
Abstract
Background
Transposable elements (TEs) are common features in eukaryotic genomes that are known to affect genome evolution critically and to play roles in gene regulation. Vertebrate genomes are dominated by TEs, which can reach copy numbers in the hundreds of thousands. To date, details regarding the presence and characteristics of TEs in camelid genomes have not been made available.
Results
We conducted a genome-wide comparative analysis of camelid TEs, focusing on the identification of TEs and elucidation of transposition histories in four species: Camelus dromedarius, C. bactrianus, C. ferus, and Vicugna pacos. Our TE library was created using both de novo structure-based and homology-based searching strategies (https://github.com/kacst-bioinfo-lab/TE_ideintification_pipeline). Annotation results indicated a similar proportion of each genomes comprising TEs (35–36%). Class I LTR retrotransposons comprised 16–20% of genomes, and mostly consisted of the endogenous retroviruses (ERVs) groups ERVL, ERVL-MaLR, ERV_classI, and ERV_classII. Non-LTR elements comprised about 12% of genomes and consisted of SINEs (MIRs) and the LINE superfamilies LINE1, LINE2, L3/CR1, and RTE clades. Least represented were the Class II DNA transposons (2%), consisting of hAT-Charlie, TcMar-Tigger, and Helitron elements and comprising about 1–2% of each genome.
Conclusions
The findings of the present study revealed that the distribution of transposable elements across camelid genomes is approximately similar. This investigation presents a characterization of TE content in four camelid to contribute to developing a better understanding of camelid genome architecture and evolution.
Similar content being viewed by others


Background
Transposable elements (TEs) are influential in determining genome structural dynamics. TEs are DNA sequences found in nearly all eukaryotes which encode various proteins which carry out the molecular mechanisms which facilitate their relocation and duplication within a host genome [103]. They can comprise substantial proportions of eukaryotic genomes, for example, 50% of the human genome, up to 90% of the maize genome, and varying from 2.7% to 47% of insect genomes [46]. The ability of a TE to proliferate within a genome contributes to it undergoing natural selection as a discrete evolutionary entity, separate from its host. Given this, TEs can be viewed as selfish intragenomic parasitic sequences [27] because their ability to undergo replicative transposition via an RNA or DNA intermediate is not seen as adaptive in most cases. The abundance and repetitive nature of TEs are among the main challenges that complicate the correct assembly of sequenced genomes [96]. However, the significantly repetitive nature of the genomes of mammals and other vertebrates also plays an essential role in various processes, contributing substantially to genome size and architecture [24, 30, 55] as well as influencing functional genomic components [14]. The technological developments in genomics and large-scale functional assays has spotlighted the multi-faceted properties of TEs and their importance in shaping genomes [15].
We are at the early stages of understanding how mobile element insertions influence specific phenotypes. TEs can disrupt host sequences and act as substrates for nonhomologous recombination, forming DNA rearrangements such as deletions, duplications, inversions, and translocations [36, 47]. Such rearrangements can be deleterious for the host through the alteration of gene-coding potential and regulation or by modifying other necessary genomic sequences [58]. TEs are, therefore, causes of mutations and genetic diseases in humans and other organisms [41, 97]. In some cases, TEs are also proposed to be involved in the rapid adaptation of invasive species to new environments [19]. Environmental stressors represent a daily challenge for some organisms, who must adapt to survive continuously changing conditions [20]. An increasing number of studies support a link between TE activity and species responsiveness to environmental conditions [35, 64]. In this context, mobile elements contribute to increasing genetic diversity, allowing organisms to better adapt to new conditions [91]. Given this, TE activity may have contributed to the high diversity of vertebrate species that colonized many habitats, from water to land and temperate to extreme environments [19].
Here, we have analyzed the global TE content in four camelid genomes, contributing to a better understanding of the genome organization and evolution in camelids. Camelus dromedarius, frequently referred to as the Arabian camel, is a heat stress-resistant animal [67] capable of living in the extremely harsh climates of the Arabian Peninsula. The adaptations of camelids to arid conditions are remarkable. Camels can fluctuate their body temperature from 34 ∘ C to 41.7 ∘ C and can conserve water by not sweating [2]. Additional members of the camelid family included in our study are the Bactrian camel (Camelus bactrianus) and the Wild Bactrian camel (Camelus ferus) of Asia, and the alpaca (Vicugna pacos) of South America [4, 38]. The extreme variation among their natural habitats opens up a series of questions about how the environment influences TEs in camelids. Such questions cannot be addressed in the absence of a high quality TE annotation and our work aims to bridge this gap.
In this work, we use a variety of bioinformatics approaches to identify and classify camelid TEs, following the system used by [103]. This system defines two classes of elements according to their transposition mechanism. Class I elements, known as retrotransposable elements (REs), can transpose themselves via an RNA intermediate, self-replicating in the process. REs are the most abundant repetitive elements in many genomes, often including many long terminal repeat REs (LTR-RTs). Class II elements, also known as DNA transposons, can move by means of a "rolling circle" Helitron, or “cut-and-paste” action characterized by terminal inverted repeats (TIRs) of variable length. Within these classes, TEs are further categorized into superfamilies based on homology or structural characteristics.
The great diversity of TEs can make their accurate detection and annotation difficult [61]. Several computational approaches have been developed for identifying TEs in assembled genomes, of which the two main strategies are homology-based and structure-based methods [12]. Additionally, TEs can be uncovered based on their repetitive nature, with queries on the structural signatures of specific TE types supporting the detection of specific types of individual full-length elements; this benefits the investigations of TE variation and evolution [102]. Combining approaches leads to increased sensitivity of detection, resulting in more comprehensive results [71, 74]. In this study, we annotated the TE fraction of the whole genome of each sequenced camelid. We present a detailed approach for the characterization of TEs in camel genomes using homology and structure-based methods, with the aim of providing a basis for future studies on TEs that expand our understanding of genomic diversity and evolution in camelid species. These sequences could be valuable tools for elucidating new genomic dynamics and making evolutionary inferences.
Methods
Data source
Four camelid species currently have draft genome sequences available from the National Center of Biotechnology Information (NCBI), assembled at the scaffold level. These sequences were downloaded from the NCBI RefSeq database [79] in FASTA format: C. dromedarius, accession GCF_000767585.1, assembled genome size ∼2004 Mb; C. bactrianus, GCF_000767855.1, ∼1992 Mb; C. ferus, GCF_000311805.1, ∼2009 Mb; and V. pacos, GCF_000164845.2, ∼2172 Mb [8, 104]. The genome completeness of the four camelid species was evaluated by Benchmarking Universal Single-Copy Orthologs (BUSCO) v3.0.2 [88], based on mammalian orthologous gene set (4,104 genes).
Identification of transposable elements
To construct reliable and comprehensive repeat libraries is a challenging task due to the variation in repeat structure and the difficulty of assembling repeats in genome sequences. As many elements vary considerably in genetic structure and sequence, the only means of achieving reliable results when identifying and annotating TEs is to practice complementary approaches [80]. A flowchart describing our overall approach to TE identification is given in Fig. 1. The specific methods for each type are detailed below. We employ de novo signature-based detection programs that rely upon prior knowledge concerning the sharing between different TEs of standard architectural features necessary for the process of transposition. Examples of classification according to similarity to known TEs include records in databases like Repbase [51] and protein profiles retrieved from the Pfam database [31]. Unfortunately, only well-described TEs that have a robust structural signature can be discovered by these methods. Some TEs do not have such characteristics and thus cannot be distinguished by this approach. In contrast to homology-based methods, signature-based methods are less biased by similarity to the set of known elements.

Flowchart for de novo identification of canonical TE sequences using both structural and homology-based approaches
Class I elements: LTR retrotransposons
Candidate LTR-RT loci were identified by employing the program LTRharvest [29], a component of GenomeTools [37], which searches the input sequence for direct repeats (LTRs) that are separated by a given distance (default 1 kb) and outside of which are apparent target site duplications (TSDs). Candidates distinguished by LTRharvest were then passed to LTRdigest [92], which annotates protein-coding domains between the LTRs of each putative element. Specifically, LTRdigest searches for homologs in the putative LTR-RTs using HMMER3 [101] and a set of TE-related pHMMs we provided from Pfam and GyDB [31, 63]. Subsequently, the EMBOSS (v.6.6.0) program getorf [84] was employed to annotate additional ORFs of at least 100 amino acids in length that do not overlap the LTRdigest predictions. Afterwards, the predicted LTR-RTs were investigated for homology to LTR-RT sequences in Dfam and Repbase through applying nhmmer and tblastx, respectively. Elements without homologs were discarded as false positives. Each retained element was considered a true positive and classified according to the superfamily of the highest-scoring entry from Dfam or Repbase. Finally, the elements were clustered using the sequence similarity method suggested by [103], based on the “80-80-80” rule, which allocates at least 80% sequence similarity in ≥80% of the element length with a minimum of 80 bp of aligned segments.
Elements were aligned using MAFFT v7.453 [57], and putative intra-element gene conversion tracts between LTRs were identified in those alignments using GENECONV v.1.81a [86], which detects stretches of greater-than-likely similarity. Putative gene conversion tracts with fewer than three total differences were not accepted. Subsequent analyses were performed on each cluster independently. Relative insertion times were calculated for each element per the method of [85]. First, the LTRs within a cluster were aligned and divergences were estimated using PAUP* [94] under the HKY85 model of nucleotide sequence evolution [42]. Since gene conversion events erase the signal of time in a LTR alignment, divergence estimates were improved under the assumption that the part(s) of an LTR alignment containing a putative gene conversion tract will exhibit the same rate of divergence as the portion of the alignment not participating in a gene conversion tract. Following the approach of [21] for calculating divergence among protein-coding genes, divergence estimates were scaled linearly relative to putative gene conversion tracts. Divergences were converted to millions of years using a previously published estimate of the C. dromedarius generational substitution rate of 2.5 x 10-8 and a generation time of five years [32]. Phylogenies were inferred for each cluster in order to investigate the evolutionary history of identified elements. Whole elements were aligned by MAFFT using 2 or three iterations of the FFT-NS-2 algorithm respectively for clusters with 50 and fewer elements or between 50 and 500 elements, and a single iteration of the FFT-NS-1 algorithm for clusters having between 500 and 1000 elements. Alignments were post-processed with trimAl [18] using the setting -automated1, and evolutionary trees were inferred using FastTree2 [77] under the GTR-CAT model. Phylogenies and LTR-RT diagrams were visualized using FigTree v1.4.4 (2018) and ETE3 [48]. We also employed Kruskal-Wallis and t-test using R stats package (R Core Team, 2019) to test for differences in underlying LTR-RT length distributions and means between species, respectively.
Class I elements: Non-LTR retrotransposons
Here, we began with the recognized genomic coordinates of LTR-RTs identified in the previous step. These candidates were masked with maskfasta from BedTools [81] to avoid conflicts or duplicate hits. Next, open reading frame sequences were extracted from the masked genome by applying the getorf tool from EMBOSS v6.4.0.0. The minimum ORF size was set to 500 bp in anticipation of detecting the apyrimidinic endonuclease (APE) gene (which is 600–800 bp in 97% of inspected non-LTR elements). Non-LTRs have been previously classified into clades or lineages [65], and subsequently into families. In only two clades, the reverse transcriptase (RT) is encoded by a single domain (R2 and CRE clades). The others have an additional coding region for an APE. Some elements, such as those belonging to clade I, have an extra RNaseH domain [106]. Accordingly, we performed an exploration of the genomic sequences with MGEScan-non-LTR [83], which identifies and classifies non-LTR TEs in genomic sequences using probabilistic models based on the structure of the 12 established non-LTR TE clades. More precisely, we used MGEScan-non-LTR and hmmsearch from HMMER 3.0 [28] with two separate hidden Markov model (HMM) profiles, one for the reverse transcriptase (RT) gene and one for the endonuclease (APE) gene, both of which are well conserved among non-LTR TEs.
Class II elements
All eukaryotic DNA transposons reported so far belong to a single category of elements which use the so-called “cut-and-paste” transposition mechanism, except Helitrons, which transpose by rolling-circle replication. Here, we employed methodologies for the detection of DNA transposons in the studied genomes based on the initial identification of TIR, and non-autonomous elements such as miniature inverted-repeat elements (MITEs) and helitron.
MITEs are DNA-based elements that have TIRs but lack a transposase gene, and their well-defined structural features make them suitable for discovery by computational approaches. We utilized an accurate, valuable tool for detecting MITEs in eukaryotic genomes, MiteFinderII [45]; this tool is capable of detecting both perfect and imperfect inverted repeats through a string matching approach [108]. It computes a new function to cluster MITE sequences into different MITE families in several steps. First, it builds a k-mer index and seeks inverted repeats. Then, all sequence candidates are distinguished by the presence of a TIR pair of default length and a TSD pair. Second, the scaffolds are divided into multiple sequence fragments that overlap by 800 bp, which is the maximum length of MITEs, to guarantee that all inverted repeats are identified. Third, pairs of TIRs having lengths in the range of 50-800 bp are retained, and the remainder used as seeds for MITE candidates in the next step. Finally, identified sequences are compared with MITEs in the Repbase database using blastn [5]. Those with high similarity are considered valid positives, and those with low similarity as false positives. For each MITE cluster, the sequence with the highest blast score was selected via an in-house script as the representative family sequence. The tool was executed with default parameters, except for the use of a confidence-score threshold of 0.5 to exclude low-confidence candidates.
Helitrons are diverse across species and even within one species. These are rolling circle eukaryotic transposons that regularly catch gene sequences and do not form target site duplications or end in TIRs. To investigate the presence of Helitrons in camelid genome, we used the structure-based tool HelitronScanner [105]. HelitronScanner relies on sequence matches between trained local combinational variables (LCVs) and genome. Specifically, it scores 5 ′ and 3 ′ termini based on a training set of published Helitrons, and then merges the coordinates and scores for putative Helitron-like sequences. We increased the threshold to 6 to avoid false positives. The predicted candidates were clustered using CD-Hit [34] at 80% similarity. Finally, we used tBlastn against Helitron sequences deposited in RepBase-20181026 to retrieve the highest scores.
Transposable element annotation, copy number and genome coverage estimation
After all libraries were generated using the programs mentioned above, the TE repeat sequences present in Camelidae species were extracted from Dfam Consensus-20170127 and RepBase-20181026 using the script “queryRepeatDatabase.pl” shipped with RepeatMasker. The results of both steps above were combined. Next, duplicates were filtered using seqKit rmdup (-s) on the basis of sequence. We then used RepeatMasker v.4.1.0 [90] to process the results for masking and annotation [87]. We used RMBlast as the search algorithm with a Smith-Waterman cutoff of 225, -no_is, -gff -s -lib, -norna and exclusion of low complexity regions -nolow; all other parameters were default. Additionally, counting the copy number of each TE and determined genome coverage obtained from the RepeatMasker output files (.out), which correspond to the number of insertions identified in the masked genomes. The remaining unmasked portion of the genome is scanned using RepeatModeler [33] with default settings to detect any unclassified TEs such as TIRs that were missed by structure-based TE identification and merge it to the library for Re-annotation.
Results
Evaluation of genome assemblies
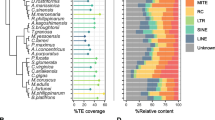
To evaluate the completeness of each of the four camelid genome assemblies, we used BUSCO mammalian lineage dataset (mammalia_odb9), which consisted of 4,104 single-copy orthologs. BUSCO results showed that 93.9–95.2% of the 4104 mammalian single-copy orthologs were complete across the four genome assemblies (Fig. 2A), suggesting the four genomes are comparable and have high-quality assemblies.

Genome assembly assessment and TE proportions. The BUSCO dataset of the mammalia_db9 including 4,104 BUSCO was utilized to evaluate the four camelid assemblies (A). TE proportions in the four camelid genomes (B)
Construction of camelid repeat libraries
TE reference libraries were generated through systematic searching procedures using both de novo signature-based and homology-based method strategies for four camelid draft genomes: C. dromedarius, C. bactrianus, C. ferus, and V. pacos. The joined libraries include related repeats deposited in Dfam and Repbase. They respectively consist of a total of 6026, 6594, 7241, and 8311 individual TE sequences, and cover both Class I (LTR-RTs, non-LTR retrotransposons) and Class II elements (TIR elements, Helitrons, and MITEs) 41,701,493 bp, 32,935,780 bp, 26,533,293 bp, and 39,127,481 bp respectively (Supplementary files S1–S4). The TE sequence annotations generated by these libraries respectively comprise 734,069,793 bp (36.63%), 718,445,875 bp (36.05%), 711,915,199 bp (35.43%), and 783,294,957 bp (36.06%) of the investigated camel genomes (Table 1, Fig. 2B, and Supplementary files S5–S8).
Our findings revealed the relative contributions to camelid genome of significant types of TEs, namely LTR, LINE, and SINE retrotransposons, as well as DNA transposons, (Fig. 2B). Here, we employed several procedures to identify each order of TEs present in four camelid genomes: C. dromedarius, C. bactrianus, C. ferus, and V. pacos. The results are classified into four main categories: 1) LTR-RTs elements identified using LTRharvest, and internal regions annotated employing LTRdigest: these comprised respective totals of 4473, 4794, 5768, and 6877 elements in the studied genomes, and open reading frames (ORFs) were simultaneously distinguished. 2) Non-LTR retrotransposons were identified by aligning reverse transcriptase accessions to ORFs predicted in the genomes, yielding 495, 475, 87, and 11 sequences, respectively. 3) Non-autonomous DNA elements (MITEs and degraded DNA transposons) were identified by their TIRs and TSDs using MITE-FinderII, yielding 96, 76, 73, and 64 families respectively. 4) Helitron-like sequences were identified using HelitronScanner, and consisted of 532, 557, 503, and 524 sequences, respectively.
LTR retrotransposons
LTR retrotransposons appear to dominate the camel genomes, being the most significant component among the identified TEs (Fig. 2B).
In the C. dromedarius genome, LTRharvest identified 11303 candidate LTR-RTs each consisting of two relatively intact LTRs and flanking TSDs. After LTRdigest annotation analyses, discarding the false-positive candidates reduced the number to 4473 putative full-length LTR-RTs, which comprise 39% of the total predicted candidates. The lengths of these elements range from 205 to 25,500 bp, with an average of 7,691.2 bp and a total genome-wide footprint of 34,402,528 bp.
In the C. bactrianus genome, LTRharvest predicted 10920 candidate LTR-RTs with two relatively intact LTRs and flanking TSDs. After discarding false positives, reduced the number to 4,794 putative full-length LTR-RTs, comprising 43% of the total predicted candidates. The lengths of these elements ranged from 203 to 15,702 bp, with an average size of 5,211 bp and a total footprint of 24,981,380 bp.
In the C. ferus genome, LTRharvest identified 17456 LTR-RTs candidates with two relatively intact LTRs and flanking TSDs. Discarding false-positive candidates reduced the list to 5768 putative full-length LTR-RTs, comprised 33% of the total predicted candidates. Their lengths ranged from 203 to 15,701 bp, with an average size of 3,480.4 bp and a total footprint of 20,075,064 bp.
In the V. pacos genome, LTRharvest predicted 24674 candidate LTR-RTs with two relatively intact LTRs and flanking TSDs. Removal of false positives reduced the total to 6877 putative full-length LTR-RTs, comprised 27% of all predicted candidates. These elements ranged from 208 to 15,887 bp in length, with an average of 4,757.2 bp and a total footprint of 32,715,308 bp.
The identified LTR-RTs were classified into 11 superfamilies according to similarity (Table 2). In all four genomes, the most abundant LTR-RT superfamily was ERVL-MaLR (1503-2031 elements). The second most prevalent was either ERV1 (1142–2455) or ERVL (987–1316), followed by ERV2 (410–679) and then Gypsy (15–56). Similar numbers were found in each genome for the ERVL, ERV1, and ERV2 families, but counts of ERV1, ERVL, and ERVL-MaLR elements were greater in genomes with successively more total LTR-RTs (Table 3).
Histograms of intra-element ages by-species are depicted in Fig. 3. When divergences were scaled based on putative gene conversion tracts, the distribution shapes remained very similar to those of the unaltered divergences, except for having long tails; therefore, the unaltered divergences are shown. In the genomes of C. dromedarius and C. bactrianus, LTR-RTs appear to have had consistently relatively low activity for the past 25 million years (mya). This contrasts with LTR-RTs in C. ferus and V. pacos, where there have been recent bursts between about 0.5 - 2.5 mya and 1.5 - 4 mya, respectively (Fig. 3). Notably, the distributions of LTR-RT lengths differed among species (Fig. 4, Tables 4 and 5). Normalized to C. dromedarius, the mean LTR-RT lengths of the other three studied species were 0.69 (C. bactrianus), 0.46 (C. ferus), and 0.63 (V. pacos).

Distributions of LTR-RT ages in each of the four camelid genomes, with speciation event interval demarcated by vertical dashed lines. Phylogenetic relationships among species are depicted by the tree on the right

LTR-RT length distributions in the four camelid genomes
Bean-plots of cluster size distributions show similar patterns for all of the species, predominantly consisting of singletons and smaller clusters (Fig. 5). The vast majority of clusters contain elements that are heterogeneous in length; relatively few contained the suite of domains necessary for transposition, as recognizable through high similarity to entries from Pfam and GyDB. However, there are generally multiple ORFs in each element; it is likely that at least some of these encode transposition machinery, but are too divergent to be detected by the pHMM search. The phylogenies of most clusters have poor bootstrap support, with the exceptions of three small clusters in the V. pacos genome, two of which ERV1 clusters (Fig. 6) and one ERVL (Fig. 7). The ERV1 LTR-RTs in V. pacos are also remarkable for the presence of multiple large clusters that almost exclusively contain short elements, most of which have no internal ORFs or identifiable LTR-RT-related protein-coding domains. These elements may be non-autonomous and rely on protein products derived from other elements.

Distributions of LTR-RT cluster sizes for clusters delineated using the 80-80-80 rule. The y-axis is log(10)-transformed. Small horizontal lines represent individual clusters; their lengths are proportional to the number of clusters with that particular size, with the exception of the line for cluster size = 2, which is shortened to save space. Thick horizontal lines are means and the dotted horizontal line is the overall mean. Singletons are included as counts at the bottom of the plot

Phylogenetic tree for LTR-RTs in cluster 6 of the ERV1 group in Vicugna pacos, with diagrams showing LTR domain structure (A) and two exemplar elements from cluster 4 (B)

Phylogenetic tree for LTR-RTs in cluster 2 of the ERVL group in V. pacos, with diagrams showing LTR domain structure
The protein sequences identified internal to LTR-RTs are summarized in Table 6. In total, 2889, 2350, 1365, and 2409 proteins were respectively identified in the four species (C. dromedarius, C. bactrianus, C. ferus, and V. pacos) (Table 6). Most of the putative LTR-RTs contained the RT-INT-ENV protein domain order (reverse transcriptase, integrase, and envelop) characteristic of ERVs.
Non-LTR retrotransposons
Non-LTR retrotransposons were identified by applying MGEScan-non-LTR to the LTR-masked genomes. This tool discovered all known full-length elements and simultaneously classified them into the following clades: CRE, I, Jockey, L1, R1, R2, and RTE. Notably, reverse transcriptase (RT) is encoded by all autonomous non-LTR retrotransposons, and therefore was used as the primary signal to distinguish and classify these elements. Previous studies have classified non-LTR retrotransposons into 11 clades based on RT phylogeny [65].
The non-LTR retrotransposons identified in camelid species are summarized in Table 7. Six clades were represented in C. dromedarius, five in C. bactrianus, four in C. ferus, and six in V. pacos. In the three Camelus species, L1 was the most abundant clade, represented by 442, 421, and 41 elements and comprising total footprints of 1,003,622 bp, 1,571,496 bp, and 490,265 bp in C. dromedarius, C. bactrianus, and C. ferus, respectively. Surprisingly, the smallest number of ORF-conserving elements was identified in V. pacos; these elements collectively occupied 13,785 bp.
DNA transposons
Miniature inverted-repeat elements (MITEs) were one such ubiquitous class, characterized by essential structural features such as TIRs and TSDs, AT-rich sequences, and a lack of coding capacity for transposases. Canonical MITE sequences with perfect TSDs, perfect or near-perfect TIR structure, and a length was between 50 and 650 bp were counted using MITEFinder software; a total of 285 families across the four examined genomes. Relative empty site analysis showed that the TSD sequences differed between families, being either 2, 3, 8, or 10 bp. Moreover, we performed homology-based repeat analysis on the library of identified camelid MITEs using a subsection of the Repbase database carrying only class II vertebrate and mammalian sequences. The resulting superfamily classifications and the number of families detected in each species are given in Table 8.
In the C. dromedarius genome, we identified a total of 69 MITE elements, which accounted for 13,922 bp of the genome and comprised 9 different families. In C. bactrianus, we identified 76 MITE elements, which accounted for 14,364 bp of the genome and clustered into 7 families, 74 of them being present in Repbase (1997 hits). In C. ferus, we identified 73 MITE elements, which account for 14,092 bp of the genome and clustered into 9 families, 72 of them being present in Repbase. Finally, in the V. pacos genome, 64 MITE elements were identified, which accounted for 12,087 bp of the genome and clustered into 9 families, 62 of them being present in Repbase (1908 hits). In all four species evaluated, the most abundant superfamily of DNA transposons was hAT, represented by 26-29 families.
Finally, we employed HelitronScanner to identify Helitron-like sequences using a structure-based approach. HelitronScanner aims to extract more definitive Helitron features than the few previously identified: the TC dinucleotide at the 5 ′ end, the hairpin structure, the CTRR (R = A or G) sequence at the 3 ′ end, and the A and T residues respectively flanking the 5 ′ and 3 ′ ends. It assigns to each identified Helitron a LCV score, which is an indicator of prediction confidence; we considered elements with scores of 6 or greater for each end. More than 500 double-ended Helitron sequences were identified in each camel genome (Table 8). These respectively accounted for 5,702,516 bp, 5,774,487 bp, 5,355,741 bp, and 5,491,739 bp total in C. dromedarius, C. bactrianus, C. ferus, and V. pacos. These candidates were classified into superfamilies based on homology (Table 9), with the most abundant being Helitron DR (225, 237, 204, and 223 total elements) and Helitron GA (31, 32, 29, and 26 total elements).
Discussion
We generated repeat libraries for each of the four camel species with available genome sequences in order to investigate the abundance and character of repeat-derived DNA within their genomes, as well as to facilitate the repeat-masking of DNA in future studies. Notably, we worked on assembled genome drafts, which frequently do not include TE-rich regions like centromeres or other heterochromatic regions. Our analysis techniques were also very conservative and may have dropped other types of TEs or elements that are ancient and divergent. To ensure the reliability of our results, we employed a method incorporating both known TEs and signature-based repeat identification tools.
TEs are abundant in almost all living organisms and closely related species have similar TE content [13, 40]. Here, our repeat analysis reveals significant similarity in total TE content (35.43–36.63%) between genome assemblies of the four species (Fig. 2B), which were consistent with lizards (34.4%) [3], carp (31.3%) [107], and western clawed frog (34.5%) [43]. Interestingly, early studies on camelid cytogenetics have evidenced a striking uniformity in the karyotypes of Asian and South American camelids, despite significant divergence times and adaptation to different environments [10, 16, 95]. In this sense, our result of similarity in the overall repeat content between camelid genomes could reflect a general evolutionary trend of genome stability in this family.
Overall, our investigation found that camelid genomes are characterized by a strong predominance of retroelements over DNA transposons. However, the genomes were similar in the relative abundances of element types (LTRs > non-LTRs > DNA) (Fig. 2B). Class I elements constituted 22–32% of the annotated genomes, with LTRs comprising 16–20%, SINEs only about 2%, and LINEs 10–11%. The observed high abundance of LTR might be a distinctive feature of camelids. However, this pattern is typical for plants compared to other mammals and vertebrates [68, 73, 93], and this could reflect the high variability of TE abundance across vertebrate genomes [22, 75]. Class II elements collectively made up 4–5% of the annotated genomes, with DNA Transposons comprising about 2–3% and Helitrons about 2%. Notably, DNA repeat proliferation is one of the key factors that influence species differences in genome size and composition; others include whole-genome duplications, segmental duplications, and deletions [72]. Our findings revealed that the examined camel genomes have similar total repetitive makeup, with unique genome content comprising 1.26, 1.27, 1.29, and 1.38 GB respectively in C. dromedarius, C. bactrianus, C. ferus, and V. pacos. [30] suggested that larger genomes might have more TEs. The observed differences between genomes could also stem from technical limitations in sequencing and assembling repeats using short-read sequencing.
LTR-RTs are usually more abundant than other types of TEs, and so the identification of full-length elements benefits research into the structural variability, diversity, and phylogenetic evolution of TEs in camelid genomes. Accordingly, we investigated full-length LTR-RTs in camelid genomes in detail. We discovered that the most abundant full-length LTRs were ERVs, including the ERVL, ERVL-MaLR, ERV classI, and ERV classII superfamilies. ERVs are remnants of past retroviral infections, which may have arisen within genomes by at least two different mechanisms: retrotransposition from a pre-existing endogenous retrovirus or the infection and integration of an exogenous source virus [50]. LTR-RTs have also been domesticated numerous times to perform roles in functions such as placental development, host defence against exogenous retroviruses, brain development, and more [70]. Among camelid ERVs, ERV-MaLR was the most frequent; this element is assumed to have been inserted into the mammalian genome about 70 million years ago [11]. Studies have revealed that ERV classII is one of the youngest members of endogenous retroviruses [56], comprising only a low percentage of animal genomes [1]. Consistent with this data, this study, found ERV Class II to have the lowest representation of all ERV elements across all four camel genomes.
To help elucidate the evolutionary history of camelid LTR-RTs, we dated insertions of each full-length LTR-RT by estimating sequence divergence (substitutions per site) between long terminal repeats and scaling them by an estimate of the C. dromedarius nuclear genome generational mutation rate [32]. Considering the timing of evolutionarily significant events such as speciations, distributions of LTR-RT abundance across time can suggest mechanisms that may have facilitated evolutionary change. For example, hybrids often face massive TE de-repression due to widespread DNA de-methylation, leading to a surge in transposition activity [60]. In C. ferus, there is a peak of LTR-RT abundance between about 0.3–2.1 mya, corresponding to the time of the speciation process that separated the C. ferus from the C. bactrianus lineage [17]. The V. pacos lineage experienced a burst of LTR-RT activity from about 1–4 mya, a range that overlaps the 0.78–1.3 mya interval during which V. pacos diverged from the extant or an extinct lineage of guanaco [25] only slightly; during a time LTR-RTs were less active than they were about a million years prior during the peak of the burst. Therefore, the increased transposition rate could reflect a process occurring in the lineage of the common ancestor of V. pacos and the extant or wild guanaco. It would be interesting to see a distribution of LTR-RT insertion dates for the exant wild guanaco. If the distribution shares the peak of LTR-RT abundances aged 1–4 mya observed in V. pacos, then it would suggest the V. pacos lineage diverged from the extant wild guanaco lineage more than 4 mya. If the distribution does not share the peak, it would suggest V. pacos is derived from an extinct wild guanaco lineage, in agreement with the model of llama domestication proposed by [26]. It would also be interesting to investigate whether genes in C. ferus and V. pacos have LTR-RT insertions near promoters of transcription factors or other genes which might be implicated in phenotypic differences between these species [7].
We determined that LINE elements were the most prevalent non-LTR repeat in camelids, contributing 10–11% of the total assembled genomes (Table 1). The LINE proportion in Camelids seems very similar to that of lizards (12.34%) and higher than in birds (6%), coelacanth (6.43%), cod (3.3%), and western clawed frog (5.4%) [3, 43, 98]. Among LINEs, the LINE1 (L1) represents the most abundant family in mammals [59], and here confirmed to be the most abundant in camelids (Table 1). Elements that depend on L1-encoded proteins for retrotransposition are responsible for new germline insertions, mostly in AT-rich regions, that can cause genetic diseases [9]. Moreover, L1 is capable of 3 ′ transduction [69]. In contrast, the RTE clade was the least abundant LINE superfamily in camelids. The RTE ORF appears most intimately related to the corresponding ORF of the CR1 autonomous element, another LINE clade which is predominantly found in avian and reptile genomes [66]. A small proportion of camelid genomes (about 0.2%) was found to consist of CR1 elements, but these were determined to have degenerated and become nonfunctional [98, 99].
The contribution of SINEs to camelid genome content was much less significant than that of LINEs, comprising only about 2% of each species’ total genome length (Table 1). SINEs evolved from RNA genes, such as 7SL and tRNA genes [89]. By definition, they are short, measuring up to 1000 base pairs long. They do not encode their retrotransposition machinery and are considered non-autonomous elements. In most cases, SINES are mobilized by L1-derived machinery [53]. Another characteristic of SINEs is that they accept RNA polymerase III transcription [24]. The Alu clade of SINE elements is mostly enriched in GC-rich or gene-rich regions, and considered an abundant and conserved repeat family in primate genomes [39]. This study observed no Alu elements in camel genomes. Instead, we identified mammalian-wide interspersed repeats (MIRs) as the predominant SINE family in camelid genomes, constituting nearly all of the identified SINE elements. MIRs are another prominent SINE clade, whose putative ancestor sequences evolved before the eutherian radiation and spread through mammalian lineages during the Mesozoic era, an estimated 130 million years ago. Accordingly, copies of MIRs have been discovered in diverse mammalian groups, including marsupials and monotremes [52]. [49] suggest that MIRs may play functional roles for their host genomes, and also positively correlate to tissue-specific gene expression. Further research using RNA-seq data could assist us in better understanding the roles of MIRs in camelid genomes.
In this study, we found that DNA transposons constitute about 3% of camelid genomes (Table 1). MITE content has explicitly been estimated in vertebrates, including mammals, birds, frog, and lizard [3, 6, 23, 43, 62, 109]. These elements are usually present in low copy numbers relative to retrotransposons, occupying less than 3% of mammalian genomes [76], consistent with our findings. Previously, scientists believed that the last activity of DNA transposons in mammalian genomes occurred at least 40 million years ago [82]. Their high copy number and structural homogeneity have served to distinguish them from most of the previously described class II elements [100]. We identified a total of 285 DNA transposon families in camelid genomes, which grouped into nine superfamilies based on their TSDs and on known associations in Repbase (Table 8). Of these families, the hAT superfamily predominated in all four studied genomes, and a particular diversity was observed for the hAT and Tc1/Mariner families (Table 1).
Another DNA-based element, Helitrons, are diverse both between species and also within one given species. These elements were first described in plants, but are also present in fungi and animals [44, 78]. They replicate using a rolling-circle mechanism, and their insertion does not result in a TSD [54]. Since their identification, the role of Helitrons in reshaping host genomes has been examined in many organisms, but their actual mechanism of transposition has remained elusive. We employed the tool HelitronScanner [105] to investigate the presence of Helitrons in camelid genomes, and found that they constitute about 2% of the total genome lengths (Table 1). Among the identified camelid Helitrons, the greatest number showed homology to Helitron-2_DR and Helitron-4_DR (Table 9).
In conclusion, the findings of this study will provide a valuable resource for further studies on camel biology. While the present study showed that the investigated genomes had similar contents and distributions of the identified repetitive regions (Fig. 2B), differences were also identified that may be associated with factors such as different evolutionary origins or discrepancies in the assembly stage of these draft genomes. Additional research into camelid repetitive elements, perhaps with more complete genome assemblies, would provide more information about and awareness of the genomic features of camels. Such additional genome-wide detail could improve strategy design for camel maintenance and breeding. Furthermore, the causes and consequences of the high degree of variability that exists in the distribution, amount, and relative proportion of TEs in different genomes are still not wholly understood; it is essential to continue characterizing this critical fraction of eukaryotic genomes. Such characterizations can bring to light evolutionary phenomena, including genomic rearrangements and other dynamic events, that have occurred in the past and may also be under way in contemporary times.
Availability of data and materials
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Abbreviations
- TEs:
-
Transposable elements
- LTR:
-
Long terminal repeat
- LINE:
-
Long interspersed element
- TIR:
-
Terminal inverted repeat
- TSD:
-
Target site duplication
- MITEs:
-
Miniature inverted–repeat transposable elements
- GyDB:
-
Gypsy database
- ORFs:
-
Open reading frames
- pHMMs:
-
Profile hidden Markov models
- APE:
-
Apurinic/apyrimidinic endonuclease
- RNaseH:
-
Ribonuclease H
- RT:
-
Reverse transcriptase
References
Adelson D, Raison J, Garber M, Edgar R. Interspersed repeats in the horse (equus caballus); spatial correlations highlight conserved chromosomal domains. Anim Genet. 2010; 41:91–9.
Al-Swailem AM, Shehata MM, Abu-Duhier FM, Al-Yamani EJ, Al-Busadah KA, Al-Arawi MS, Al-Khider AY, Al-Muhaimeed AN, Al-Qahtani FH, Manee MM, Al-Shomrani BM, Al-Qhtani SM, Al-Harthi AS, Akdemir KC, Inan MS, Otu HH. Sequencing, Analysis, and Annotation of Expressed Sequence Tags for Camelus dromedarius. PLoS ONE. 2010; 5:e10720.
Alföldi J, Di Palma F, Grabherr M, Williams C, Kong L, Mauceli E, Russell P, Lowe CB, Glor RE, Jaffe JD, et al.The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature. 2011; 477:587–91.
Alshehri MA, Manee MM, Al-Fageeh MB, Al-Shomrani BM. Genomic analysis of intrinsically disordered proteins in the genus camelus. Int J Mol Sci. 2020; 21:4010.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990; 215:403–10.
Amemiya CT, Alföldi J, Lee AP, Fan S, Philippe H, MacCallum I, Braasch I, Manousaki T, Schneider I, Rohner N, et al.The african coelacanth genome provides insights into tetrapod evolution. Nature. 2013; 496:311–6.
Auvinet J, Graça P, Belkadi L, Petit L, Bonnivard E, Dettaï A, Detrich W, Ozouf-Costaz C, Higuet D. Mobilization of retrotransposons as a cause of chromosomal diversification and rapid speciation: the case for the antarctic teleost genus trematomus. BMC Genomics. 2018; 19:1–18.
Bactrian Camels Genome Sequencing and Analysis Consortium, Jirimutu, Wang Z, Ding G, Chen G, Sun Y, Sun Z, Zhang H, Wang L, Hasi S, Zhang Y, Li J, Shi Y, Xu Z, He C, Yu S, Li S, Zhang W, Batmunkh M, Ts B, Narenbatu U, Bat-Ireedui S, Gao H, Baysgalan B, Li Q, Jia Z, Turigenbayila, Subudenggerile, Narenmanduhu, Wang Z, Wang J, Pan L, Chen Y, Ganerdene Y, Dabxilt, Erdemt, Altansha, Altansukh, Liu T, Cao M, Aruuntsever, Bayart, Hosblig, He F, Zha-ti A., Zheng G, Qiu F, Sun Z, Zhao L, Zhao W, Liu B, Li C, Chen Y, Tang X, Guo C, Liu W, Ming L, Temuulen, Cui A, Li Y, Gao J, Li J, Wurentaodi, Niu S, Sun T, Zhai Z, Zhang M, Chen C, Baldan T, Bayaer T, Li Y, Meng H. Genome sequences of wild and domestic bactrian camels. Nat Commun. 2012; 3:1202.
Beck CR, Garcia-Perez JL, Badge RM, Moran JV. Line-1 elements in structural variation and disease. Ann Rev Genomics Hum Genet. 2011; 12:187–215.
Benirschke K. Sterility and Fertility of Interspecific Mammalian Hybrids In: Benirschke K, editor. Comparative Aspects of Reproductive Failure. Berlin, Heidelberg: Springer: 1967. https://doi.org/10.1007/978-3-642-48949-5_12.
Bénit L, Lallemand J-B, Casella J-F, Philippe H, Heidmann T. Erv-l elements: a family of endogenous retrovirus-like elements active throughout the evolution of mammals. J Virol. 1999; 73:3301–8.
Bergman C, Quesneville MH. Discovering and detecting transposable elements in genome sequences. Brief Bioinform. 2007; 8:382–92.
Bonchev G, Parisod C. Transposable elements and microevolutionary changes in natural populations. Mol Ecol Resour. 2013; 13:765–75.
Bourque G. Transposable elements in gene regulation and in the evolution of vertebrate genomes. Curr Opin Genet Dev. 2009; 19:607–12.
Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvák Z, Levin HL, Macfarlan TS, et al.Ten things you should know about transposable elements. Genome Biol. 2018; 19:1–12.
Bunch TD, Foote WC, Maciulis A. Chromosome banding pattern homologies and nors for the bactrian camel, guanaco, and llama. J Hered. 1985; 76:115–8.
Burger PA. The history of old world camelids in the light of molecular genetics. Trop Anim Health Prod. 2016; 48:905–13.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimal: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009; 25:1972–3.
Carducci F, Biscotti M, Barucca M, Canapa A. Transposable elements in vertebrates: species evolution and environmental adaptation. Eur Zool J. 2019; 86:497–503.
Casacuberta E, González J. The impact of transposable elements in environmental adaptation. Mol Ecol. 2013; 22:1503–17.
Casola C, Ganote CL, Hahn MW. Nonallelic gene conversion in the genus drosophila. Genetics. 2010; 185:95–103.
Chalopin D, Naville M, Plard F, Galiana D, Volff J-N. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol Evol. 2015; 7:567–80.
The International Aphid Genomics Consortium. Genome Sequence of the Pea Aphid Acyrthosiphon pisum. PLoS Biol. 2010; 8(2):e1000313. https://doi.org/10.1371/journal.pbio.1000313.
Deininger PL, Moran JV, Batzer MA, Kazazian HH. Mobile elements and mammalian genome evolution. Curr Opin Genet Dev. 2003; 13:651–8.
Diaz-Lameiro AM. Evolutionary origins and domestication of South American camelids, the alpaca (Vicugna pacos) and the llama (Lama glama) explained through molecular DNA methods. Binghamton: State University of New York; 2016.
Diaz-Maroto P, Rey-Iglesia A, Cartajena I, Núñez L, Westbury MV, Varas V, Moraga M, Campos PF, Orozco-terWengel P, Marin JC, et al.Ancient dna reveals the lost domestication history of south american camelids in northern chile and across the andes. Elife. 2021; 10:e63390.
Doolittle W, Sapienza FC. Selfish genes, the phenotype paradigm and genome evolution. Nature. 1980; 284:601.
Eddy SR. Accelerated profile hmm searches. PLoS Comput Biol. 2011; 7:e1002195.
Ellinghaus D, Kurtz S, Willhoeft U. Ltrharvest, an efficient and flexible software for de novo detection of ltr retrotransposons. BMC Bioinformatics. 2008; 9:18.
Elliott TA, Gregory TR. What’s in a genome? the c-value enigma and the evolution of eukaryotic genome content. Phil Trans R Soc B Biol Sci. 2015; 370:20140331.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al.Pfam: the protein families database. Nucleic Acids Res. 2013; 42:D222—D230.
Fitak RR, Mohandesan E, Corander J, Burger PA. The de novo genome assembly and annotation of a female domestic dromedary of north african origin. Mol Ecol Resour. 2016; 16:314–24.
Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. Repeatmodeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci. 2020; 117:9451–7.
Fu L, Niu B, Zhu Z, Wu S, Li W. Cd-hit: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012; 28:3150–2.
Fujino K, Hashida S. -n., Ogawa T, Natsume T, Uchiyama T, Mikami T, Kishima Y. Temperature controls nuclear import of tam3 transposase in antirrhinum. Plant J. 2011; 65:146–55.
Gray YH. It takes two transposons to tango: transposable-element-mediated chromosomal rearrangements. Trends Genet. 2000; 16:461–8.
Gremme G, Steinbiss S, Kurtz S. Genometools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinforma (TCBB). 2013; 10:645–56.
Groeneveld LF, Lenstra JA, Eding H, Toro MA, Scherf B, Pilling D, Negrini R, Finlay EK, Jianlin H, Groeneveld E, Weigend S, GLOBALDIV Consortium. Genetic diversity in farm animals–a review. Anim Genet. 2010; 41(Suppl 1):6–31.
Gu Z, Wang H, Nekrutenko A, Li W-H. Densities, length proportions, and other distributional features of repetitive sequences in the human genome estimated from 430 megabases of genomic sequence. Gene. 2000; 259:81–8.
Guner B, Gozukirmizi CN. Origin and distribution of different retrotransposons in different taxa. Genet Appl. 2018; 2:13–9.
Hancks DC, Kazazian Jr HH. Active human retrotransposons: variation and disease. Curr Opin Genet Dev. 2012; 22:191–203.
Hasegawa M, Kishino H, Yano T-A. Dating of the human-ape splitting by a molecular clock of mitochondrial dna. J Mol Evol. 1985; 22:160–74.
Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V, Ovcharenko I, Putnam NH, Shu S, Taher L, et al.The genome of the western clawed frog xenopus tropicalis. Science. 2010; 328:633–6.
Hood ME. Repetitive dna in the automictic fungus microbotryum violaceum. Genetica. 2005; 124:1–10.
Hu TT, Pattyn P, Bakker EG, Cao J, Cheng J-F, Clark RM, Fahlgren N, Fawcett JA, Grimwood J, Gundlach H, et al.The arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nature Genet. 2011; 43:476.
Hua-Van A, Le Rouzic A, Boutin TS, Filée J, Capy P. The struggle for life of the genome’s selfish architects. Biol Direct. 2011; 6:19.
Huang CRL, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, et al.Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010; 141:1171–82.
Huerta-Cepas J, Serra F, Bork P. Ete 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol. 2016; 33:1635–8.
Jjingo D, Conley AB, Wang J, Mariño-Ramírez L, Lunyak VV, Jordan IK. Mammalian-wide interspersed repeat (mir)-derived enhancers and the regulation of human gene expression. Mobile DNA. 2014; 5:14.
Johnson WE. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol. 2019; 17:355–70.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005; 110:462–7.
Jurka J, Zietkiewicz E, Labuda D. Ubiquitous mammalian-wide interspersed repeats (mirs) are molecular fossils from the mesozoic era. Nucleic Acids Res. 1995; 23:170–5.
Kajikawa M, Okada N. Lines mobilize sines in the eel through a shared 3 ′ sequence. Cell. 2002; 111:433–44.
Kapitonov V, Jurka VJ. Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci. 2001; 98:8714–9.
Kapusta A, Suh A, Feschotte C. Dynamics of genome size evolution in birds and mammals. Proc Natl Acad Sci. 2017; 114:E1460—E1469.
Katoh I, Kurata S-I. Association of endogenous retroviruses and long terminal repeats with human disorders. Front Oncol. 2013; 3:234.
Katoh K, Standley DM. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013; 30:772–80.
Kazazian HH. Science. 2004; 303:1626–32.
Kramerov D, Vassetzky N. Origin and evolution of sines in eukaryotic genomes. Heredity. 2011; 107:487.
Laporte M, Le Luyer J, Rougeux C, Dion-Côté AM, Krick M, Bernatchez L. Dna methylation reprogramming, te derepression, and postzygotic isolation of nascent animal species. Sci Adv. 2019; 5:eaaw1644.
Lerat E. Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity. 2010; 104:520.
Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, et al.Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005; 438:803–19.
Llorens C, Futami R, Covelli L, Domínguez-Escribá L, Viu J. M, Tamarit D, Aguilar-Rodríguez J, Vicente-Ripolles M, Fuster G, Bernet GP, et al.The gypsy database (gydb) of mobile genetic elements: release 2.0. Nucleic Acids Res. 2010; 39:D70—D74.
Makarevitch I, Waters AJ, West PT, Stitzer M, Hirsch CN, Ross-Ibarra J, Springer NM. Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 2015; 11:e1004915.
Malik HS, Burke WD, Eickbush TH. The age and evolution of non-ltr retrotransposable elements. Mol Biol Evol. 1999; 16:793–805.
Malik H, Eickbush STH. The rte class of non-ltr retrotransposons is widely distributed in animals and is the origin of many sines. Mol Biol Evol. 1998; 15:1123–34.
Manee MM, Alharbi SN, Algarni AT, Alghamdi WM, Altammami MA, Alkhrayef MN, Alnafjan BM. Molecular cloning, bioinformatics analysis, and expression of small heat shock protein beta-1 from Camelus dromedarius, Arabian camel. PLoS ONE. 2017; 12:e0189905.
McCarthy E, McDonald MJF. Long terminal repeat retrotransposons of mus musculus. Genome Biol. 2004; 5:1–8.
Moran JV, DeBerardinis RJ, Kazazian HH. Exon shuffling by l1 retrotransposition. Science. 1999; 283:1530–4.
Naville M, Warren I, Haftek-Terreau Z, Chalopin D, Brunet F, Levin P, Galiana D, Volff J-N. Not so bad after all: retroviruses and long terminal repeat retrotransposons as a source of new genes in vertebrates. Clin Microbiol Infect. 2016; 22:312–23.
Permal E, Flutre T, Quesneville H. Roadmap for Annotating Transposable Elements in Eukaryote Genomes In: Bigot Y, editor. Mobile Genetic Elements. Methods in Molecular Biology (Methods and Protocols), vol 859. Totowa: Humana Press: 2012. p. 53–68. https://doi.org/10.1007/978-1-61779-603-6_3.
Petrov DA. Evolution of genome size: new approaches to an old problem. Trends Genet. 2001; 17:23–8.
Pisano MP, Grandi N, Tramontano E. Human endogenous retroviruses (hervs) and mammalian apparent ltrs retrotransposons (malrs) are dynamically modulated in different stages of immunity. Biology. 2021; 10:405.
Platt RN, Blanco-Berdugo L, Ray DA. Accurate transposable element annotation is vital when analyzing new genome assemblies. Genome Biol Evol. 2016; 8:403–10.
Platt RN, Vandewege MW, Ray DA. Mammalian transposable elements and their impacts on genome evolution. Chromosom Res. 2018; 26:25–43.
Platt II, Ray RNDA. A non-ltr retroelement extinction in spermophilus tridecemlineatus. Gene. 2012; 500:47–53.
Price MN, Dehal PS, Arkin AP. FastTree 2 Ű Approximately Maximum Likelihood Trees for Large Alignments. PloS ONE. 2010; 5(3):e9490. https://doi.org/10.1371/journal.pone.0009490.
Pritham E, Feschotte JC. Massive amplification of rolling-circle transposons in the lineage of the bat myotis lucifugus. Proc Natl Acad Sci. 2007; 104:1895–900.
Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012; 40:D130—D135.
Quesneville H, Bergman CM, Andrieu O, Autard D, Nouaud D, Ashburner M, Anxolabehere D. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput Biol. 2005; 1:e22.
Quinlan A, Hall RIM. Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–2.
Ray DA, Feschotte C, Pagan HJ, Smith JD, Pritham EJ, Arensburger P, Atkinson PW, Craig NL. Multiple waves of recent dna transposon activity in the bat, myotis lucifugus. Genome Res. 2008; 18:717–28.
Rho M, Tang H. Mgescan-non-ltr: computational identification and classification of autonomous non-ltr retrotransposons in eukaryotic genomes. Nucleic Acids Res. 2009; 37:e143—e143.
Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000; 16(6):276–7.
SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. The paleontology of intergene retrotransposons of maize. Nat Genet. 1998; 20:43.
Sawyer S. Geneconv: a computer package for the statistical detection of gene conversion. 1999. http://www.math.wustl.edu/sawyer. Accessed 15 Mar 2020.
Shen W, Le S, Li Y, Hu F. Seqkit: a cross-platform and ultrafast toolkit for fasta/q file manipulation. PloS ONE. 2016; 11:e0163962.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. Busco: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015; 31:3210–2.
Singer MF. Highly repeated sequences in mammalian genomes. In: International review of cytology, vol. 76. Amsterdam: Elsevier: 1982. p. 67–112.
Smit AFA, Hubley R, Green P. Repeatmasker at http://repeatmasker.org. Nucleic Acids Res. 2016. Accessed 7 Feb 2020.
Stapley J, Santure AW, Dennis SR. Transposable elements as agents of rapid adaptation may explain the genetic paradox of invasive species. Mol Ecol. 2015; 24:2241–52.
Steinbiss S, Willhoeft U, Gremme G, Kurtz S. Fine-grained annotation and classification of de novo predicted ltr retrotransposons. Nucleic Acids Res. 2009; 37:7002–13.
Sultana N, Menzel G, Seibt KM, Garcia S, Weber B, Serçe S, Heitkam T. Genome-wide analysis of long terminal repeat retrotransposons from the cranberry vaccinium macrocarpon. bioRxiv. 2021.
Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (and other methods) 4.0 b8. Sunderland: Sinauer; 2001.
Taylor K, Hungerford D, Snyder R, F Ulmer Jr. Uniformity of karyotypes in the camelidae. Cytogenet Genome Res. 1968; 7:8–15.
Treangen TJ, Salzberg SL. Repetitive dna and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012; 13:36–46.
Vorechovsky I. Transposable elements in disease-associated cryptic exons. Human Genet. 2010; 127:135–54.
Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A, Searle S, White S, Vilella AJ, Fairley S, et al.The genome of a songbird. Nature. 2010; 464:757–62.
Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al.Initial sequencing and comparative analysis of the mouse genome. Nature. 2002; 420:520–62.
Wessler SR, Bureau TE, White SE. Ltr-retrotransposons and mites: important players in the evolution of plant genomes. Curr Opin Genet Dev. 1995; 5:814–21.
Wheeler TJ, Eddy SR. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013; 29:2487–9.
Wicker T, Gundlach H, Spannagl M, Uauy C, Borrill P, Ramirez-Gonzalez RH, De Oliveira R, Mayer KF, Paux E, Choulet F. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018; 19:103.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al.A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007; 8:973.
Wu H, Guang X, Al-Fageeh MB, Cao J, Pan S, Zhou H, Zhang L, Abutarboush MH, Xing Y, Xie Z, et al.Camelid genomes reveal evolution and adaptation to desert environments. Nat Commun. 2014; 5:1–10.
Xiong W, He L, Lai J, Dooner HK, Du C. Helitronscanner uncovers a large overlooked cache of helitron transposons in many plant genomes. Proc Natl Acad Sci. 2014; 111:10263–8.
Xiong Y, Eickbush TH. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990; 9:3353–62.
Xu P, Zhang X, Wang X, Li J, Liu G, Kuang Y, Xu J, Zheng X, Ren L, Wang G, et al.Genome sequence and genetic diversity of the common carp, cyprinus carpio. Nat Genet. 2014; 46:1212.
Ye C, Ji G, Li L, Liang C. detectir: A novel program for detecting perfect and imperfect inverted repeats using complex numbers and vector calculation. PloS ONE. 2014; 9:e113349.
Yuan Y-W, Wessler SR. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci. 2011; 108:7884–9.
Acknowledgments
The authors would like to thank Casey Bergman and Guilherme Dias at the Department of Genetics and Institute of Bioinformatics, University of Georgia, for their valuable comments and suggestions to improve the quality of the manuscript; and Tyler Elliott at the Centre for Biodiversity Genomics, Biodiversity Institute of Ontario, University of Guelph, for his critical review of the draft. The authors would also thank Amer S. Alharthi at the General Directorate for Research and Innovation, King Abdulaziz City for Science and Technology, for his technical support.
Funding
This work was supported by the National Centre for Biotechnology, Life Science and Environment Research Institute, and the Center of Excellence for Genomics (grant 20-0078), King Abdulaziz City for Science and Technology, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
MMM, MAI, and MBA conceived and designed the experiments; MAI, MS, BMA, SNA, and MMM carried out the experiments; MAI, MMM, MS, SNA, BMA, and FHA analyzed the data; MAI, MMM, SNA, and MS wrote the manuscript. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval for this study was obtained from the Institute Review Board in King Abdulaziz City for Science and Technology (HAP-01-R-086), with the ethics number (IRB 20005).
Consent for publication
Not applicable
Competing interests
The authors declare there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ibrahim, M.A., Al-Shomrani, B.M., Simenc, M. et al. Comparative analysis of transposable elements provides insights into genome evolution in the genus Camelus. BMC Genomics 22, 842 (2021). https://doi.org/10.1186/s12864-021-08117-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08117-9