing in endoplasmic reticulum, spliceosome, cAMP signaling pathway, calcium signaling pathway, and pathways in cancer (Table 2, Additional file 2: Table S8) are important for phytonematode survival inside the host [16, 27].
Enzyme regulator function was found enriched in M. javanica secretomes (Fig. 3) which are associated with the regulation of enzymes such as peptidases which are important virulence factors in nematodes [30]. Peptidases and peptidase inhibitors constitute a significant component of PPNs-secreted proteins [27, 55]. In our secretome analysis, 276 proteins were annotated as peptidases mapped into five major catalytic classes distributed across 43 families. Similarly, the various peptidase classes are spread in the secretome of various PPN groups including the genus Meloidogyne [27]. The most abundant peptidase classes in this study were cysteine and serine peptidases (Fig. 4) which were supported by our domain analysis (Table 1). Both catalytic classes play important roles in PPN infection processes such as host tissue invasion, nematode development, reproduction, and pathogenicity. Furthermore, it has been demonstrated that silencing cysteine and serine peptidases in parasitic nematodes is detrimental to nematode fecundity and parasitism [56,57,58,59]. Peptidase inhibitors, on the other hand, are implicated in defense mechanisms that avoid peptidase-mediated defense, manipulate host defense responses, and facilitate nematode feeding [30, 60]. The present study shows that peptidase inhibitors are important for nematode-host interactions where several peptidase inhibitors were identified and distributed across 18 families of known peptidase inhibitor classes.
Although there are parallels between M. javanica and M. incognita secretomes, it was noteworthy that secondary and pigment metabolic processes, developmental processes, and anatomical developmental structure were enriched in M. javanica secretome (Fig. 3) but not in M. incognita secretome [16]. Secondary metabolites and pigment compounds are crucial for parasite survival and pathogenicity [61]. Though precise functions in RKN infection are unknown. However, comparable roles may be hypothesized for RKN parasitism. Enrichment of developmental process and anatomical developmental structure implies that RKN-secreted proteins function in the development and maintenance of giant cells, which provide nutrients to the developing nematode after host colonization [62].
Further comparative analysis with different Meloidogyne species secretomes was carried out to uncover species-specific M. javanica secretome. The species-specific proteins may reveal the parasite-specific molecular mechanism of host adaptation and specialization [40]. Here, 382 M. javanica-specific secreted proteins were discovered that may help to explain nematode lifestyle, virulence, and host specificity and could eventually be applied to design control strategies for M. javanica infestations.
These secreted proteins might be critical in mediating M. javanica parasitic survival, as shown by functional groups identified such as defense and cell death. Remarkably, positive regulation of siRNA production and regulation of miRNA-mediated gene silencing were among the represent GO terms. Current research indicates that pathogen small RNAs play an active role in virulence [63]. However, their regulatory role in PPNs is unknown. Molecular activities associated with these secreted protein sets include hydrolase and aspartic-type endopeptidase activity which are related to typical function of secreted proteins. Many of these predicted sets of secreted proteins (Additional file 2: Table S10 and Table S13) can be exploited for functional characterization and can also be utilized as a panel of effectors in root-knot nematode ‘effectorome’ investigations.
Conclusions
In conclusion, this study sheds light on what constitutes the secretome of M. javanica. To the best of our knowledge, this is the first account of M. javanica secretome being identified and functionally annotated. In silico analysis of secretomes encoded by parasites’ genomes can precede and complement the laborious and complex experimental methodologies applied in deciphering the function of putative excreted-secreted proteins.
Materials and methods
Prediction of excretory-secretory proteins from the M. javanica genome
The proteome deduced from M. javanica genome under BioProject ID PRJEB8714 [20] was retrieved from Wormbase parasite database version WBPS14. Our bioinformatics workflow integrated various computational tools to identify and annotate ESPs from the proteome is summarized in Fig. 1. Firstly, the SignalP v5.0 [64] was applied to predict signal peptides of classically secreted proteins, with parameters set for eukaryote organisms, cleavage site determined by the software. SecretomeP v1.0 with default option for mammalian organisms and cut-off over 0.9 NN- scores was used to predict proteins secreted through unconventional secretory protein pathways accounting for a set of non-classically secreted proteins [65]. Both sets of predicted secretory proteins were scanned with TMHMM (version 2.0) [66] and Phobius [67] for the presence of transmembrane helices using default parameters. All the proteins with a transmembrane region were discarded from this dataset. Mitochondrial targeting proteins predicted by TargetP v1.1 [68] with a probability score over 0.90 were eliminated from the dataset. The resulting list of proteins was subsequently scanned for the presence of endoplasmic reticulum retention signal and glycosylphosphatidylinositol (GPI) anchor signals by PS SCAN [69] (Prosite pattern: PS00014) and PredGPI [70], respectively using default parameters. Finally, all the protein sequences exhibiting ER signals and GP-anchor signals were excluded from the dataset and the remaining proteins were considered secretory proteins for further investigation. To determine the proportion of secretory proteins supported at the transcriptional level, the predicted secretory proteins were matched to M. javanica 7,734 expressed sequence tags (EST) retrieved from the NCBI dbEST database (https://www.ncbi.nlm.nih.gov/genbank/dbest/) using tblastn at a cut-off value of 1e-05 and percentage identity of ≥ 50%.
All the secretory proteins were scanned for the presence of the recently identified core motif Mel-DOG (Meloidogyne DOrsal Gland; TGCMCTT) associated with the expression of dorsal gland effectors [29]. The 2 kb putative promoter regions upstream of the coding region were retrieved from the Wormbase database [71]. FIMO webserver (v5.4.1) [72] was used to search for the core motif occurrence (p-value ˂ 0.0001) in the promoter regions of the predicted secretory proteins.
Functional annotation of secretory proteins
Functional annotation of the secretory proteins comprised assignment of Gene Ontology (GO) terms, GO enrichment analysis, mapping protein domains, and pathway associations using KEGG pathways. The secretory proteins were assigned to Gene Ontology (GO) terms into three functional categories of Biological Process (BP), Molecular Function (MF), and Cellular Component (CC) using the PANNZER2 functional annotation web server [73]. GO visualization and enrichment analysis was performed by WEGO 2.0 web tool https://wego.genomics.cn/view/WEGOID72126237138727 [74]. Significantly enriched GO terms were considered at a P-value ˂ 0.05 using the entire proteome GO terms as the reference group. Additionally, peptidases (proteolytic enzymes) and peptidase inhibitors were identified by scanning against the MEROPS database, v11.0 [75] using BLASTP (e-value ˂1e-5). Secretory proteins coding for carbohydrate-active enzymes (CAZymes) were identified using the dbCAN web server v10.0 HMMER (e-value ˂ 1e-15, coverage ˃ 0.35) [76]. Protein families and domains analyses were mined from the Blast2GO suite (version 6.03) [77].
The KAAS (KEGG [Kyoto Encyclopedia of Gene and Genomes], Automatic Annotation Server, v2.1) [78] was used to map all the secretory proteins to KEGG pathways and KEGG BRITE using BBH (bi-directional best hit) method to assign the orthologs [79]. The eukaryotes and nematode datasets were used as reference datasets in KAAS mapping.
Functional analysis of specific M. javanica secretome
To identify putative orthologs in the secretome of closely related Meloidogyne species including M. incognita, M. enterolobii, and M. floridensis we performed orthologous gene clusters using orthovenn2 with the following parameters; e-value 1e-05 and an inflation value 1.5 [80]. The species-specific predicted secretory proteins were searched for sequence similarities against the NCBI nr database using BLASTP (e-value < 1e-05).
Discovery of M. javanica proteins with potential for functional analysis using RNA interefence (RNAi) tool
The predicted M. javanica secretome was searched for similarity against the host proteomes (Solanum tuberosum, Arabidopsis thaliana, Oryza sativa, and Zea mays) using Orthovenn 2 default parameters; e-value 1e-05 and an inflation value 1.5 [80]. The resulting RKN-specific proteins were screened for the presence of Caenorhabditis elegans lethal RNA interference (RNAi) phenotypes genes retrieved from the WormMine database (WS284) using the tblastn program and filtered using an e-value threshold of 1e-05 and a percentage identity of 50%.
Data Availability
All data generated or analyzed during this study are available in the figshare repository, https://doi.org/10.6084/m9.figshare.21641726.
References
Onkendi EM, Kariuki G, Marais M, Moleleki LN. The threat of root-knot nematodes (Meloidogyne spp.) in Africa: a review. Plant Pathol. 2014;63(4):727–37.
Coyne DL, Cortada L, Dalzell JJ, Claudius-Cole AO, Haukeland S, Luambano N, Talwana H. Plant-parasitic nematodes and food security in Sub-Saharan Africa. Annu Rev Phytopathol. 2018;56:381–403.
Saucet SB, Van Ghelder C, Abad P, Duval H, Esmenjaud D. Resistance to root-knot nematodes Meloidogyne spp. in woody plants. New Phytol. 2016;211(1):41–56.
Cooper D, Eleftherianos I. Parasitic nematode immunomodulatory strategies: recent advances and perspectives. Pathogens. 2016;5(3):58.
Bobardt SD, Dillman AR, Nair MG. The two faces of nematode infection: virulence and immunomodulatory molecules from nematode parasites of mammals, insects and plants. Front Microbiol. 2020;11:577–846.
Ranganathan S, Garg G. Secretome: clues into pathogen infection and clinical applications. Genome Med. 2009;1(11):1–7.
Forghani F, Hajihassani A. Recent advances in the development of environmentally benign treatments to control root-knot nematodes. Front Plant Sci 2020, 11.
Hajihassani A, Marquez J, Woldemeskel M, Hamidi N. Identification of four populations of Meloidogyne incognita in Georgia, United States, capable of parasitizing Tomato-Bearing Mi-1.2 gene. Plant Dis. 2022;106(1):137–43.
Mejias J, Truong NM, Abad P, Favery B, Quentin M. Plant proteins and processes targeted by parasitic nematode effectors. Front Plant Sci. 2019;10:970.
Bellafiore S, Shen Z, Rosso M-N, Abad P, Shih P, Briggs SP. Direct identification of the Meloidogyne incognita secretome reveals proteins with host cell reprogramming potential. PLoS Pathog. 2008;4(10):e1000192.
Huang G, Gao B, Maier T, Allen R, Davis EL, Baum TJ, Hussey RS. A profile of putative parasitism genes expressed in the esophageal gland cells of the root-knot nematode Meloidogyne incognita. Mol Plant Microbe Interact. 2003;16(5):376–81.
Rutter WB, Hewezi T, Abubucker S, Maier TR, Huang G, Mitreva M, Hussey RS, Baum TJ. Mining novel effector proteins from the esophageal gland cells of Meloidogyne incognita. Mol Plant Microbe Interact. 2014;27(9):965–74.
Haegeman A, Bauters L, Kyndt T, Rahman MM, Gheysen G. Identification of candidate effector genes in the transcriptome of the rice root knot nematode M eloidogyne graminicola. Mol Plant Pathol. 2013;14(4):379–90.
Petitot AS, Dereeper A, Agbessi M, Da Silva C, Guy J, Ardisson M, Fernandez D. Dual RNA-seq reveals Meloidogyne graminicola transcriptome and candidate effectors during the interaction with rice plants. Mol Plant Pathol. 2016;17(6):860–74.
Shukla N, Yadav R, Kaur P, Rasmussen S, Goel S, Agarwal M, Jagannath A, Gupta R, Kumar A. Transcriptome analysis of root-knot nematode (Meloidogyne incognita)‐infected tomato (Solanum lycopersicum) roots reveals complex gene expression profiles and metabolic networks of both host and nematode during susceptible and resistance responses. Mol Plant Pathol. 2018;19(3):615–33.
Gahoi S, Gautam B. Genome-wide analysis of excretory/secretory proteins in root-knot nematode, Meloidogyne incognita provides potential targets for parasite control. Comput Biol Chem. 2017;67:225–33.
Grynberg P, Coiti Togawa R, Dias de Freitas L, Antonino JD, Rancurel C, Mota do Carmo Costa M, Grossi-de-Sa MF, Miller RN, Brasileiro ACM. Messenberg Guimaraes P: comparative genomics reveals novel target genes towards specific control of plant-parasitic nematodes. Genes. 2020;11(11):1347.
Abad P, Gouzy J, Aury J-M, Castagnone-Sereno P, Danchin EG, Deleury E, Perfus-Barbeoch L, Anthouard V, Artiguenave F, Blok VC. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat Biotechnol. 2008;26(8):909–15.
Szitenberg A, Salazar-Jaramillo L, Blok VC, Laetsch DR, Joseph S, Williamson VM, Blaxter ML, Lunt DH. Comparative genomics of apomictic root-knot nematodes: hybridization, ploidy, and dynamic genome change. Genome Biol Evol. 2017;9(10):2844–61.
Blanc-Mathieu R, Perfus-Barbeoch L, Aury J-M, Da Rocha M, Gouzy J, Sallet E, Martin-Jimenez C, Bailly-Bechet M, Castagnone-Sereno P, Flot J-F. Hybridization and polyploidy enable genomic plasticity without sex in the most devastating plant-parasitic nematodes. PLoS Genet. 2017;13(6):e1006777.
Phan NT, Orjuela J, Danchin EG, Klopp C, Perfus-Barbeoch L, Kozlowski DK, Koutsovoulos GD, Lopez‐Roques C, Bouchez O, Zahm M. Genome structure and content of the rice root‐knot nematode (Meloidogyne graminicola). Ecol Evol. 2020;10(20):11006–21.
Koutsovoulos GD, Poullet M, Elashry A, Kozlowski DK, Sallet E, Da Rocha M, Perfus-Barbeoch L, Martin-Jimenez C, Frey JE, Ahrens CH. Genome assembly and annotation of Meloidogyne enterolobii, an emerging parthenogenetic root-knot nematode. Sci Data. 2020;7(1):1–13.
Susič N, Koutsovoulos GD, Riccio C, Danchin EG, Blaxter ML, Lunt DH, Strajnar P, Širca S, Urek G, Stare BG. Genome sequence of the root-knot nematode Meloidogyne luci. J nematology 2020, 52.
Phan NT, Besnard G, Ouazahrou R, Sánchez WS, Gil L, Manzi S, Bellafiore S. Genome sequence of the coffee root-knot nematode Meloidogyne exigua. J Nematology. 2021;53(1):1–6.
Gomez S, Adalid-Peralta L, Palafox-Fonseca H, Cantu-Robles VA, Soberon X, Sciutto E, Fragoso G, Bobes RJ, Laclette JP. Yauner LdP: genome analysis of Excretory/Secretory proteins in Taenia solium reveals their abundance of antigenic regions (AAR). Sci Rep. 2015;5(1):1–11.
Wang S, Wei W, Cai X. Genome-wide analysis of excretory/secretory proteins in Echinococcus multilocularis: insights into functional characteristics of the tapeworm secretome. Parasites & vectors. 2015;8(1):1–13.
Gahoi S, Singh S, Gautam B. Genome-wide identification and comprehensive analysis of Excretory/Secretory proteins in nematodes provide potential drug targets for parasite control. Genomics. 2019;111(3):297–309.
Goverse A, Smant G. The activation and suppression of plant innate immunity by parasitic nematodes. Annu Rev Phytopathol. 2014;52:243–65.
Da Rocha M, Bournaud C, Dazenière J, Thorpe P, Bailly-Bechet M, Pellegrin C, Péré A, Grynberg P, Perfus-Barbeoch L. Eves-van den Akker S: genome expression Dynamics reveal the Parasitism Regulatory Landscape of the Root-Knot Nematode Meloidogyne incognita and a promoter Motif Associated with Effector genes. Genes. 2021;12(5):771.
Malagón D, Benítez R, Kasny M, Adroher FJ, Erzinger G. Peptidases in parasitic nematodes. A review. Parasites: Ecology, diseases and management Erzinger GS, Editor United Kingdom 2013:61–102.
Ali MA, Azeem F, Li H, Bohlmann H. Smart parasitic nematodes use multifaceted strategies to parasitize plants. Front Plant Sci 2017:1699.
Lavezzo E, Falda M, Fontana P, Bianco L, Toppo S. Enhancing protein function prediction with taxonomic constraints–the Argot2. 5 web server. Methods. 2016;93:15–23.
Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6(7):e21800.
Kelley LA, Sternberg MJ. Protein structure prediction on the web: a case study using the Phyre server. Nat Protoc. 2009;4(3):363–71.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58.
Gabaldón T, Koonin EV. Functional and evolutionary implications of gene orthology. Nat Rev Genet. 2013;14(5):360–6.
Moens M, Perry RN, Starr JL. Meloidogyne species - a diverse group of novel and important plant parasites. Root-knot Nematodes 2009:1–17.
Rutter WB, Hewezi T, Maier TR, Mitchum MG, Davis EL, Hussey RS, Baum TJ. Members of the Meloidogyne avirulence protein family contain multiple plant ligand-like motifs. Phytopathology. 2014;104(8):879–85.
Bournaud C, Gillet F-X, Murad AM, Bresso E, Albuquerque EV, Grossi-de-Sá MF. Meloidogyne incognita PASSE-MURAILLE (MiPM) gene encodes a cell-penetrating protein that interacts with the CSN5 subunit of the COP9 signalosome. Front Plant Sci. 2018;9:904.
Schulze-Lefert P, Panstruga R. A molecular evolutionary concept connecting nonhost resistance, pathogen host range, and pathogen speciation. Trends Plant Sci. 2011;16(3):117–25.
Shinya R, Morisaka H, Kikuchi T, Takeuchi Y, Ueda M, Futai K. Secretome analysis of the pine wood nematode Bursaphelenchus xylophilus reveals the tangled roots of parasitism and its potential for molecular mimicry. PLoS ONE. 2013;8(6):e67377.
Jones JT, Haegeman A, Danchin EG, Gaur HS, Helder J, Jones MG, Kikuchi T, Manzanilla-López R, Palomares‐Rius JE, Wesemael WM. Top 10 plant‐parasitic nematodes in molecular plant pathology. Mol Plant Pathol. 2013;14(9):946–61.
Jing L, Guo D, Hu W, Niu X. The prediction of a pathogenesis-related secretome of Puccinia helianthi through high-throughput transcriptome analysis. BMC Bioinformatics. 2017;18(1):1–13.
Neu E, Debener T. Prediction of the Diplocarpon rosae secretome reveals candidate genes for effectors and virulence factors. Fungal biology. 2019;123(3):231–9.
Rybarczyk-Mydłowska K, Maboreke HR, van Megen H, van den Elsen S, Mooyman P, Smant G, Bakker J, Helder J. Rather than by direct acquisition via lateral gene transfer, GHF5 cellulases were passed on from early Pratylenchidae to root-knot and cyst nematodes. BMC Evol Biol. 2012;12(1):1–10.
Wu G-L, Kuo T-H, Tsay T-T, Tsai IJ, Chen PJ. Glycoside hydrolase (GH) 45 and 5 candidate cellulases in Aphelenchoides besseyi isolated from bird’s-nest fern. PLoS ONE. 2016;11(7):e0158663.
Rai KM, Balasubramanian VK, Welker CM, Pang M, Hii MM, Mendu V. Genome wide comprehensive analysis and web resource development on cell wall degrading enzymes from phyto-parasitic nematodes. BMC Plant Biol. 2015;15(1):1–15.
Fitoussi N, Borrego E, Kolomiets MV, Qing X, Bucki P, Sela N, Belausov E, Miyara SB. Oxylipins are implicated as communication signals in tomato–root-knot nematode (Meloidogyne javanica) interaction. Sci Rep. 2021;11(1):1–16.
Chen J, Li Z, Lin B, Liao J, Zhuo K. A Meloidogyne graminicola pectate lyase is involved in virulence and activation of host defense responses. Front Plant Sci. 2021;12:401.
Caza M, Kronstad JW. The cAMP/protein kinase A pathway regulates virulence and adaptation to host conditions in Cryptococcus neoformans. Front Cell Infect Microbiol. 2019;9:212.
Zhuo K, Naalden D, Nowak S, Xuan Huy N, Bauters L, Gheysen G. A Meloidogyne graminicola C-type lectin, Mg01965, is secreted into the host apoplast to suppress plant defence and promote parasitism. Mol Plant Pathol. 2019;20(3):346–55.
Zhao J, Sun Q, Quentin M, Ling J, Abad P, Zhang X, Li Y, Yang Y, Favery B, Mao Z. A Meloidogyne incognita C-type lectin effector targets plant catalases to promote parasitism. New Phytol. 2021;232(5):2124–37.
Lin B, Zhuo K, Chen S, Hu L, Sun L, Wang X, Zhang LH, Liao J. A novel nematode effector suppresses plant immunity by activating host reactive oxygen species-scavenging system. New Phytol. 2016;209(3):1159–73.
Gross R, Zhang S, Wei L, Caplan A, Kuhl J, Dandurand L-M, Wang X, Xiao F. The Globodera pallida effector GpPDI1 is a functional thioredoxin and triggers defense-related cell death independent of its enzymatic activity. Phytopathology®. 2020;110(11):1838–44.
Vieira P, Gleason C. Plant-parasitic nematode effectors—insights into their diversity and new tools for their identification. Curr Opin Plant Biol. 2019;50:37–43.
Dutta TK, Papolu PK, Banakar P, Choudhary D, Sirohi A, Rao U. Tomato transgenic plants expressing hairpin construct of a nematode protease gene conferred enhanced resistance to root-knot nematodes. Front Microbiol. 2015;6:260.
Huang X, Xu C-L, Chen W-Z, Chen C, Xie H. Cloning and characterization of the first serine carboxypeptidase from a plant parasitic nematode, Radopholus similis. Sci Rep. 2017;7(1):1–9.
Xue Q, Wu X-Q, Zhang W-J, Deng L-N, Wu M-M. Cathepsin L-like cysteine proteinase genes are associated with the development and pathogenicity of pine wood nematode, Bursaphelenchus xylophilus. Int J Mol Sci. 2019;20(1):215.
Huang X, Chi Y-k, Birhan AA, Wei Z, QI, R-d. Peng D-l: the new effector AbSCP1 of foliar nematode (Aphelenchoides besseyi) is required for parasitism rice. J Integr Agric. 2022;21(4):1084–93.
Coghlan A, Tyagi R, Cotton JA, Holroyd N, Rosa BA, Tsai IJ, Laetsch DR, Beech RN, Day TA, Hallsworth-Pepin K, et al. Comparative genomics of the major parasitic worms. Nat Genet. 2019;51(1):163–74.
van Baarlen P, Van Belkum A, Summerbell RC, Crous PW, Thomma BP. Molecular mechanisms of pathogenicity: how do pathogenic microorganisms develop cross-kingdom host jumps? FEMS Microbiol Rev. 2007;31(3):239–77.
Palomares-Rius JE, Escobar C, Cabrera J, Vovlas A, Castillo P. Anatomical alterations in plant tissues induced by plant-parasitic nematodes. Front Plant Sci 2017, 8:1987.
Islam W, Islam Su, Qasim M, Wang L. Host-Pathogen interactions modulated by small RNAs. RNA Biol. 2017;14(7):891–904.
Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, Nielsen H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420–3.
Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Selection. 2004;17(4):349–56.
Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80.
Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338(5):1027–36.
Emanuelsson O, Nielsen H, Brunak S, Von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300(4):1005–16.
De Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34(suppl2):W362–5.
Pierleoni A, Martelli PL, Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinformatics. 2008;9(1):1–11.
Howe KL, Bolt BJ, Shafie M, Kersey P, Berriman M. WormBase ParaSite – a comprehensive resource for helminth genomics. Mol Biochem Parasitol. 2017;215:2–10.
Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27(7):1017–8.
Törönen P, Medlar A, Holm L. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. 2018;46(W1):W84–8.
Ye J, Zhang Y, Cui H, Liu J, Wu Y, Cheng Y, Xu H, Huang X, Li S, Zhou A. WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018;46(W1):W71–5.
Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018;46(D1):D624–32.
Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46(W1):W95–W101.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–35.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35(suppl2):W182–5.
Kanehisa M, Sato Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020;29(1):28–35.
Xu L, Dong Z, Fang L, Luo Y, Wei Z, Guo H, Zhang G, Gu YQ, Coleman-Derr D, Xia Q. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019;47(W1):W52–8.
Acknowledgements
Not applicable.
Funding
The National Research Foundation (NRF) Grant 120858 provided funding for this research. All findings, viewpoints, deductions, and recommendations in this material are those of the author(s), and the NRF does not accept any liability in this regard.
Author information
Authors and Affiliations
Contributions
L.N.M: conceptualization, funding acquisition, project administration, writing-review, and editing. T.N.M: conceptualization, data curation, bioinformatic analysis, formal data analysis, prepared figures and writing original draft. T.A.D: conceptualization, data curation, bioinformatic analysis, review, and editing. The authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All methods were carried out in accordance with the institution’s relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Macharia, T.N., Duong, T.A. & Moleleki, L.N. In silico secretome analyses of the polyphagous root-knot nematode Meloidogyne javanica: a resource for studying M. javanica secreted proteins. BMC Genomics 24, 296 (2023). https://doi.org/10.1186/s12864-023-09366-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09366-6