
Abstract
Background
Recent studies have demonstrated the utility of scRNA-seq SNVs to distinguish tumor from normal cells, characterize intra-tumoral heterogeneity, and define mutation-associated expression signatures. In addition to cancer studies, SNVs from single cells have been useful in studies of transcriptional burst kinetics, allelic expression, chromosome X inactivation, ploidy estimations, and haplotype inference.
Results
To aid these types of studies, we have developed a tool, SCReadCounts, for cell-level tabulation of the sequencing read counts bearing SNV reference and variant alleles from barcoded scRNA-seq alignments. Provided genomic loci and expected alleles, SCReadCounts generates cell-SNV matrices with the absolute variant- and reference-harboring read counts, as well as cell-SNV matrices of expressed Variant Allele Fraction (VAFRNA) suitable for a variety of downstream applications. We demonstrate three different SCReadCounts applications on 59,884 cells from seven neuroblastoma samples: (1) estimation of cell-level expression of known somatic mutations and RNA-editing sites, (2) estimation of cell- level allele expression of biallelic SNVs, and (3) a discovery mode assessment of the reference and each of the three alternative nucleotides at genomic positions of interest that does not require prior SNV information. For the later, we applied SCReadCounts on the coding regions of KRAS, where it identified known and novel somatic mutations in a low-to-moderate proportion of cells. The SCReadCounts read counts module is benchmarked against the analogous modules of GATK and Samtools. SCReadCounts is freely available (https://github.com/HorvathLab/NGS) as 64-bit self-contained binary distributions for Linux and MacOS, in addition to Python source.
Conclusions
SCReadCounts supplies a fast and efficient solution for estimation of cell-level SNV expression from scRNA-seq data. SCReadCounts enables distinguishing cells with monoallelic reference expression from those with no gene expression and is applicable to assess SNVs present in only a small proportion of the cells, such as somatic mutations in cancer.
Similar content being viewed by others


Background
Single cell RNA sequencing (scRNA-seq) brings major advantages over bulk RNA-seq analyses, especially the ability to distinguish cell populations and to assess cell-type specific phenotypes [1]. Connecting these phenotypes to cell-level genetic variants (such as Single Nucleotide Variants, SNVs) is essential for phenotype interpretation. In cancer, studies on cellular genetic heterogeneity have been instrumental in tracing lineages and resolving sub-clonal tumor architecture [2,3,4,5,6,7,8,9] In addition to cancer studies, SNV observations from single cells have been useful in studies of transcriptional burst kinetics, allelic expression, chromosome X inactivation, ploidy estimations, haplotype inference, and quantitative trait loci (QTL) [10,11,12,13,14,15,16,17,18,19,20,21].
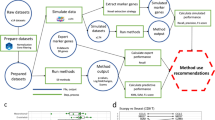
To aid with the considerable data-analysis and data-management demands of such studies, we have developed a tool, SCReadCounts, for cell-level quantitation of SNV expression. Provided with barcoded scRNA-seq alignments, list of barcodes and genomic loci and alleles of interest, SCReadCounts tabulates, for each cell, the reference and variant read counts (nref and nvar, respectively), and expressed Variant Allele Fraction (VAFRNA = nvar/(nvar + nref)). SCReadCounts generates a cell-SNV matrix with the absolute nvar and nref counts, and a cell-SNV matrix with the VAFRNA estimated at a user-defined threshold of minimum number of required sequencing reads (minR) (Fig. 1). Particular strengths of SCReadCounts include its named, explicit, flexible, and configurable read-filtering and cell-barcode extraction capabilities; accounting for all reads overlapping each locus, whether counted or ignored; and straightforward input and output formats – these features make SCReadCounts easy to integrate in multi-tool analysis pipelines. The cell-SNV matrices then can be used for a wide range of downstream analyses.

SCReadCounts workflow using publicly available tools
Unlike variant callers (i.e. GATK, Samtools [22, 23]), SCReadCounts estimates the read counts per allele and per cell, across all cells, including cells in which the position of interest is covered with only reference reads. This is particularly useful in scRNA-seq settings, where it enables distinguishing cells with monoallelic reference expression from those with no gene expression. The later can be used to assess cell-level allele dynamics, and to correlate variant expression to gene expression [24].
Implementation
SCReadCounts is freely available (https://github.com/HorvathLab/NGS) as a self-contained binary package for 64-bit Linux and MacOS (Darwin) systems, and as Python source. The self-contained binary packages are appropriate for most Linux and MacOS users. The pythonic version requires pysam, numpy and scipy along with other packages (detailed instructions at https://github.com/HorvathLab/NGS#SCReadCounts).
Currently, SCReadCounts has three programs. The program scReadCounts manages the sequential execution of programs readCounts and readCountsMatrix, collects the necessary arguments for successful execution, and avoids unnecessary execution of the expensive readCounts tool if possible. readCounts requires three input files: a pooled single cell alignment, a list of cell-barcodes, and a list of genomic positions of interest. Optionally, readCounts can be user-configured for read filtering. readCounts utilizes the barcode information from the pooled single cell alignments and outputs the variant and reference read counts (nvar and nref, respectively), for each barcode (cell), in a tab separated text file. This file is then used as an input for the second program - readCountsMatrix - which, upon providing an output prefix, generates two outputs: (1) a cell-position matrix with absolute nvar and nref counts, and (2) a cell-position matrix with the expressed VAFRNA. VAFRNA is estimated at a user-defined threshold of minimum required sequencing reads (minR); default minR = 5. readCountsMatrix is time-efficient and can be re-run multiple times at various minR thresholds.
SCReadcounts provides explicit configuration for alignments barcoded through STARsoloUMItools. Additional cellular barcode extraction logic can be configured for SCReadCounts software, based on BAM file tags or RNA sequence name and delimited tokens or regular expressions (see the “SCReadCounts Read Grouping” documentation).
Performance
To assess SCReadCounts performance we compared the variant and reference read counts tabulations of SCReadCounts with the analogous modules of the mpileup utility of Samtools and the haplotype caller of GATK [22, 23]. SCReadCounts default options generate nearly identical values to mpileup and GATK (Fig. 2). SCReadCounts uses, by default, a very simple read-filtering criteria, but it can also be readily configured to achieve scenario-specific, mpileup-consistent, or GATK-consistent results, with optional explicit output of the number of reads discarded by each filtering rule. On our system (2 × 14 core CPUs with 1.5 TB RAM compute node) processing of a file containing ~ 5000 cells, ~150mln reads, and ~ 80 K SNVs, requires approximately 4 h for the tabulation of nvar and nref, and up to 2 min for the generation of the cell-SNV matrices. The later enables the users to quickly generate VAFRNA matrices at various minR.

Concordance between nvar, nref and VAFRNA estimates across SCReadCounts, the mpileup utility of Samtools, and the haplotype caller of GATK in individual cell scRNA-seq alignments (a) and pooled scRNA-seq alignments (b) of sample. SRR10156295. Top: VAFRNA middle: nvar, bottom: nref.
Results
We have explored a variety of SCReadCounts applications on over 300,000 single cells from six different studies on normal and tumor human samples, including adipose tissue, adrenal neuroblastoma, acute myeloid leukemia, non-small lung cancer, prostate carcinoma, and the MCF7 cell line derived from breast adenocarcinoma [3,4,5,6, 25,26,27]. Here we demonstrate three different SCReadCounts applications on 59,884 cells derived from seven neuroblastoma samples [3]: (1) estimation of cell level expression of known somatic mutations and RNA-editing sites, (2) estimation of cell level allele expression from biallelic positions as called in the pooled scRNA-seq data, and (3) a discovery mode assessment of the reference and each of the three alternative nucleotides at genomic positions of interest. The discovery mode does not require prior knowledge on existing genetic variants and is particularly convenient for a quick focused assessment of a gene or a group of genes or regions of interest.
For all three applications the scRNA-seq data was processed using publicly available tools. In the exemplified workflow (See Fig. 1), the raw sequencing reads are aligned to the reference human genome (GRCh38) using STARsolo (v.2.7.7a) which processes the cellular barcodes, generates a list of error corrected barcodes, and deduplicates the alignments, retaining the reads with the highest mapping quality using the unique molecular identifiers (UMI) [28]. In addition to STARsolo, SCReadCounts accepts barcoded alignments and barcode lists generated by UMI-tools [29, 30]. The alignments can be filtered to correct for allele-mapping bias by removing reads mapped ambiguously due to the variant nucleotide (WASP); this filtering utilizes the same list of positions to be used as input for SCReadCounts [31].
SCReadCounts on known variant loci
SCReadCounts can be applied to assess known genetic variation loci such as somatic mutational hotspots or RNA-editing sites. We first asked if we could assess known somatic mutations in the set of seven neuroblastoma scRNA-seq samples (S_Table 1). Known somatic mutations (118,401 loci) were extracted from COSMIC by selecting loci from the Cancer Census Genes positioned outside repetitive regions (to exclude alignment bias) and not present in dbSNP (to ensure germline variants are excluded (dbSNPv.154) [32]. The resulting list of COSMIC somatic mutations (S_Table 2) was provided to SCReadCounts. A minimum of 3 sequencing reads (nvar + nref > = 3, minR = 3) was required for loci to be considered for further analysis. SCReadCounts identified 450 distinct COSMIC mutations expressed in at least 4 individual cells in one or more of the 7 neuroblastoma samples (S_Table 3). Examples include COSV99055840 in CENPF, COSV55220443 in STMN2, COSV85221362 in TXNIP, COSV10111219 in SYNJ2BP, COSV101287113 in PRAME, COSV100451465 in MRPS24, and COSV104673712 in SERPINA1 (Fig. 3). To assess the consistency between SCReadCounts and the variant callers across all cells, we split the alignments based on cell barcodes [33] and performed variant call on all the individual cell alignments using GATK in samples SRR10156295, SRR10156296, SRR10156299 and SRR10156300. GATK called all the SNVs for which 2 or more reads bearing the variant nucleotide were tabulated by scReadCounts (relaxed filtering, See 1) in the corresponding cells (See Fig. 3). Importantly, because SCReadCounts excludes loci not covered with required number of reads (minR), it allows to distinguish cells with only reference reads from cells with low or no expression of the SNV-harboring gene (See Fig. 3). Accordingly, based on the relative expression of variant reads and the SNV locus, SCReadCounts can discern different gene-SNV-expression patterns. For example, some genes and their harbored SNV are expressed primarily in one cell type (i.e. COSV99055840 in CENPF in erythrocytes, COSV101287113 in PRAME in neurons, and COSV104673712 in SERPINA1 in monocytes, See Fig. 3). Other SNVs are expressed primarily in one cell type even when the gene is expressed across different cell types (i.e COSV55220443 in STMN2 and COSV10111219 in SYNJ2BP in neurons, see Fig. 3).

Two-dimensional UMAP clusters of samples SRR10156295 and SRR10156296 showing cells classified by type (left) and visualizing cell-level expression of somatic mutations (right): COSV99055840 in CENPF, COSV55220443 in STMN2, COSV85221362 in TXNIP, COSV10111219 in SYNJ2BP, COSV101287113 in PRAME, COSV100451465 in MRPS24, and COSV104673712 in SERPINA1. Each pair of panels shows VAFRNA, (top) where the intensity of the red color corresponds to the relative expression of the somatic mutation and the green color indicates that all the reads (minR = 3) covering the position in the cell carried the reference nucleotide, compared to variant call (bottom), where the black color indicates presence of the mutation, and grey indicates either lack of variant or lack of gene expression. Hence, SCReadCounts allows to distinguish cells with only reference reads from cells with low or no expression of the SNV-harboring gene
Next, we demonstrate that SCReadCounts can quantify cell-specific RNA-editing in the same neuroblastoma samples. For this analysis we used the previously described single nucleotide RNA-editing events catalogued in the REDI database [34], after excluding genomic positions in repetitive regions or that coincide with a potential germline variant. A total of 107,053 distinct RNA-editing sites were provided to SCReadCounts (S_Table 4) along with the corresponding scRNA-seq alignments. At minR = 5, SCReadCounts identified 72 positions which were edited in at least 2 cells in one or more of the 7 neuroblastoma samples (S_Table 5). We investigated the A > G RNA-editing event at 14:100846310 in the cancer-implicated lincRNA MEG3; this position was edited in 6 of the 7 samples. Cells with MEG3 RNA-editing were predominantly clustered in neurons, where the proportion of edited RNA molecules (as reflected through the VAFRNA), suggested variable degrees of RNA editing (S_Figure 2).
SCReadCounts after variant call
SCReadCounts can be applied in conjunction with variant callers to estimate the cell-specific allele expression of germline or somatic SNVs. To explore this application, we called variants from the pooled alignments using GATK (v4.1.7.0, [22]), and filtered the calls retaining high quality biallelic positions for which both the variant and the reference allele were supported by a minimum of 50 sequencing reads, as we have previously described [24, 26]. The variant lists were then provided to SCReadCounts together with the corresponding alignments and the STARsolo generated list of error corrected barcodes. The resulting VAFRNA estimates can be used to explore a variety of cell-level allelic features. For example, the distribution of the VAFRNA at minR = 5 across the cells for each of the seven neuroblastoma samples is plotted on S_Figure 3, which shows that many of the SNVs have skewed or mono-allelic expression. VAFRNA estimates for positions covered by at least 10 total reads (minR = 10) in 20 and more cells per sample are summarized in S_Table 6. A systematic analysis of the distribution VAFRNA of heterozygous SNVs at different minR is analyzed in our recent work [26]. In addition, VAFRNA estimates can be used to explore correlations between allele- and gene-expression in single cells using scReQTL. Our previous research applying scReQTL on normal adipose datasets has shown that scReQTLs are substantially enriched in GWAS-significant SNVs and in known gene-gene interactions [24].
SCReadCounts in discovery mode
As mentioned earlier, SCReadCounts can be applied in a discovery mode which does not require any prior knowledge of SNVs. In this use-case, SCReadCounts considers positions of interest where the reference nucleotide is substituted with each of the three alternative nucleotides. Such SCReadCounts inputs can be generated for a gene, region or a group of genes/regions of interest, either manually, or using a script (provided at https://github.com/HorvathLab/NGS#SCReadCounts). Herein, we demonstrate this approach using an enumeration of each position in the coding region of KRAS (S_Table 7), mutations in which have been implicated in neuroblastoma. Across the seven samples, SCReadCounts identified a total of 30 distinct SNVs that do not coincide with known germline variants in the coding sequence of KRAS (S_Table 8). The SNVs included missense, nonsense and synonymous substitutions in up to 15 individual cells per sample. Eight of the 30 SNVs were seen in more than one sample; for example, the synonymous substation 12:25227293_G > A (Gly77Gly, Fig. 4) was seen in 4 out of the 7 samples. Seven of the 30 mutations were previously catalogued in the COSMIC database – the remaining 23 substitutions represent novel KRAS variants. To assess the consistency of SCReadCounts with variant calls, we applied the above-described strategy of GATK variant call on barcode-split individual cell alignments. Similarly to the COSMIC mutations, GATK called all the SNVs for which 2 or more reads bearing the variant nucleotide were tabulated by scReadCounts in the corresponding cells (See Fig. 4). In addition, we examined the IGV visualization of the alignments from 12:25227293_G > A positive cells; alignments from cells with different proportion of variant reads are shown on S_Figure 4.

Two-dimensional UMAP clusters of samples SRR10156299 and SRR10156300 showing cells classified by type (left panel) and visualizing cell-level expression of
the novel somatic mutation Gly77Gly (12:25227293_G > A) in the gene KRAS (middle panel) in comparison with results from variant call in barcode-split individual alignments (12:25227293_G > A – positive cells are indicated in black).
Discussion
SCReadCounts joins the handful of currently emerging approaches for analysis of genetic variation from scRNA-seq data [7, 8, 16, 35,36,37].
SCReadCounts supports several use-cases specific to scRNA-seq. First, and perhaps most importantly, SCReadCounts can detect and quantify SNVs present in a low proportion of cells, such as somatic and mosaic SNVs. SNVs present in a low proportion of cells are known to be frequently missed by variant calls carried out on pooled scRNA-seq alignments [35, 36, 38]. Indeed, the majority of the somatic mutations detected by SCReadCounts were not called on the pooled scRNA-seq data by GATK at its default setting but were called after extracting individual cell alignments by barcode (See S_Tables 3, 8). Splitting alignments by barcodes and calling variants from thousands of individual cells however is computationally expensive [39]. The statistical significance of novel cell-level somatic mutations can be readily assessed by tools such as SCmut [35] from SCReadCounts matrices, when the experimental design and availability of bulk sequencing data permit. However, even without such sophisticated approaches, SCReadCounts minR criteria provides some measure of control over false positives in discovery mode, and surfaces read counts for cells with only reference reads. Thereby SCReadCounts enables exploration of gene-SNV expression patterns and outlining SNVs specific for a cell type for cell-specific or ubiquitously expressed genes (See Fig. 3).
In the herein presented analysis, SCReadCounts detected novel somatic mutations occurring in individual cells or in a small number of cells in one or more of the neuroblastoma samples. Second, SCReadCounts provides per cell quantitation of absolute and relative number of variant reads across all cells, including those where the position is covered with only reference reads, allowing the identification of cells with preferential expression of the variant or the reference allele. Furthermore, the flexible VAFRNA minR enables tuning of SCReadCounts to the particular application and depth of sequencing. Here, a major consideration is the balance between inclusivity (low minR) and higher-confidence VAFRNA estimates (high minR). In our analyses we use different minR depending on the application [24, 26]. For example, to confidently estimate allele expression of germline variants in highly transcribed genes, high minR is needed. In contrast, assessments of somatic mutations would benefit from high inclusivity using low minR. Finally, SCReadCounts generates cell-SNV matrices that are analogous to the cell-gene matrices generated by popular scRNA-seq tools, which streamlines down- stream applications combining SNV and gene expression.
Conclusions
In conclusion, we believe that SCReadCounts supplies a fast and efficient solution for estimation of scRNA-seq genetic variation. Importantly, SCReadCounts enables distinguishing cells with monoallelic reference expression from those with no gene expression and is applicable to assess SNVs present in only a small proportion of the cells, such as somatic mutations in cancer.
Materials and methods
Sequencing datasets
The sequencing datasets were freely available from the NCBI Sequence Read Archive (SRA) under the accession numbers SRR10156295, SRR10156296, SRR10156297, SRR10156299, SRR10156300, SRR10156302, and SRR10156303. The patients demographics, neuroblastoma (NB) phenotype, obtaining of the samples, and the specifics of the 10x Genomics sequencing protocol are described in details in the original study [3]. Briefly, all the samples were processed using 10x Genomics 3-UTR v2 workflow, and the produced libraries were sequenced to a 150 nt sequencing length on NovaSeq 6000 (Illumina) at a depth of approximately 400 M reads per sample.
Data processing
For alignment of the scRNA sequencing reads, read-to-gene assignment, cell barcode demultiplexing, error correction, and unique molecular identifier (UMI) collapsing, we used the STARsolo module of STAR v.2.7.7a with transcript annotations from the assembly GRCh38.79 [28] as previously described [24]. To generate individual cell alignments we adopted a publicly available python script which splits the pooled scRNA-seq alignments based on cellular barcode [40].
Variant call
SNV were called using the HaplotypeCaller module of GATK v.4.2.0.0 [22], filtered using bcftools v.1.10.2 [23] and annotated using SeattleSeq v.16.00 (dbSNP build 154), as previously described [24]. When calling SNVs from the individual alignments, no filtering was applied on the SNV calls in order to retain calls where the variant nucleotide is present in a single read.
Gene expression estimation from scRNA-seq data
Gene expression was estimated from raw read count matrices output by STARsolo, and normalized and scaled using the SCTransform function of Seurat v.3.0 [41, 42], as previously described [24, 43]. Based on the cells’ and features’ distribution, we have filtered out: (1) cells with mitochondrial gene expression over between 7.5 and 15%, (2) cells with less than between 500 and 1000 genes, and (3) cells with more than between 2600 and 4500 detected genes (to remove potential doublets, examples on S_Figure 5). Batch effects and cell cycle effects were removed as previously described [24].
Cell type assessments
Likely cell types were assigned to cell clusters using SingleR v.1.0.5 [44] as previously described [24]; examples are shown on S_Figure 6.
Statistical analyses
Statistical assessments were performed using the statistical modules implemented in the used software packages [41, 44], with built-in multiple testing corrections, where p-value of 0.05 was considered significant.
Availability of data and materials
All the data analyzed in this study are supplied with the supplemental material or available as indicated in the cited publications. The sequencing datasets are downloaded from the NCBI Sequence Read Archive (SRA) under the accession numbers SRR10156295, SRR10156296, SRR10156297, SRR10156299, SRR10156300, SRR10156302, and SRR10156303.
Abbreviations
- scRNA-seq:
-
Single cell RNA sequencing
- SNV:
-
Single nucleotide variant
- SNP:
-
Single nucleotide polymorphism
- VAF:
-
Variant allele fraction
- scReQTL:
-
Single cell RNA expressed quantitative trait loci
References
Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356(6335). https://doi.org/10.1126/science.aah4573.
Brady SW, McQuerry JA, Qiao Y, Piccolo SR, Shrestha G, Jenkins DF, et al. Combating subclonal evolution of resistant cancer phenotypes. Nat Commun. 2017;8(1):1231. https://doi.org/10.1038/s41467-017-01174-3.
Dong R, Yang R, Zhan Y, Lai H-D, Ye C-J, Yao X-Y, et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell. 2020;38(5):716-33. https://doi.org/10.1016/j.ccell.2020.08.014.
Chen J, Tan Y, Sun F, Hou L, Zhang C, Ge T, et al. Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biol. 2020;21(1):152. https://doi.org/10.1186/s13059-020-02064-6.
Ma X, Guo J, Liu K, Chen L, Liu D, Dong S, et al. Identification of a distinct luminal subgroup diagnosing and stratifying early stage prostate cancer by tissue-based single-cell RNA sequencing. Mol Cancer. 2020;19(1):147. https://doi.org/10.1186/s12943-020-01264-9.
Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018;560(7718):325-30. https://doi.org/10.1038/s41586-018-0409-3.
Petti AA, Williams SR, Miller CA, Fiddes IT, Srivatsan SN, Chen DY, et al. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun. 2019;10(1):3660. https://doi.org/10.1038/s41467-019-11591-1.
Poirion O, Zhu X, Ching T, Garmire LX. Using single nucleotide variations in single-cell RNA-seq to identify subpopulations and genotype-phenotype linkage. Nat Commun. 2018;9(1):4892. https://doi.org/10.1038/s41467-018-07170-5.
Lee JK, Wang J, Sa JK, Ladewig E, Lee HO, Lee IH, et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat Genet. 2017;49(4):594-9. https://doi.org/10.1038/ng.3806.
Van Der Wijst MGP, Brugge H, De Vries DH, Deelen P, Swertz MA, Franke L. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat Genet. 2018;50(4):493-7. https://doi.org/10.1038/s41588-018-0089-9.
van der Wijst MG, de Vries DH, Groot HE, Trynka G, Hon C-C, Bonder M-J, et al. The single-cell eQTLGen consortium. Elife [Internet] eLife Sciences Publications, Ltd. 2020;9 [cited 2020 Apr 6] Available from: http://www.ncbi.nlm.nih.gov/pubmed/32149610.
Edsgärd D, Reinius B, Sandberg R. Scphaser: Haplotype inference using single-cell RNA-seq data. Bioinformatics. 2016;32(19):3038-40. https://doi.org/10.1093/bioinformatics/btw484.
Gallivan CP, Ren H, Read EL. Analysis of Single-Cell Gene Pair Coexpression Landscapes by Stochastic Kinetic Modeling Reveals Gene-Pair Interactions in Development. Front Genet. 2020;10:1387. https://doi.org/10.3389/fgene.2019.01387.
La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature. 2018;560(7719):494-8. https://doi.org/10.1038/s41586-018-0414-6.
Larsson AJM, Johnsson P, Hagemann-Jensen M, Hartmanis L, Faridani OR, Reinius B, et al. Genomic encoding of transcriptional burst kinetics. Nature. 2019;565(7738):251-4. https://doi.org/10.1038/s41586-018-0836-1.
Santoni FA, Stamoulis G, Garieri M, Falconnet E, Ribaux P, Borel C, et al. Detection of imprinted genes by single-cell allele-specific gene expression. Am J Hum Genet. 2017;100(3):444–53. https://doi.org/10.1016/j.ajhg.2017.01.028.
Deng Q, Ramsköld D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343(6167):193-6. https://doi.org/10.1126/science.1245316.
Kim JK, Kolodziejczyk AA, Illicic T, Teichmann SA, Marioni JC. Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression. Nat Commun. 2015;6:8687. https://doi.org/10.1038/ncomms9687.
De Mello JCM, Fernandes GR, Vibranovski MD, Pereira LV. Early X chromosome inactivation during human preimplantation development revealed by single-cell RNA-sequencing. Sci Rep. 2017;7(1):10794. https://doi.org/10.1038/s41598-017-11044-z.
D’Antonio-Chronowska A, Donovan MKR, Young Greenwald WW, Nguyen JP, Fujita K, Hashem S, et al. Association of Human iPSC Gene Signatures and X Chromosome Dosage with Two Distinct Cardiac Differentiation Trajectories. Stem Cell Reports. 2019;13(5):924-38. https://doi.org/10.1016/j.stemcr.2019.09.011.
Griffiths JA, Scialdone A, Marioni JC. Mosaic autosomal aneuploidies are detectable from single-cell RNAseq data. BMC Genomics. 2017;18(1):904. https://doi.org/10.1186/s12864-017-4253-x.
Auwera Mauricio O. GAV der C, Hartl C, Poplin R, Angel G del, Levy-Moonshine A, Jordan T, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2002;43(1110):11.10.1-11.10.33. https://doi.org/10.1002/0471250953.bi1110s43.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078-9. https://doi.org/10.1093/bioinformatics/btp352.
Liu H, Prashant NM, Spurr LF, Bousounis P, Alomran N, Ibeawuchi H, et al. scReQTL: an approach to correlate SNVs to gene expression from individual scRNA-seq datasets. BMC Genomics [internet]. 2021;22:40 Available from: https://doi.org/10.1186/s12864-020-07334-y.
X. L, Q. X, F. X, J. H, N. Y, Q. Z, et al. Single-cell RNA-seq of cultured human adipose-derived mesenchymal stem cells. Sci Data. 2019;6:190031. https://doi.org/10.1038/sdata.2019.31.
Prashant NM, Liu H, Bousounis P, Spurr L, Alomran N, Ibeawuchi H, et al. Estimating the allele-specific expression of snvs from 10× genomics single-cell rna-sequencing data. Genes (Basel). 2020;11(3):240. https://doi.org/10.3390/genes11030240.
Johnston G, Ramsey HE, Liu Q, Wang J, Stengel KR, Sampathi S, et al. Nascent transcript and single-cell RNA-seq analysis defines the mechanism of action of the LSD1 inhibitor INCB059872 in myeloid leukemia. Gene. 2020;752:144758. https://doi.org/10.1016/j.gene.2020.144758.
Kaminow B, Yunusov D, Dobin A. STARsolo: accurate, fast and versatile mapping/quantification of single-cell and single-nucleus RNA-seq data. BioRxiv. https://doi.org/10.1101/2021.05.05.442755.
Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. https://doi.org/10.1038/ncomms14049.
Smith T, Heger A, Sudbery I. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 2017;27(3):491–9. https://doi.org/10.1101/gr.209601.116.
Van De Geijn B, Mcvicker G, Gilad Y, Pritchard JK. WASP: Allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods. 2015;12(11):1061-3. https://doi.org/10.1038/nmeth.3582.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47(D1):D941-7. https://doi.org/10.1093/nar/gky1015.
Kretzschmar WW. split_bams_by_barcodes; 2021.
Picardi E, D’Erchia AM, Lo GC, Pesole G. REDIportal: A comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017;45(D1):D750-7. https://doi.org/10.1093/nar/gkw767.
Vu TN, Nguyen HN, Calza S, Kalari KR, Wang L, Pawitan Y, et al. Cell-level somatic mutation detection from single-cell RNA sequencing. Bioinformatics. 2019;35(22):4679-87. https://doi.org/10.1093/bioinformatics/btz288.
Schnepp PM, Chen M, Keller ET, Zhou X. SNV identification from single-cell RNA sequencing data. Hum Mol Genet. 2019;28(21):3569-83. https://doi.org/10.1093/hmg/ddz207.
Liu F, Zhang Y, Zhang L, Li Z, Fang Q, Gao R, et al. Systematic comparative analysis of single-nucleotide variant detection methods from single-cell RNA sequencing data. Genome Biol. 2019;20(1):242. https://doi.org/10.1186/s13059-019-1863-4.
Tools VD. MuTect2. GATK Man; 2017.
Prashant NM, Liu H, Dillard C, Ibeawuchi H, Alsaeedy T, Chan KH, et al. Improved SNV discovery in barcode-stratified scRNA-seq alignments. bioRxiv [Internet]. 2021;2021(06):12.448184 Available from: http://biorxiv.org/content/early/2021/06/13/2021.06.12.448184.abstract.
SplitBamsScript. https://gist.github.com/winni2k/978b33d62fee5e3484ec757de1a00412.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20. https://doi.org/10.1038/nbt.4096.
Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20(1):296. https://doi.org/10.1186/s13059-019-1874-1.
Prashant NM, Liu H, Bousounis P, Spurr L, Alomran N. Estimating allele-specific expression of SNVs from 10x Genomics Single-Cell RNA-Sequencing Data. Genes (Basel). 2020;11(3):240. https://doi.org/10.3390/genes11030240.
Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163-72. https://doi.org/10.1038/s41590-018-0276-y.
Acknowledgements
Not Applicable.
Availability and requirements
Project name: SCReadCounts
Project home page: https://github.com/HorvathLab/NGS#SCReadCounts
Operating system(s): Linux, MacOS
Programming language: Python
Other requirements: N/A
License: MIT License
Any restrictions to use by non-academics: no restrictions
Funding
This work was supported by MGPC, The George Washington University; [MGPC_PG2020 to AH]. The funding body did not play any role in the design of the study, the collection, analysis, and interpretation of data, or in the writing the manuscript.
Author information
Authors and Affiliations
Contributions
PNM, NA, YC and NE developed and tested the software; HL, PB and MM performed the analyses and contributed to the visualization; NE conceptually developed and implemented the tool, AH devised and supervised the study and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable (The study uses only previously published and freely available datasets).
Consent for publication
Not Applicable (The study uses only previously published and freely available datasets).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Fig. 1.
CCND1. Two-dimensional UMAP clusters of samples SRR10156296, SRR10156297 and SRR10156299 showing cells classified by type (left) and visualizing COSV57121427 in CCND1, which was seen primarily in neurons in all three samples. The intensity of the red color corresponds to the proportion variant reads) of the VAFRNA of the indicated somatic mutations in the corresponding cells of the neuroblastoma samples; green color indicates that all the reads (minR = 3) covering the position in the cell carried the reference nucleotide.
Additional file 2: Supplementary Fig. 2.
RNA-editing. Two-dimensional UMAP clusters of samples SRR10156297 and SRR10156299 showing cells classified by type (left) and visualizing RNA-editing levels (right) in the gene MEG3, where the intensity of the red color corresponds to the proportion of edited reads, and the green color indicates that all the reads (minR = 3) covering the position in the cell carried the reference nucleotide.
Additional file 3: Supplementary Fig. 3.
scVAFRNA distribution. scVAFRNA estimated at genomic positions covered by a minimum of 5 sequencing reads (minR = 5) at the sites with bi-allelic calls (GATK) in the 7 neuroblastoma samples; the positions are sorted by VAFRNA (y-axis). For the majority of positions, VAFRNA showed predominantly mono-allelic expression, with a substantial proportion of the scVAFRNA estimations in the intervals 0–0.2 (orange) and 0.8–1.0 (purple). The percentage of cells with the corresponding VAFRNA is displayed on the x-axis.
Additional file 4: Supplementary Fig. 4.
KRAS. IGV visualization of variable scVAFRNA of the novel somatic mutation Gly77Gly (12:25227293_G > A) in the gene KRAS in three individual cells of sample SRR10156295.
Additional file 5: Supplementary Fig. 5.
Before and after filtering features distribution. Examples of density plots showing the distribution of cells based on proportion of transcripts of mitochondrial origin and number of genes, plotted against the counts of sequencing reads before (top) and after (bottom) filtering. The selected QC thresholds are: mitochondrial gene expression above between 6 and 15%, and number of genes below between 800 and 1000. To remove potential doublets/multiples we also filtered out signals with more than between 2600 and 4500 genes.
Additional file 6: Supplementary Fig. 6.
SingleR Heatmaps. Heatmaps of SingleR scores for top correlated cell types from each of Seurat generated clusters. SingleR uses expression data to regenerate the clusters, and for each cluster, calculates the Spearman coefficient for the genes in the reference dataset. Then, it uses multiple correlation coefficient to collect a single value per cell type per cluster.
Additional file 7: Supplementary Table 1.
Samples. Samples used in this study.
Additional file 8: Supplementary Table 2.
COSMIC input. List COSMIC somatic mutations used as input to SCReadCounts.
Additional file 9: Supplementary Table 3.
COSMIC output. 450 distinct COSMIC mutations found by SCReadCounts to be expressed in at least 4 individual cells in one or more of the 7 neuroblastoma samples.
Additional file 10: Supplementary Table 4.
REDI input. 107,053 distinct RNA-editing sites provided as input to SCReadCounts.
Additional file 11: Supplementary Table 5.
REDI output. 72 positions found by SCReadCounts to be edited in at least 2 cells in one or more of the 7 neuroblastoma samples.
Additional file 12: Supplementary Table 6.
scVAFRNA estimates. scVAFRNA estimates for positions covered by at least 10 total reads (minR = 10) in 20 and more cells per sample.
Additional file 13: Supplementary Table 7.
KRAS input in Discovery mode. Genomic positions used as a discovery mode input for scReadCounts.
Additional file 14: Supplementary Table 8.
KRAS output in Discovery mode. 30 distinct SNVs that do not coincide with known germline variants in the coding sequence of KRAS.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Prashant, N.M., Alomran, N., Chen, Y. et al. SCReadCounts: estimation of cell-level SNVs expression from scRNA-seq data. BMC Genomics 22, 689 (2021). https://doi.org/10.1186/s12864-021-07974-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07974-8