
Abstract
Objectives
ST11 KPC-producing Klebsiella pneumoniae (Kp) is highly prevalent in China. We investigated the inter- and intra- host transmission and evolution characteristics of ST11 KPC-producing Kp.
Methods
A retrospective study was conducted in a hospital. The clinical data and antimicrobial resistance (AMR) phenotypes were collected. Whole genome sequencing was performed. The transmission route was reconstructed by combining single nucleotide polymorphisms (SNPs) with the clinical information. Hypervirulent Kp (HvKp) was defined as the presence of some combination of peg-344, iroB, iucA, rmpA, or rmpA2.
Results
Fifty-eight Kp strains isolated from thirty-five patients were enrolled. The information of one isolate was missing. The mean age of the patients was 74.3 ± 18.0 years, and 18 (50.0%) were female. Fifteen patients (41.7%, 15/36) presented with poor prognosis. All the strains were identified as ST11, and 57 strains harbored blaKPC-2. Two distinguished clades were identified based on the 1,325 high quality SNPs. In clade 1, carbapenem-resistant (CR)-hvKp accounted for 48.3% of the strains (28/58), which mostly presented as KL64 subclones, whereas CR-classical Klebsiella pneumoniae (cKp) commonly possessing KL47 were clustered in Clade 2. One CR-hvKp strain might have originated from the CR-cKp strain from within-host evolution. Even worse, a prolonged transmission of CR-hvKp has led to its spread into healthcare institutes.
Conclusion
Two endemic subclones of ST11 KPC-producing Kp, KL64-CR-hvKp and KL47-CR-cKp, were transmitted in parallel within the hospital and/or the healthcare institute, suggesting that the ongoing genomic surveillance should be enhanced.
Similar content being viewed by others

Introduction
Klebsiella pneumoniae (Kp) is increasingly emerging in China and has become the dominant pathogen in hospital-acquired infections (HAIs) [1]. Kp is a common cause of various fatal infections, posing a great threat to healthcare services [2]. Notably, Kp has evolved into two main pathotypes: hypervirulent Klebsiella pneumoniae (hvKp) and classical Klebsiella pneumoniae (cKp) [3, 4]. Previous studies demonstrated that hvKp is highly associated with community-acquired infection (CAI) and is sensitive to most antibiotics, whereas cKp is closely related to HAI and is resistant to most antimicrobial agents [3, 5].
Initially, the string test was used to distinguish hvKp and cKp [6]. However, some studies confirmed that the string test presented a lower prediction for identifying hvKp [7,8,9]. Recently, a combination of five virulence-associated genes, peg-344, iroB, iucA, rmpA, and rmpA2, showed higher diagnostic accuracy for hvKp than the string test or other virulence-associated genes [10, 11]. In our previous study, we found that hvKp replaced cKp as the dominant pathotype in the HAI, which primarily contributed to the acquisition of key virulence genes within the prevalent cKp (ST11) [11]. In China, the ST23 clone was common in the hvKp group, and ST11 was the prevalent cKp [1, 6, 12, 13]. ST11 cKp was highly associated with the Mulitdrug-resistance (MDR) or Carbapenem-resistance (CR) phenotype due to the acquisition of blaKPC-2 [1, 14]. The serotypes KL64 and KL47 were common within the ST11 Kp strain [11], but the epidemiological trends showed that KL64, not KL47, is prevalent among ST11 strains [15]. However, ST11 cKp is becoming hypervirulent by acquiring various virulence genes or a pLVKP-like plasmid [11, 16, 17]. These ST11 MDR hvKp strains have caused serious nosocomial infections and outbreaks worldwide [11, 17, 18]. Therefore, deeply understanding the genomic variation of ST11 during transmission might be a good choice to optimize surveillance.
Previous studies have mostly focused on the Kp strains isolated from different hosts. However, it is important to enroll the strains during in vivo evolution to construct the comprehensive transmission route by whole genome sequencing [19]. Therefore, our group conducted a retrospective study to clearly understand the genomic characteristics and evolution within the ST11 strains. Surprisingly, we found that ST11 has successfully evolved into two clades in which CR-hvKp that presented with KL64 and CR-cKp that was associated with KL47 were circulated in parallel within the hospital and the healthcare institute, suggesting that enhanced genomic surveillance is essential.
Materials and methods
Enrolled patients
A retrospective study was conducted from December 2017 to December 2020 at a hospital, and the isolates were selected. The distance between the branch of Peking University Third Hospital and headquarters is approximately 27 km. The clinical information of patients with ST11 infection was collected, including the basic demographic characteristics, admitted department, isolated time, specimen type, underlying diseases, antibiotic exposure within 90 days, Charlson comorbidity index (CCI), usage of invasive catheters and sequential organ failure assessment (SOFA). The presence of > 1 infection site in the same patient was defined as a metastatic infection. The infection types of the patients were divided into three groups: community-acquired infection, healthcare-associated infection (HCAI) and hospital-acquired infection (HAI). The definitions were as previously described [11]. The primary endpoint was poor prognosis within 30 days (death and withheld life-sustaining therapy).
The protocol for this study was approved by the Peking University Third Hospital Ethics Committee (M2021545). Due to the retrospective nature of the study, informed consent was not essential, and all the patient data in this study were anonymized.
Clinical Klebsiella pneumoniae strains
All the Kp strains were stored at -80 °C. The strains were initially inoculated and cultured on Columbia blood agar. The monoclonal strain was selected for identification by MALDI-TOF mass spectrometry and then by the Vitek compact 2 system. The definition of hvKp was the presence of some combination of peg-344, iroB, iucA, rmpA, or rmpA2 [10].
Antimicrobial susceptibility testing (AST)
AST was performed by a Vitek 2 system (bioMérieux, Marcy-l’Étoile, France). If necessary, we also used the disk diffusion method. The results were interpreted according to the 2020 Clinical and Laboratory Standards Institute (CLSI) guidelines. The antibiotics used for AST included Imipenem (IPM), Meropenem (MEM), Ertapenem (ETP), Minocycline (MNO), Amikacin (AMK), Cefepime (FEP), Ceftazidime (CAZ), Levofloxacin (LVX), Piperacillin/tazobactam (TZP), Cefoperazone/sulbactam (CSL) and trimethoprim/sulfamethoxazole (SXT). Strains resistant to three or more different antimicrobial categories were defined as having a multidrug resistance (MDR) phenotype [12]. Additionally, the definition of carbapenem resistance (CR) was the presence of resistance to IPM, MEM, or ETP [20].
DNA extraction and whole genome sequencing
Whole genomic DNA was extracted by the MagaBio Bacterium DNA Fast Purification Kit (BSC45S1E, Bioer Technology, Hangzhou, Zhejiang, China), and all strains were sequenced using the NovaSeq platform by constructing paired-end libraries to obtain 150 bp reads as previously described. We used fastQC software to obtain the clean data from the raw data and assembled the clean data by using SPAdes v3.15.2 [11]. Then, the draft genome sequences were annotated using Prokka [14].
To clearly determine the subtypes of these isolates, the sequencing type (ST) and capsular types were analyzed using Kleborate [21]. Antimicrobial resistance genes, virulence genes, and plasmid replicon types were annotated by comparison with relevant databases (ResFinder, Virulence Factor Database, plasmidFinder) using BLAST software. The thresholds of 90% identity and minimum length coverage of 80% for identifying antimicrobial resistance and virulence genes were determined.
To perform phylogenetic analysis, we applied the complete genome sequence of K.pneumoniae HS11286 (accession no.: NC_016845) as the reference. The sequencing reads were mapped to the reference using Bowtie 2 v2.2.8, and the single nucleotide polymorphisms (SNPs) were analyzed by using Samtools v1.9 and were combined together according to the reference using the iSNV-calling pipeline that we previously constructed [22, 23]. The high-quality SNPs supported by more than 5 reads of a mapping quality > 20 remained. Then, the recombination sites were detected by Gubbins [11, 24]. The concatenated sequences of the filtered polymorphic sites that were conserved in all the Kp strains (core genome SNPs, cgSNPs) were used to perform phylogenetic analysis using the maximum likelihood method by FastTree as previously described [11].
Results
Clinical characteristics of the ST11 Kp-infected patients
To perform a potential transmission route, Kp strains isolated from inter- and intrahosts were enrolled in the study. In total, fifty-eight Kp strains isolated from thirty-five patients were incorporated into this study. Four Kp strains were isolated from one of the 36 patients. Among seven patients, three isolates were detected from each patient, and two strains were obtained from each of the other six patients. Additionally, the information of one strain was missing.
One strain was isolated at December 2017, while three strains were isolated at September 2018, and the other strains were all from 2020 (Additional file 1: Figure S1). Most of the strains (56.9%, 33/58) were isolated from the respiratory tract. Fourteen strains (24.1%, 14/58) were from urine (Additional file 1: Figure S2).
The mean age of the enrolled patients was 74.3 ± 18.0 years, and 18 (50.0%) were female. Thirty-four patients were admitted to six departments of the headquarters, including the emergency department (17/36), ICU (9/36), geriatric ward (3/36), outpatient department (1/36), orthopedics department (1/36), cardiac surgery ward (1/36) and general surgery department (1/36). The other two patients were hospitalized in the branch of the hospital. Most of the patients presented with cardiovascular disease (32/36, 88.9%). More than half of the patients had cerebrovascular disease (20/36, 55.6%), and fourteen patients (38.9%, 14/36) had diabetes. Importantly, all patients had antibiotic exposure within 90 days. The mean CCI of the enrolled patients was 3.4 ± 1.6. Notably, all patients underwent invasive catheter procedures, among which 77.8% (28/36), 91.7% (33/36), 38.9% (14/36), 86.1% (31/36) and 30.6% (11/36) of patients had central intravenous catheters, urinary catheters, endotracheal tubes, gastrostomy tubes, and drainage tubes, respectively. Furthermore, ten patients experienced metastatic infections. Thirty-two patients (88.9%, 32/36) were identified as HAI, and three (8.3%, 3/36) patients were associated with HCAI. The median SOFA score of the enrolled patients was 4 (percentiles 25 = 1, percentiles 75 = 9). Fifteen patients (41.7%, 15/36) presented with poor prognosis. Specifically, 13 patients died within 30 days, and two patients withheld life-sustaining therapy (Table 1).
Subtyping, antimicrobial resistance and virulence profiles
All Kp strains were identified as ST11 (gapA3-infB3-mdh1-pgi1-phoE1-rpoB1-tonB4). Twenty-eight strains were identified as hvKp. Most of the Kp strains (98.3%, 57/58) carried yersiniabactin. IucA was positive in 28 strains (48.3%, 28/58), and 17 isolates (29.3%, 17/58) harbored rmpA. Sixteen isolates contained peg-344 (27.9%, 16/58). Only one strain was associated with iroB. Surprisingly, none of the strains harbored colibactin or rmpA2. The common key virulence genotype was iucA + peg-344 + rmpA. For the serotype, KL64 was positive in 27 strains (46.6%, 27/58), and KL47 was positive in 28 isolates (48.3%, 28/58). The other three strains presented with KL25.
All the ST11 Kp strains exhibited resistance against more than 3 classes of antimicrobial agents and presented with MDR, especially CR, phenotypes. More specifically, all the ST11 strains were resistant to TZP, CAZ, IPM, MEM, ETP and FEP. Additionally, only one isolate showed an intermediary for the LVX and CSL. Seven strains (12.1%, 7/58) were susceptible to AMK. Fortunately, 58.6% (34/58) of the strains were susceptible to SXT, and the 30 isolates were associated with a nonresistance phenotype for MNO (Additional file 1: Table S1).
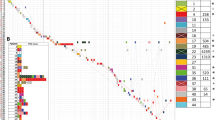
All the ST11 strains harbored fosA before fosfomycin exposure. Only one Kp strain did not present with blaKPC-2. Among the blaCTX-M-like positive strains, most of the strains (60.3%, 35/58) were associated with blaCTX-M-65, distributed in CR-hvKp (12 strains) and CR-cKp (23 strains). The strains that harbored blaCTX-M-14 (4 strains) and blaCTX-M-3 (1 strain) belonged to cKp. Even worse, blaNDM-1 emerged in the hospital. Another common ESBL, blaTEM-1B, existed in 53 isolates. Furthermore, twenty-four strains carried tet(A) (Fig. 1).

Phylogenetic tree of the ST11 Klebsiella pneumoniae strains. Orange: KL47 Blue: KL25. Pink: KL64
Combined with virulence and AST, 28 strains were defined as CR-hvKp, and 30 isolates were CR-cKp. The common genomic characteristics of CR-hvKp presented with KL64 + iucA + peg-344 + blaKPC-2 + blaTEM-1B, whereas CR-cKp frequently harbored KL47 + blaKPC-2 + blaTEM-1B + tet(A) + blaCTX-M-65.
All strains harbored the IncFII plasmid replicon, and 98.3% (57/58) contained the ColRNAI plasmid replicon. Fifty strains (86.2%, 50/58) carried IncR plasmid replicons, and 48.3% (28/58) were positive for IncHI1B plasmid replicons. More than four types of replicons were present in 3/58 (5.2%) isolates (Fig. 1).
Phylogenetic relationships and distribution of the CRKP
In total, 1,325 high quality SNPs were identified and applied for phylogenetic analysis (Fig. 1). Among all the ST11 CRKP strains, two distinguished clades were identified. Clade 1 contained 27 strains belonging to the CR-hvKp and KL64 serotypes, most of which were collected from the emergency department (14 isolates) and ICU (6 isolates). Clade 1 consisted of two clusters, clade 1a and clade 1b. Notably, all 15 strains of clade 1a harbored peg-344 + rmpA, while only one strain of clade 1b (PEkp146) possessed rmpA and no strain possessed peg-344. Additionally, all 12 isolates of clade 1b harbored blaCTX-M-65, which wasn’t present in clade 1a. Clade 2 was comprised of 31 isolates, which contained four clusters, clade 2a, clade 2b, clade 2c and clade 2d, and could be separated into two large branches. Clades 2a, 2b and 2c belonged to the KL47 branch, and clade 2d belonged to the KL25 branch. Clade 2d comprised of three isolates, which possessed blaCMY-42 and carried IncI1 plasmid replicon. Three strains of Clade 2b harbored catA2, aph(3’)-IIa, aadA1, arr-3 and aac(6’)Ib-cr. All isolates of clade 1 were hvKp. Notably, all strains of clade 2 were identified as CR-cKp, except only one strain, PEkp193, which might become CR-hvKp from CR-cKp during within-host evolution.
Transmission route of CR-hvKp and CR-cKp
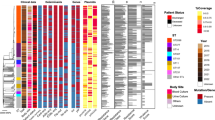
After combining the SNPs with epidemiological information, we reconstructed the potential transmission routes among these patients (Fig. 2). It is estimated that the hvKp ancestor might have separated from an ancestor and then spread into the emergency department and ICU (Fig. 2A). Briefly, the common ancestor of Clade 1 evolved into two main subclones (PEkp183 and PEkp017), which resulted in circulation within the emergency department and then spread to the outpatient department and other departments. Importantly, the PEkp125 strain was isolated from respiratory clinics and triggered HCAI.

Evolutionary and transmission route of the isolated Klebsiella pneumoniae strains. A clade 1 strains. B clade 2 strains. Gray circle, strains that were not captured during transmission in this study. The numbers on the arrows are SNPs. Gray shadow, strains were from the same patient. Different Colors of strain number represent different collection date. White: 2020; Yellow: 2018; Gray, 2017; Pink: Missing
Most of the strains (34.5%, 20/58) in Clade 2 were clustered within Clade 2a (Fig. 2B). The common ancestor of clade 2a underwent slight changes in SNPs during inter- and intrahost evolution, suggesting that the MDR ST11 Kp strains are circulating within the ICU and emergency department. More importantly, MDR-cKp has been transmitted into the branch of the hospital, alarming that enhancing surveillance is essential. Additionally, PEkp099 resulted in HCAI, suggesting that ST11 CR-cKp might successfully transmit into the healthcare-associated institution. All strains from the same patients belonged to the same clade, which presented high similarity in SNPs. Notably, although tiny differences in SNPs were shown among PEkp050, PEkp182 and PEkp193 within the same host, PEkp193 acquired iucA + peg-344 + peg-589 + rmpA and subsequently conferred CR-hvKp.
Discussion
In this study, we characterized the ST11 genome during inter- and intrahost evolution and transmission in the hospital. CR-hvKp presented with KL64, and CR-cKp harboring KL47 was distinctly distributed into two major clades, resulting in prolonged circulation within the ICU and emergency department, and might subsequently be transmitted into healthcare-associated institutes, indicating that genomic surveillance should be enhanced.
A recent epidemiological survey in China confirmed the rapid rise of MDR strains attributed to the ST11 clone [1, 25], which is a common cause of hospital infection outbreaks [26, 27]. Different from the classical hvKp (i.e., ST23), the loss of the “thick wall” of cKp enhanced the acquisition of external genetic elements [28]. Notably, all the ST11 strains were MDR and CR in our study, regardless of cKp or hvKp. A previous study in our country reported an outbreak due to the hypermucoviscous ST11 CRKP with an unclear serotype that occurred in China by PFGE [29] and then was the fatal outbreak of ST11-CR-hvKp, which was confirmed by whole genome sequencing and an in vivo model [17]. In this study, we reported that ST11 CRKP has evolved into KL64-CR-hvKp and KL47-CR-cKp, with parallel spreading in the hospital.
A previous study reported that the pandemic spread of blaKPC-2 among ST11 in China is mediated by the horizontal genetic element IncFII-like plasmids [30]. Similarly, all the ST11 strains except one in our group harbored IncFII plasmid replications carrying blaKPC-2 and were resistant to carbapenems. One strain, PEkp154, did not harbor blaKPC-2 but presented with a CR phenotype, which might be due to the presence of blaNDM-1. Based on the blaKPC-2 genetic context, ST11 frequently presented with MDR or CR phenotypes. Importantly, ST11, the epidemic sequence type in China, tends to take on various virulence-associated genes, becoming MDR-hvKp [11, 16]. A previous study reported that the CRKP ST11 clone with coproduction of blaCTX-M-65 and blaKPC-2 disseminated in the ICU and suggested that the CRKP presented with hypermucoviscosity [31]. Even more disturbing is that a fatal outbreak due to CR-ST11-hvKp occurred in the ICU, resulting in poor prognosis in all patients [17]. In the study, almost all the ST11 CR-hvKp strains coharbored blaKPC-2 and blaTEM-1B, and approximately half of them also carried blaCTX-M-65, suggesting that the trend of convergence of the AMR and virulence genes is forming. Moreover, our previous study demonstrated that various virulence-associated genes and AMR genes converged within the ST11 clone during interhost evolution and transmission [14]. More importantly, CAI caused by MDR-ST11-hvKp was also detected [14]. Whole-genome sequencing may provide improved resolution to define transmission pathways and characterize outbreaks, instructing the prevention and treatment of infection control. Previous study reported that WGS could effectively investigate MRSA or CR-hvKp outbreak and timely enhance intervention of infection control, liking disinfection and unoccupied period [17, 32,33,34]. In the study, CR-hvKp might be successfully transmitted within the hospital and/or healthcare institutes, and CR-cKp has diffused into two branches, suggesting that genomic surveillance is essential and should cover healthcare institutes.
Our previous study demonstrated that cKp, primary ST11, acquiring the pVir-like plasmid (iucA + rmpA2) or virulence-associated genes is the common reason for shaping MDR-hvKp, resulting in a major shift in hvKp epidemiology [11]. A study conducted in multiple centers in Japan also demonstrated that hvKp are frequently involved in HCAI and HAI [35]. ST11-KL64 still frequently presents with blaKPC-2 [36]. Additionally, ST11-KL64 CR-hvKp could rapidly gain and lose genes during interhospital transmission [37]. Recently, an important reason for enhanced transmission is that the fused plasmid converged with AMR and virulence genes [38]. However, the key virulence gene, iucA, was predominant within the ST11 CR-hvKp group, but none of the strains possessed rmpA2, suggesting that new plasmids might emerge and may have a tendency to spread. Long-read sequencing should be used for further study.
The KL64 and KL47 serotypes were common in the ST11 clone [11]. Interestingly, a previous study conducted in a single center in Zhe Jiang concluded that the ST11-KL64 clone is replacing ST11-K47 due to a new fused plasmid and that the ST11-KL64 capsule type may be more virulent [15]. Our previous study reported that KL47 was the dominant serotype in ST11 [14]. In this study, KL64 and KL47 accounted for nearly half. Moreover, all the KL64-positive strains carried various key virulence-associated genes, whereas only one KL47 strain possessed iroB + peg-344 + iucA + rmpA, which might be similar to the pLVPK-like plasmid [3]. Interestingly, 8 patients (50%, 8/16) with KL64-ST11 Kp infections presented with poor outcomes, whereas 6 patients (35.3%, 6/17) who had ST11-KL47 Kp died.
The main limitation of our study was that it was a retrospective study conducted at a single center. Furthermore, the number of strains was small, and they were randomly selected. A larger sample size originating from multiple centers is needed to deeply understand the genetic epidemiology. Additionally, long-read sequencing may assist us in fully characterizing the virulence-associated plasmid context. Last, an animal model should be selected to identify the virulence phenotype.
In conclusion, KL64-CR-hvKp and KL47-CR-cKp within ST11 were spread in parallel in the hospital. Importantly, ST11-CR-hvKp presenting with KL64 might successfully transmit within the hospital and/or healthcare institutes, and ST11-CR-cKp associated with KL47 spread to the two branches of the hospital, suggesting that ongoing surveillance of the endemic ST11 clone is critical.
Availability of data and materials
All ST11 K. pneumoniae genome sequences have been deposited in the NCBI Sequencing Read Archive database under the accession number PRJNA806574. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Wang Q, Wang X, Wang J, Ouyang P, Jin C, Wang R, Zhang Y, Jin L, Chen H, Wang Z, Zhang F, Cao B, Xie L, Liao K, Gu B, Yang C, Liu Z, Ma X, Jin L, Zhang X, Man S, Li W, Pei F, Xu X, Jin Y, Ji P, Wang H. Phenotypic and genotypic characterization of carbapenem-resistant enterobacteriaceae: data from a longitudinal large-scale CRE study in China (2012–2016). Clin Infect Dis. 2018;67:S196–205.
Magill SS, O’Leary E, Janelle SJ, Thompson DL, Dumyati G, Nadle J, Wilson LE, Kainer MA, Lynfield R, Greissman S, Ray SM, Beldavs Z, Gross C, Bamberg W, Sievers M, Concannon C, Buhr N, Warnke L, Maloney M, Ocampo V, Brooks J, Oyewumi T, Sharmin S, Richards K, Rainbow J, Samper M, Hancock EB, Leaptrot D, Scalise E, Badrun F, Phelps R, Edwards JR, Survey EIPHP, T. Changes in prevalence of health care-associated infections in U.S. Hospitals N Engl J Med. 2018;379:1732–44.
Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32(3):e00001-19.
Catalan-Najera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: two different but complementary Klebsiella spp. phenotypes? Virulence. 2017;8:1111–23.
Marr CM, Russo TA. Hypervirulent Klebsiella pneumoniae: a new public health threat. Expert Rev Anti Infect Ther. 2018. https://doi.org/10.1080/14787210.2019.1555470.
Li W, Sun G, Yu Y, Li N, Chen M, Jin R, Jiao Y, Wu H. Increasing occurrence of antimicrobial-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae isolates in China. Clin Infect Dis. 2014;58:225–32.
Lin YC, Lu MC, Tang HL, Liu HC, Chen CH, Liu KS, Lin C, Chiou CS, Chiang MK, Chen CM, Lai YC. Assessment of hypermucoviscosity as a virulence factor for experimental Klebsiella pneumoniae infections: comparative virulence analysis with hypermucoviscosity-negative strain. BMC Microbiol. 2011;11:50.
Zhang Y, Zeng J, Liu W, Zhao F, Hu Z, Zhao C, Wang Q, Wang X, Chen H, Li H, Zhang F, Li S, Cao B, Wang H. Emergence of a hypervirulent carbapenem-resistant Klebsiella pneumoniae isolate from clinical infections in China. J Infect. 2015;71:553–60.
Zhang Y, Zhao C, Wang Q, Wang X, Chen H, Li H, Zhang F, Li S, Wang R, Wang H. High prevalence of Hypervirulent Klebsiella pneumoniae Infection in China: geographic distribution, clinical characteristics, and antimicrobial resistance. Antimicrob Agents Chemother. 2016;60:6115–20.
Russo TA, Olson R, Fang CT, Stoesser N, Miller M, MacDonald U, Hutson A, Barker JH, La Hoz RM, Johnson JR. Identification of Biomarkers for Differentiation of Hypervirulent Klebsiella pneumoniae from Classical K. pneumoniae. J Clin Microbiol. 2018;56(9):e00776–18.
Liu C, Du P, Xiao N, Ji F, Russo TA, Guo J. Hypervirulent Klebsiella pneumoniae is emerging as an increasingly prevalent K. pneumoniae pathotype responsible for nosocomial and healthcare-associated infections in Beijing. China Virulence. 2020;11:1215–24.
Liu C, Shi J, Guo J. High prevalence of hypervirulent Klebsiella pneumoniae infection in the genetic background of elderly patients in two teaching hospitals in China. Infect Drug Resist. 2018;11:1031–41.
Lam MMC, Wyres KL, Duchene S, Wick RR, Judd LM, Gan YH, Hoh CH, Archuleta S, Molton JS, Kalimuddin S, Koh TH, Passet V, Brisse S, Holt KE. Population genomics of hypervirulent Klebsiella pneumoniae clonal-group 23 reveals early emergence and rapid global dissemination. Nat Commun. 2018;9:2703.
Liu C, Du P, Zhao J, Li B, Wang C, Sun L, Lu B, Wang Y, Liu Y, Cao B. Phenotypic and genomic characterization of virulence heterogeneity in multidrug-resistant ST11 Klebsiella pneumoniae during inter-host transmission and evolution. Infect Drug Resist. 2020;13:1713–21.
Zhou K, Xiao T, David S, Wang Q, Zhou Y, Guo L, Aanensen D, Holt KE, Thomson NR, Grundmann H, Shen P, Xiao Y. Novel subclone of carbapenem-resistant Klebsiella pneumoniae sequence type 11 with enhanced virulence and transmissibility, China. Emerg Infect Dis. 2020;26:289–97.
Zhang Y, Jin L, Ouyang P, Wang Q, Wang R, Wang J, Gao H, Wang X, Wang H. Evolution of hypervirulence in carbapenem-resistant Klebsiella pneumoniae in China: a multicentre, molecular epidemiological analysis. J Antimicrob Chemother. 2019. https://doi.org/10.1093/jac/dkz446.
Gu D, Dong N, Zheng Z, Lin D, Huang M, Wang L, Chan EW, Shu L, Yu J, Zhang R, Chen S. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis. 2018;18:37–46.
de Campos TA, Goncalves LF, Magalhaes KG, de Paulo MV, Pappas Junior GJ, Peirano G, Pitout JDD, Goncalves GB, Furlan JPR, Stehling EG, Pitondo-Silva A. A fatal bacteremia caused by hypermucousviscous KPC-2 producing extensively drug-resistant K64-ST11 Klebsiella pneumoniae in Brazil. Front Med (Lausanne). 2018;5:265.
Didelot X, Walker AS, Peto TE, Crook DW, Wilson DJ. Within-host evolution of bacterial pathogens. Nat Rev Microbiol. 2016;14:150–62.
Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18:268–81.
Wyres KL, Nguyen TNT, Lam MMC, Judd LM, van Vinh CN, Dance DAB, Ip M, Karkey A, Ling CL, Miliya T, Newton PN, Lan NPH, Sengduangphachanh A, Turner P, Veeraraghavan B, Vinh PV, Vongsouvath M, Thomson NR, Baker S, Holt KE. Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from South and Southeast Asia. Genome Med. 2020;12:11.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Ni M, Chen C, Qian J, Xiao HX, Shi WF, Luo Y, Wang HY, Li Z, Wu J, Xu PS, Chen SH, Wong G, Bi Y, Xia ZP, Li W, Lu HJ, Ma J, Tong YG, Zeng H, Wang SQ, Gao GF, Bo XC, Liu D. Intra-host dynamics of Ebola virus during 2014. Nat Microbiol. 2016;1:16151.
Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:e15.
Qi Y, Wei Z, Ji S, Du X, Shen P, Yu Y. ST11, the dominant clone of KPC-producing Klebsiella pneumoniae in China. J Antimicrob Chemother. 2011;66:307–12.
Voulgari E, Gartzonika C, Vrioni G, Politi L, Priavali E, Levidiotou-Stefanou S, Tsakris A. The Balkan region: NDM-1-producing Klebsiella pneumoniae ST11 clonal strain causing outbreaks in Greece. J Antimicrob Chemother. 2014;69:2091–7.
Voulgari E, Zarkotou O, Ranellou K, Karageorgopoulos DE, Vrioni G, Mamali V, Themeli-Digalaki K, Tsakris A. Outbreak of OXA-48 carbapenemase-producing Klebsiella pneumoniae in Greece involving an ST11 clone. J Antimicrob Chemother. 2013;68:84–8.
Wyres KL, Wick RR, Judd LM, Froumine R, Tokolyi A, Gorrie CL, Lam MMC, Duchene S, Jenney A, Holt KE. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019;15:e1008114.
Zhan L, Wang S, Guo Y, Jin Y, Duan J, Hao Z, Lv J, Qi X, Hu L, Chen L, Kreiswirth BN, Zhang R, Pan J, Wang L, Yu F. Outbreak by Hypermucoviscous Klebsiella pneumoniae ST11 Isolates with carbapenem resistance in a tertiary hospital in China. Front Cell Infect Microbiol. 2017;7:182.
Fu P, Tang Y, Li G, Yu L, Wang Y, Jiang X. Pandemic spread of blaKPC-2 among Klebsiella pneumoniae ST11 in China is associated with horizontal transfer mediated by IncFII-like plasmids. Int J Antimicrob Agents. 2019;54:117–24.
Yu F, Hu L, Zhong Q, Hang Y, Liu Y, Hu X, Ding H, Chen Y, Xu X, Fang X, Yu F. Dissemination of Klebsiella pneumoniae ST11 isolates with carbapenem resistance in integrated and emergency intensive care units in a Chinese tertiary hospital. J Med Microbiol. 2019;68:882–9.
Jernigan JA, Hatfield KM, Wolford H, Nelson RE, Olubajo B, Reddy SC, McCarthy N, Paul P, McDonald LC, Kallen A, Fiore A, Craig M, Baggs J. Multidrug-resistant bacterial infections in U.S. hospitalized patients, 2012–2017. N Engl J Med. 2020;382:1309–19.
Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, Bernard H, Fruth A, Prager R, Spode A, Wadl M, Zoufaly A, Jordan S, Kemper MJ, Follin P, Müller L, King LA, Rosner B, Buchholz U, Stark K, Krause G. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med. 2011;365:1771–80.
Köser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, Hsu LY, Chewapreecha C, Croucher NJ, Harris SR, Sanders M, Enright MC, Dougan G, Bentley SD, Parkhill J, Fraser LJ, Betley JR, Schulz-Trieglaff OB, Smith GP, Peacock SJ. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med. 2012;366:2267–75.
Harada S, Aoki K, Yamamoto S, Ishii Y, Sekiya N, Kurai H, Furukawa K, Doi A, Tochitani K, Kubo K, Yamaguchi Y, Narita M, Kamiyama S, Suzuki J, Fukuchi T, Gu Y, Okinaka K, Shiiki S, Hayakawa K, Tachikawa N, Kasahara K, Nakamura T, Yokota K, Komatsu M, Takamiya M, Tateda K, Doi Y. Clinical and molecular characteristics of Klebsiella pneumoniae causing bloodstream infections in Japan: occurrence of hypervirulent infections in healthcare. J Clin Microbiol. 2019. https://doi.org/10.1128/JCM.01206-19.
Wang B, Pan F, Wang C, Zhao W, Sun Y, Zhang T, Shi Y, Zhang H. Molecular epidemiology of Carbapenem-resistant Klebsiella pneumoniae in a paediatric hospital in China. Int J Infect Dis. 2020;93:311–9.
Zhang X, Ouyang J, He W, Zeng T, Liu B, Jiang H, Zhang Y, Zhou L, Zhou H, Liu Z, Liu L. Co-occurrence of rapid gene gain and loss in an interhospital outbreak of Carbapenem-Resistant Hypervirulent ST11-K64 Klebsiella pneumoniae. Front Microbiol. 2020;11:579618.
Yang X, Wai-Chi Chan E, Zhang R, Chen S. A conjugative plasmid that augments virulence in Klebsiella pneumoniae. Nat Microbiol. 2019. https://doi.org/10.1038/s41564-019-0566-7.
Acknowledgements
Not applicable.
Funding
This work was supported by the Clinical Cohort Construction Program of Peking University Third Hospital (BYSYDL2019007) and Beijing Key Clinical Specialty Program (010071).
Author information
Authors and Affiliations
Contributions
ML and NS designed the study, supervised the whole project and revised the manuscript. CL and PY performed the bioinformatic analysis and data collection and contributed to writing and revising the manuscript. JZ performed the experiments, collected the clinical data and revised the manuscript. JY participated in the experimental performance. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All methods have been performed in accordance with the Declaration of Helsinki. The protocol for this study was approved by the Peking University Third Hospital Medical Science Research Ethics Committee (M2021545). Due to the retrospective nature of the study, the need for approval was waived by the Peking University Third Hospital Medical Science Research Ethics Committee, and all the patient data enrolled in this study were anonymized.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Collection date of these ST11-Kp strains. Figure S2. Specimen type of these ST11-Kp strains. Table S1. Antimicrobial resistance profile of these ST11-Kp strains.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, C., Yang, P., Zheng, J. et al. Convergence of two serotypes within the epidemic ST11 KPC-producing Klebsiella pneumoniae creates the “Perfect Storm” in a teaching hospital. BMC Genomics 23, 693 (2022). https://doi.org/10.1186/s12864-022-08924-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08924-8