
Abstract
Background
Species in the genus Armillaria (fungi, basidiomycota) are well-known as saprophytes and pathogens on plants. Many of them cause white-rot root disease in diverse woody plants worldwide. Mitochondrial genomes (mitogenomes) are widely used in evolutionary and population studies, but despite the importance and wide distribution of Armillaria, the complete mitogenomes have not previously been reported for this genus. Meanwhile, the well-supported phylogeny of Armillaria species provides an excellent framework in which to study variation in mitogenomes and how they have evolved over time.
Results
Here we completely sequenced, assembled, and annotated the circular mitogenomes of four species: A. borealis, A. gallica, A. sinapina, and A. solidipes (116,443, 98,896, 103,563, and 122,167 bp, respectively). The variation in mitogenome size can be explained by variable numbers of mobile genetic elements, introns, and plasmid-related sequences. Most Armillaria introns contained open reading frames (ORFs) that are related to homing endonucleases of the LAGLIDADG and GIY-YIG families. Insertions of mobile elements were also evident as fragments of plasmid-related sequences in Armillaria mitogenomes. We also found several truncated gene duplications in all four mitogenomes.
Conclusions
Our study showed that fungal mitogenomes have a high degree of variation in size, gene content, and genomic organization even among closely related species of Armillara. We suggest that mobile genetic elements invading introns and intergenic sequences in the Armillaria mitogenomes have played a significant role in shaping their genome structure. The mitogenome changes we describe here are consistent with widely accepted phylogenetic relationships among the four species.
Similar content being viewed by others


Background
The genus Armillaria consists of common saprophytic and pathogenic fungi that belong to the basidiomycete family Physalacriaceae. Armillaria parasitizes numerous tree species in forests of the Northern and Southern hemispheres. Armillaria species vary in virulence level and host spectrum and play important role in carbon cycling in forests [1, 2]. The life cycle of Armillaria is unique among basidiomycetes in that the vegetative phase is diploid, rather than dikaryotic [3]. Due to their capacity for vegetative growth and persistence through the production of rhizomoprhs, individuals of Armillaria are among the largest and oldest organisms on Earth [4,5,6,7].
Mitochondrial DNA (mtDNA) restriction maps of A. solidipes (formerly known as A. ostoyae) from different geographic regions were previously shown to differ greatly in size [8]. The interpretation was that biparental inheritance could increase cytoplasmic mixing and allow recombination in mitogenome. Although Armillaria mitogenome in natural populations is inherited uniparentally, the potential for transient cytoplasmic mixing, heteroplasmy, and recombination exists with each mating event [9]. Indeed the actual signature of recombination in the mitogenome of A. gallica has been detected [10]. No Armillaria mitogenomes, however, have been completely annotated and described previously. In this study, we report the complete sequences of the mitogenomes of A. borealis, A. gallica, A. sinapina, and A. solidipes, and describe their organization, gene content and a comparative analysis.
The main function of mitochondria is energy production via the oxidative phosphorylation. In addition to the primary function in respiratory metabolism and energy production, mitochondria are also involved in many other processes such as cell aging and apoptosis [11]. The limited number of genes in current mitogenomes can be likely explained by past transfer of many of their original genes into the eukaryotic nuclear genome, which occurred after a free-living ancestral bacterium was incorporated into an ancient cell as an endosymbiont [12,13,14]. According to the comparative mitogenome and proteome data, the organelle ancestor was likely related to Alphaproteobacteria [15,16,17]. In general, 14 conserved protein-coding genes involved in electron transport and respiratory chain complexes (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L and nad6), one ribosomal protein gene (rps3), two genes encoding ribosomal RNA subunits - small (rns) and large (rnl) - and a set of tRNA genes have been found in fungal mitogenomes [18, 19]. Despite the relatively conserved gene content, however, fungal mitogenomes vary greatly in size: from 18,844 bp in Hanseniaspora uvarum [20] up to 235,849 bp in Rhizoctonia solani [21]. This wide size range might be explained in part by variation in length of intergenic regions, differences in number of introns (group I and II) and their various sizes [22]. For example, large mitogenome size of Phlebia radiata (156 Kbp) was explained by a large number of intronic and intergenic regions [23].
Mitogenomes may provide clues into the evolutionary biology and systematics of eukaryotes. Mitogenomes could be especially helpful to establish phylogenetic relationships when nuclear genes do not provide clear or substantial phylogenetic data to solve conflicting phylogenies [24]. Moreover, the high degree of polymorphism is found in some mitochondrial introns and intergenic regions making these DNA regions also useful in population studies [25, 26].
Most of the mitochondrial group I introns contain ORFs with GIY-YIG or LAGLIDADG homing endonucleases (HEGs) motifs [27,28,29]. HEGs represent one of the types of mobile genetic elements that are able to insert themselves into specific genome positions [30]. As shown, HEGs can expand mitogenome size, may cause genome rearrangements, gene duplications and import of exogenic nucleotide sequences through horizontal gene transfer (HGT) [31,32,33,34]. HEGs may be involved in the spread of group I introns between distant species [35, 36]. However, the scale, rate, and direction of intron transfer have not yet been sufficiently studied. According to one hypothesis, a common evolutionary trajectory is from an ancestor of high intron content to derivatives of low intron content via progressive loss [37,38,39,40], but further testing of this possibility is needed. More studies of intron losses and acquisitions in closely related lineages are required to shed light on their evolution.
The number of evolutionary and systematic studies based on comparative analysis of complete fungal mitogenome sequences has substantially increased recently [41,42,43,44,45,46], but the mitogenome of only one member (Flammulina velutipes) in the Physalacriaceae family (Agaricales, Basidiomycota) is now available [47]. Here, we describe the complete mitogenomes of four Armillaria species.
Results
Mitogenome organization
The mitogenomes of Armillaria are 116,433 (A. borealis; GenBank accession number MH407470), 98,896 (A. gallica; MH878687), 103,563 (A. sinapina; MH282847), and 122,167 (A. solidipes; MH660713) bp circular DNAs (Fig. 1). The sequences were all AT-rich with similar AT content: 70.7% for A. borealis, 70.8% for both A. gallica and A. solidipes, and 71.5% for A. sinapina. We detected 16 tandem repeat or minisatellite loci in A. borealis and A. sinapina, 17 in A. gallica, and 11 in A. solidipes (Additional file 1: Table S1) using Tandem Repeats Finder (https://tandem.bu.edu/trf/trf.html). The length of the longest tandem motif was 41 bp in A. borealis, 27 bp in A. gallica, 23 bp in A. sinapina, and 37 bp in A. solidipes with two repeats in each species. In general, most tandem repeat loci contained two or three repeats. In addition, we also searched for microsatellite or simple sequence repeat (SSR) loci using SciRoKo (https://kofler.or.at/bioinformatics/SciRoKo) and found 8 SSR loci in A. borealis, 12 in A. gallica, 15 in A. sinapina, and 10 in A. solidipes (Additional file 2: Table S2). The comparisons of the whole mitogenomes using MAUVE identified conserved genomic blocks, as well as sequences rearrangements in several locations (Figs. 2 and 3).

Circular complete graphic mitogenome maps of four Armillaria species: A. borealis, A. solidipes, A. sinapina, and A. gallica. Genes are transcribed in a clockwise direction. The inner gray rings show the GC content of these genomes

Linear complete graphic mitogenome gene maps of four Armillaria species: A. borealis, A. solidipes, A. sinapina, and A. gallica with tRNA gene locations highlighted by red ovals emphasizing clustering of some of them

Gene order and rearrangements in mitogenomes of four Armillaria species: A. borealis, A. solidipes, A. sinapina, and A. gallica
Each mitogenome contained 15 protein-coding genes: three ATP-synthase complex F0 subunit genes (atp6, atp8, and atp9), three complex IV subunits genes (cox1, cox2, and cox3), one complex III subunit gene (cob), seven electron transport complex I subunits genes (nad1, nad2, nad3, nad4, nad4L, nad5, and nad6), one ribosomal protein gene (rps3), as well as large and small ribosomal subunits RNA genes (rnl, and rns) that are encoded on both strands. In all four mitogenomes the nad2 and nad3 and nad4L and nad5 genes were linked with a slight overlap: the stop-codon of nad2 overlapped the following start codon of nad3 by one nucleotide, and the stop codon of nad4L also overlapped the following start codon of nad5 by one nucleotide. All of these protein-coding genes are encoded on the same DNA strand, except for nad2 and nad3 that start with the typical translation initiation codon ATG, but are encoded on the opposite strand in A. borealis and A. solidipes (Fig. 3).
Some exons in protein-coding genes were difficult to annotate using MFannot due to their particularly small size. The smallest exons were found in the cob, cox1 and cox2 genes, such as 15 bp long 10th exon in cox1 and 12 bp long exon 6 in cob in A. borealis, 12 bp long exon 5 in cob in A. sinapina, 15 bp long exon 9 in cox1 and 15 bp long exon 3 in cox2 in A. solidipes. Therefore, these exons were annotated manually.
In total, 26, 24, 25, and 26 tRNA genes were annotated in the mitogenomes of A. borealis, A. gallica, A. sinapina, and A. solidipes, respectively. Similar to most fungal mitogenomes studied so far, the tRNA genes in all four mitogenomes were mainly clustered (Fig. 2), except the tRNA-Tyr gene (trnY), which was located between rnl and nad4 in all four Armillaria mitogenomes, and the tRNA-Phe gene (trnF) that was located along outside of clusters in all mitogenomes except A. sinapina. A. borealis and A. solidipes had the same five clusters. A. gallica and A. sinapina had four similar clusters that were only slightly different from five clusters in A. borealis and A. solidipes. The clusters were only slightly different in composition and location. All different tRNA genes were presented by a single copy except the tRNA-Pro gene (trnP) that had two copies in A. borealis and A. solidipes.
Gene order
The whole-genome alignments of the mitogenomes of A. borealis, A. gallica, A. sinapina, and A. solidipes revealed a predominant pattern of conservation of gene order and orientation, but with distinct variations (Figs. 2 and 3). A. borealis and A. solidipes had the same gene order and orientation, while A. gallica and A. sinapina contained gene rearrangements between nad3 and atp9 genes. A. gallica is different from A. borealis and A. solidipes only by a single inversion having the nad2-nad3-cox3 gene order vs. cox3-nad3-nad2. In addition, nad3 and nad2 are translated in the opposite direction from the opposite strand in A. borealis and A. solidipes. In A. sinapina the cox3 and atp6 genes were transposed and rearranged. The rearrangements are consistent with A. borealis and A. solidipes being sister species and A. sinapina and A. gallica being more distantly related [48, 49].
Codon usage
The codon usage frequencies for 14 protein-coding mitochondrial genes were determined for each Armillaria species (Additional file 3: Table S3). The start codon ATG was detected across all four species in all genes ended with the TAA stop codon except atp9 gene, which ended with TAG. The AT-rich codons were predominant, and the most-frequently used codons were invariant: TTA (Leu,10.77–11.03%), TTT (Phe, 5.63–5.92%), ATA (Ile, 5.18–5.28%), ATT (Ile 5.14–5.30%), GGT (Gly 3.09–3.19%). On the other hand, the СGC (Arg) codon was universally absent in all four species. Moreover, several codons were under-represented (having frequency < 0.5%), such as TGC (Cys, 0.02%), AGG (Arg, 0.02–0.05%), CGG (Arg, 0.10–0.14%), CGA (Arg, 0.17%), CGT (Arg, 0.05–0.07%), AGC (Ser, 0.17–0.19%), TGG (Trp, 0.29–0.36%), CAG (Gln, 0.24–0.26%), and CCC (Pro, 0.43–0.50%). Similar to other fungal studies, mitochondrial genes of Armillaria had a high number of AT-rich codons, and similar codon frequencies are found in other fungal mitogenomes [22].
Introns and plasmid-related sequences
In total, 26 introns were found in seven out of 15 protein-coding genes in A. borealis, 27 introns in six genes in A. solidipes, and 18 introns in six genes in A. sinapina and A. gallica (Table 1).
The size of the introns ranged from 189 bp (intron in atp9 in A. gallica) to 2615 bp (intron 2 in nad1 in A. solidipes). The average length of introns in all four species was 1902 bp. All introns were classified into group I, and some of them were further classified into subgroups IA (1), IB (10), and I-derived (7) in A. borealis, IB (10) and I-derived (6) in A. gallica, IB (5), ID (1), and I-derived (5) in A. sinapina, and IB (10) and I-derived (8) in A. solidipes (Additional file 4: Table S4).
Some introns in the same genes demonstrated only partial identity or orthology. For example, intron 2 in cox1 had 100% sequence similarity and the same insertion point in A. borealis and A. solidipes, but it showed no sequence similarity with intron 2 in cox1 of A. gallica. Intron 5 in cox1 had the same insertion point in A. borealis and A. solidipes, but had different insertion point in A. gallica and was completely identical (with 100% sequence similarity) to intron 3 in this species, but was not found in A. sinapina. However, all introns in cox1 of A. sinapina seemed orthologous to those in A. borealis and A. solidipes. In total, nine orthologous introns could be identified for cox1 between A. borealis and A. solidipes, four such introns among A. borealis, A. solidipes and A. sinapina, four introns among A. borealis, A. solidipes and A. gallica, and only one orthologous intron between A. sinapina and A. gallica (Fig. 4). Therefore, due to the presence and absence of various introns, the size of the cox1 gene varied from 8132 bp in A. sinapina to 15,987 bp in A. borealis. Here again, the pattern of change is consistent with A. borealis and A. solidipes as sister species and A. gallica and A. sinapina as more distantly related.

Introns (1–9) of the cox1 gene in four Armillaria species: A. borealis, A. solidipes, A. sinapina, and A. gallica. Black boxes represent exons. Arrows depict homologous or orthologous introns
Overall, A. borealis shared 25, 15 and 15 homologous or orthologous introns with A. solidipes, A. sinapina and A. gallica, respectively; A. solidipes 25, 15 and 16 with A. borealis, A. sinapina and A. gallica, respectively; A. sinapina 15, 15 and 9 with A. borealis, A. solidipes and A. gallica, respectively. A. gallica 16, 15 and 9 introns with A. solidipes, A. borealis and A. sinapina, respectively. The unique introns from each mitogenome were blasted against the NCBI GenBank database and revealed some similar sequences even in distantly related fungal mitogenomes (Table 2). In total, 11 unique introns were found in the four species: three in A. borealis (introns 1 and 6 in cob and intron 2 in cox2 that were 2288, 551 and 2585 bp long, respectively); five in A. solidipes (intron 1 in nad5, intron 3 in cob, introns 2 and 3 in cox2, and intron 1 in cox3 that were 1199, 1560, 1567, 381 and 1668 bp long, respectively). A. sinapina contained one unique intron 2 in nad1 (2547 bp), and A. gallica contained one unique intron 2 in cox1 (1320 bp).
Many introns contained ORFs encoding proteins which have similarities with homing endonucleases of LAGLIDADG (12 ORFs) and GIY-YIG (7 ORFs) families in A. sinapina, 15 and 9 in A. borealis, 17 and 8 in A. solidipes, 13 and 4 in A. gallica (Table 3). Among free-standing ORFs, we found two possible homing endonuclease genes in A. sinapina, the first was located between rnl and nad4 (LAGLIDADG) and the second was between atp6 and cox3 (GIY-YIG). One possible free-standing homing endonuclease was found in each A. borealis and A. gallica (LAGLIDADG) next to atp9.
We found ORFs in all four species that had homology with another type of mobile genetic elements – plasmid-like elements: five ORFs in A. sinapina, eight in A. borealis, six in A. solidipes, and two in A. gallica. In A. borealis and A. solidipes three plasmid ORFs were located between rps3 and cox3, two of them were similar to the DNA polymerase and RNA polymerase genes, and one ORF had unknown function. These ORFs were not present in mitogenomes of A. gallica and A. sinapina. Regions located between rps3 and cox3 in the mitogenomes of A. borealis and A. solidipes contained also ORFs that encode a 2034 bp (in A. solidipes) and 2646 bp (in A. borealis) long fragment of the DNA polymerase gene and a nearby located 1053 bp (in A. solidipes) and 1080 bp (in A. borealis) long fragments of the RNA polymerase gene. They were not present in the A. sinapina mitogenome.
In A. gallica, two plasmid-related ORFs (1173 and 681 bp) were located between nad3 and cox3 and one (375 bp) between cox3 and nad6. All of them were similar to the RNA polymerase genes.
In A. sinapina, two plasmid-related ORFs were located between nad3 and nad6 and represented 774 and 549 bp long RNA-polymerase genes. In addition, four ORFs were located between nad6 and atp6 and represented two 606 and 609 bp long genes that may encode hypothetical proteins with unknown function and other two 534 and 1707 bp long genes that were similar to the DNA-polymerase genes and arranged one after another.
Gene duplications
The mitogenomes of A. solidipes and A. sinapina contained a common region with homology to atp9 and located on a complementary strand in the rnl gene. It consisted of an 89 bp long sequence of the atp9 gene with 87% identity with the 89 bp long fragment of the 222 bp long original gene in both species. Although A. borealis and A. gallica lacked copies in these regions, they contained 47 bp and 54 bp long copies of the exon 2 of the atp9 gene, respectively, which were located upstream to the atp9 222 bp long coding sequence, next to the LAGLIDADG free-standing ORF.
Mitogenome size variation
The mitogenomes described in this study showed substantial size variation, with A. solidipes having the largest (122,167 bp) and A. gallica the smallest (98,896 bp) mitogenomes. Different numbers and sizse of introns and intergenic regions are the simplest explanation for this variation. The mitogenomes with 27 introns in A. solidipes and 26 in A. borealis were larger than mitogenomes in A. sinapina and A. gallica with only 18 introns. The largest gene in A. borealis, A. solidipes and A. gallica was cox1 that contained 9, 9 and 5 introns, respectively, contributing to its large size (15,955, 15,986 and 9624 bp, respectively). In A. sinapina, the largest gene was cob, which had 6 introns and was 9649 bp. The longest intron (2615 bp) was observed in the A. solidipes mitogenome (intron 2 of the nad1 gene), and the shortest intron was 189 bp long in the atp9 gene of the A. gallica mitogenome. Exons of the protein-coding genes and sequences of the rRNA genes covered 29% (29,159 bp) of mitogenome in A. gallica, 30% (31,139 bp) in A. sinapina, 26% (30,781 bp) in A. borealis and 24% (29,241 bp) in A. solidipes. The total length (and percentage) of intergenic sequences together with all introns and intergenic ORFs was 69,737 (71%), 72,424 (70%), 85,652 (74%) and 92,921 (76%) bp in A. gallica, A. sinapina, A. borealis and A. solidipes, respectively. These estimates were confirmed by the whole mitogenome comparative alignments generated by MAUVE, which showed variation in the intronic and intergenic regions (Fig. 5).

Structural alignments of Armillaria mitochondrial genomes based on the Mauve analysis. The color blocks indicate the homologous regions between the sequences shared by all mitogenomes. Gaps show unique sequences for each mitogenome. Blocks drawn under the horizontal line represent inverted sequence orientations
Mapping RNA-seq reads to mitogenomes
The annotation of conserved protein-coding genes and rRNA genes was validated by mapping RNA-seq reads to mitogenomes. After filtering, 2,371,666 and 1,844,578 high quality reads of A. borealis and A. sinapina, respectively, were retained for mapping to their mitogenomes. In total, 258,471 reads were mapped to the A. borealis mitogenome and 227,565 reads to the A. sinapina mitogenome. In A. borealis and A. sinapina 17 and 16 genes were covered by the RNA reads, respectively (Additional file 5: Table S5). Much less number of reads were mapped to the A. gallica and A. solidipes mitogenomes using RNA-seq data for these two species downloaded from NCBI Sequence Read Archive (SRA). Only 11 genes were covered by 566 reads in the A. gallica mitogenome and eight genes by 442 reads in the A. solidipes mitogenome. Low mapping coverage data for these two species can be likely explained by a very low quality of transcriptome reads for A. gallica and A. solidipes, long stretches of which contained anonymous nucleotides (N). When A. borealis RNA reads were used for mapping to the A. gallica and A. solidipes mitogenomes, 16 genes were covered in both species by 251,265 and 255,744 reads, respectively.
Phylogeny
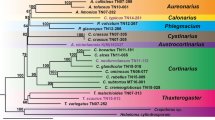
Phylogenetic analyses were performed using protein sequences consisting of totally 3645 amino acids of 14 concatenated protein-coding mitochondrial genes representing 25 fungal taxa. The maximum likelihood (ML) phylogenetic tree (Fig. 6) demonstrated that Agaricales formed a monophyletic group with a strong bootstrap support (100%). Within this clade, four families, Physalacriaceae, Marasmiaceae, Omphalotaceae and Pleurotaceae can be recognized as very strongly supported subclades also with 100% bootstrap support. Physalacriaceae that includes Armillaria species together with Flammulina velutipes was a sister lineage to Marasmiaceae. A. solidipes grouped with A. borealis, while A. sinapina and A. gallica seemed to be more distantly related. The protein changes are consistent with the established phylogenetic relationships of the four species [48, 49].

Maximum Likelihood phylogenetic tree based on protein sequences representing totally 3645 amino acids of 14 concatenated protein-coding mitochondrial genes from 25 fungal taxa. The numbers next to the cluster nodes represents bootstrap values based on 10,000 replicates
Discussion
A. borealis, A. solidipes, A. sinapina and A. gallica are closely related, but their mitogenomes vary substantially in size. Some fungal mitogenomes contain multiple repeat sequences that represent mobile elements capable of inflating genome size, as has been observed, for example, in the mitogenome of Ophiocordyceps sinensis [44] and Rhizoctonia solani [21]. However, sequence repeats were not found in all four Armillaria mitogenomes, reflecting, that accumulation of repeats is not the reason for size variation between Armillaria species.
Although gene duplication could not explain genome size variation, we found that the mitogenomes of Armillaria were rich in mobile genetic elements that could be involved in increasing genome size. In earlier publications (see [50] for review) it was suggested that fungal mitogenome size variation could result, at least in part, from variation in the number and size of introns and in the length of intergenic sequences. Our data on variation of mitogenome sizes are consistent with this suggestion and with published studies that attributed expansions of fungal mitogenomes to intron sequences [24, 26, 37, 51], as well as plasmid-related sequences [32, 52]. The extent to which the intron sizes and numbers can affect mitogenome size is evident from the cox1 gene in A. borealis and A. solidipes, which was the largest reservoir of 9 introns. This gene in these two species was more than double the size of this gene in A. sinapina due to additional introns. This observation was not limited to the cox1 gene, but was true also for all other mitochondrial genes harboring introns. Long stretches of intergenic sequences also seemed to affect the mitogenome sizes of these species. In other fungi, these regions also harbor ORFs that have been associated with mitogenome size variation [29, 31, 37].
Introns in the four Armillaria mitogenomes confirmed the diversity of mitochondrial introns within the same genes reported in other published studies [53, 54]. Intron content of the Armillaria mitochondrial genes was notably diverse within the cox1, cob, nad1, nad2 and nad5 genes. Fungal mitochondrial introns showed a wide range of diversity even among closely-related species from the same genus [55, 56]. Thus, the diversity of introns in the mitogenomes of Armillaria is a common fungal feature, which can also explain mitogenome size variations.
In fungal mitogenomes, intron acquisition can occur through vertical and horizontal transmissions, and insertions do not necessarily occur at homologous gene positions [38, 54]. A high level of sequence similarity was observed between the introns of the A. borealis and A. solidipes mitogenomes, most of which encoded similar HEGs and had similar insertion points. Introns that lacked sequence similarities may be examples of independent evolutionary histories implying multiple acquisitions [24, 27]. For the few introns identified in these mitogenomes that did not share sequence similarities, similar sequences could be identified from other fungal species. This suggests that these introns were probably acquired by horizontal transfer. Our study confirmed published data that some fungal mitochondrial introns can contain ORFs that encode HEGs of the LAGLIDADG or GIY-YIG families [57]. These ORFs did not have the same start codons that are common in other fungal mitochondrial genes. Similarly to Stone et al. [24] we also observed in A. sinapina, A. borealis and A. gallica that these ORFs were free standing in mitogenomic regions between typical oxidative phosphorylation genes.
We also found that Armillaria mitogenomes contained rearrangements. Based on the genome-wide alignment, we surmise that mitogenome rearrangements are related to the presence of non-conserved plasmid-related sequences and homing endonucleases ORFs, which are common within intergenic regions of fungi [28, 58]. Most of these sequences represented truncated sequences of DNA polymerase and RNA polymerase genes, while some ORFs had unknown functions in Armillaria. In addition to the introns, plasmids represent mobile genetic elements often found in the mitochondria of fungi and plants and contain two ORFs, one of which encodes a family B DNA polymerase, and the other of which encodes the RNA polymerase subunit [59]. These genetic element insertions have been shown to be associated with or promote genomic rearrangements through non-homologous recombination [60, 61]. Plasmid-related DNA polymerase genes were found in mobile mitochondrial plasmids that occured either as free linear or circular DNAs, and have been shown to also insert into mitogenomes [30, 62, 63]. In the comparison of four closely related Armillaria species the presence of plasmid-related sequence insertions in the rps3-atp9 region was observed. In A. sinapina and A. gallica they were sufficiently divergent from each other and from those in A. borealis and A. solidipes, which had more similarity between plasmid-related sequences in their mitogenomes, and consequently were most likely resulted from plasmid insertion events independent to those in A. sinapina and A. gallica. The sequences of the two DNA polymerase and RNA polymerase genes in A. borealis and A. solidipes had a high similarity (99% nucleotide identity), suggesting the common origin of these insertions. Other plasmid-related ORFs in the A. gallica and A. sinapina mitogenomes had low sequence similarity (< 45%) with each other and with A. borealis and A. solidipes indicating that these genes could have diverse origins. Therefore, acquisition of these genes in A. sinapina and A. gallica seems to have independent evolutionary origins which were then followed by the accumulation of mutations.
Mitogenomes of Armillaria species contained truncated copies of some genes, such as atp9. The high nucleotide identity (90–95%) between copies in all three genomes was accompanied by the high level of synteny around the duplicated gene regions. In A. borealis and A. gallica the copy was located nearby original atp9 gene, but in A. solidipes and A. sinapina it was on the minus strand within rnl gene. In A. borealis and A. gallica the second exon of atp9 was duplicated, while atp9 in A. sinapina and A. solidipes did not have an intron, but had a truncated copy that corresponded to the first exon of atp9 in A. borealis and A. sinapina. Duplications and copies of atp9 were found also in mitogenomes of other fungal species, which implies that gene duplications (often accompanied or followed by their truncation) is a frequent process in fungal mitogenomes [24, 41].
According to the ML phylogenetic tree A. solidipes was very closely related to A. borealis, apart from A. sinapina and A. gallica that were less related to each other, which is consistent with previous phylogenetic studies based on a few genetic markers such as ITS, tef1-α and β-tubulin [64] or on analysis of six gene regions such as 28S, EF1α, RPB2, TUB, gpd and actin-1 [48]. Moreover, it is important to point out that there was an agreement between the phylogenetic grouping and the mitogenome organization of these species considering the fact that A. borealis and A. solidipes had the same gene order while A. sinapina and A. gallica each had the different unique gene order.
What is the impact of the mitogenome changes observed here on phenotype and fitness? The simplest hypothesis is that the mitogenome changes are neutral with respect to fitness and that their random accumulation parallels the species phylogeny [48, 49]. There may be an experimental test of this hypothesis. If mitogenome variation exists within species, then mitogenome recombinants could be obtained in laboratory matings [65]. Mitogenomes variants with precisely the same nuclear genome could then be tested for differences in phenotype and/or fitness traits in the laboratory.
Conclusions
The mitogenomes of A. borealis, A. solidipes, A. sinapina and A. gallica had similar gene content. They contained 14 protein-coding conserved genes involved in oxidative phosphorylation and electron transport. The rnl, rns, rps3 and tRNA genes were also found in all four mitogenomes. The genes order was the same in A. borealis and A. solidipes, but different in A. sinapina and A. gallica, consistent with the widely accepted interpretation of species phylogeny. Comparative analyses showed high size variation of these mitogenomes, which appeared due to the different number and size of introns and intergenic regions. Several introns seemed to have been acquired independently through intron-encoded homing endonucleases ORFs mobility. The frequent lack of sequence identity between introns identified in this study but high sequence identity with the sequences of other fungi available in the NCBI GenBank suggests their possible acquisition via horizontal transfer between even distantly related fungal species. However, a further comparative evolutionary analysis is required for these genes. The studied mitogenomes provide useful resources for these and other comparative studies.
Methods
DNA isolation
Mitogenomes were assembled from DNA sequences obtained using the total genomic DNA isolated from fungal mycelium without prior mtDNA isolation or enrichment. A. borealis and A. sinapina mycelia were collected in Western Siberia from Abies sibirica trees. The mycelia were fixed and stored for 2 days at 4 °C in the RNA stabilization solution RNAlater (Thermo Fisher Scientific Company, Waltham, Massachusetts, USA). The RNAlater-fixed mycelium was then quickly ground in acid-washed and autoclaved mortar. DNA was isolated using a modified version of the hot-CTAB extraction at 65 °C [66] followed by chloroform double-wash. Total DNA was precipitated within one hour with isopropanol at 4 °C, centrifuged at 6500 g for 30 min at 4 °C, washed twice with 70% ethanol, and eluted with 50 μl RNase-free water. Integrity and amount of the isolated total DNA were examined by 1.5% (wt/vol) agarose gel electrophoresis and using the NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific Company, Waltham, Massachusetts, USA). DNA was quantified on the Qubit 2.0 Fluorimeter (Thermo Fisher Scientific Company, Waltham, Massachusetts, USA).
DNA sequencing and de novo assembly
The total DNA isolated from A. borealis and A. sinapina was sequenced in the Laboratory of Forest Genomics (Genome Research and Education Center, Siberian Federal University, Krasnoyarsk, Russia) using the Illumina MiSeq platform. The pair-end (PE) libraries with a mean insert size of 250 bp were subjected to 2 × 250 cycles of PE sequencing. Adapter sequences were trimmed and short reads were filtered using Trimmomatic v. 0.36 [67] with minimum quality of 19 and minimum length of 35 bp. Quality was assessed using FASTQC v. 0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The obtained sequence reads were assembled into contigs and scaffolds using the CLC Assembly Cell v. 5.0.0 (QIAGEN Bioinformatics, Hilden, Germany; https://www.qiagenbioinformatics.com/products/clc-assembly-cell). The whole-genome assemblies of A. borealis and A. sinapina consisted of 23,459 contigs with a total length of 72,723,723 bp (N50 = 13,708 bp) and 34,632 contigs with a total length of 94,366,584 bp (N50 = 7681 bp), respectively. To find among these contigs those that represent mitochondrial sequences, they were blasted against all mitochondrial basidiomycete sequences available in the NCBI GenBank, and contigs that matched these mitochondrial sequences were selected for further analysis.
The mitogenomes of A. solidipes and A. gallica were assembled as part of the whole genome sequencing project at the Joint Genome Institute. The complete sequences of their mitogenomes were available as two scaffolds deposited in the JGI Genome portal (http://genome.jgi.doe.gov/Armost1/Armost1.home.html and https://genome.jgi.doe.gov/Armga1/Armga1.home.html, respectively). These scaffolds were retrieved for annotation and comparative analysis in this study.
Mitochondrial contigs found in the total A. borealis and A. sinapina contig assemblies were verified by mapping these contigs to the A. solidipes mitogenome using CLC Genomics Workbench v. 9.0.1 (QIAGEN Bioinformatics, Hilden, Germany; https://www.qiagenbioinformatics.com/products/clc-genomics-workbench). Identified mitochondrial contigs were additionally further confirmed by BLASTn searches against non-redundant nucleotide sequences in the NCBI GenBank database. After identification and confirmation of mitochondrial contigs those of them that displayed overlaps at both ends were used to circularize the mitogenomes using Cyclic DNA Sequence Aligner [68]. Finally, two 116,433 and 103,563 bp single contigs representing the whole mitogenomes of A. borealis and A. sinapina were arranged and circularized into mitogenomes, respectively. For final verification, raw paired-end sequence reads were mapped to them using CLC Genomics Workbench v 9.0.1.
RNA isolation, sequencing and mapping
The total RNA was extracted from grown mycelia fixed in RNAlater (Thermo Fisher Scientific Company, Waltham, Massachusetts, USA) using Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA). The quality and concentration of the RNA were measured using Agilent 2100 Bioanalyzer and Agilent RNA 6000 Nano kit. High quality purified RNA was selected for cDNA library construction. Isolation of mRNA from total RNA was performed using Oligo (dT) magnetic beads. The mRNA treated with fragmentation buffer was used as template for cDNA synthesis. Double-stranded cDNA libraries were constructed using the TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA). End-repair, A-tailing, adapter ligation, and library amplification were done for the cDNA library construction followed by cluster generation and sequencing on the Illumina MiSeq platform in the Laboratory of Forest Genomics (Genome Research and Education Center, Siberian Federal University, Krasnoyarsk, Russia) using MiSeq Reagent Kit v2 (2 × 150). Raw sequence data were processed, and adapters were removed. High quality reads were mapped to A. borealis and A. sinapina mitogenomes using the RNA-Seq module of CLC Genomics Workbench v. 9.0.1 (QIAGEN Bioinformatics, Hilden, Germany; https://www.qiagenbioinformatics.com/products/clc-genomics-workbench).
RNA-seq data for A. solidipes and A. gallica were downloaded from the NCBI Sequence Read Archive (accession numbers SRR4063418 and SRX5202894, respectively) and used for mapping these two mitogenomes.
Gene annotation and bioinformatic analyses
Mitogenomes for three Armillaria species were checked for homology with other fungal mitogenomes existing in the NCBI GenBank database using the NCBI BLAST algorithm. The mitogenomes were annotated using the MFannot program (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl) with default settings. Multiple ORFs were analyzed by a BLASTx homology search against protein database in the NCBI GenBank database. Intron-exon boundaries were verified using RNAweasel (http://megasun.bch.umontreal.ca/RNAweasel). Large (rnl) and small (rns) subunits of the mitochondrial ribosome genes were predicted using both BLASTn and RNAweasel. The tRNA genes were discovered using ARAGORN [69] and tRNAscan-SE [70] tools. Duplicated mtDNA sequences were identified by local BLASTn searches of mtDNAs against themselves with the e-value cut-off of 10− 5. To identify sequence repeats in intergenic regions of the mitogenomes SciRoKo [71] and Tandem Repeat Finder [72] were used with defaults parameters. Whole mitogenome alignments to identify syntenic blocks in mitogenomes of A. borealis, A. solidipes, A. sinapina and A. gallica were performed using MAUVE 2.3.1 [73]. We also determined the frequency of codon usage in 14 mitochondrial genes using the web-based Sequence Manipulation Suite (http://www.bioinformatics.org/sms2/codon_usage.html) with the fungal mitochondrial genetic code 4.
Phylogeny
Phylogenetic analysis was performed using protein sequences representing totally 3645 amino acids of 14 concatenated protein-coding mitochondrial genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, cob, cox1, cox2, cox3, atp6, atp8, atp9) partitioned from mitogenomes of four Armillaria species described in this study and 21 fungal taxa downloaded from GenBank (their names and accession numbers are provided in Additional file 6: Table S6. The maximum likelihood (ML) method in the IQTree v. 1.5.6 software was used to generate a phylogenetic tree [74]. Sequence alignments were produced using the MAFFT algorithm implemented in SeaView v. 4.7 ([75]; http://doua.prabi.fr/software/seaview). Poorly aligned regions were trimmed using GBlocks [76]. The best models of evolution for amino acid sequences were selected by PartitionFinder2 v. 2.1.1 [77]. LG + G + F were determined as the best models for the atp6, atp8, cox3, nad2, nad3, nad4, nad5, and nad6 genes and LG + I + G + F models for the atp9, cob, cox1, cox2, nad1, and nad4L genes, where LG – one of general amino-acid replacement models introduced by [78], +I – invariable site model (one of the common rate heterogeneity across sites models allowing for a proportion of invariable sites) [79], +G - discrete Gamma model [80], and + F - empirical base frequencies. To determine the statistical support of the recovered nodes in the ML phylogenetic tree, the ultrafast bootstrap approximation was performed with 10,000 replicates.
Abbreviations
- atp6 and atp8 :
-
Genes for ATP synthase subunits 6 and 8
- bp :
-
Base pair
- cob :
-
Gene for cytochrome b
- cox1, cox2 and cox3 :
-
Genes for cytochrome c oxidase subunits 1, 2 and 3;
- kb :
-
Kilo base pair
- Mt :
-
Mitochondrial
- nad1, nad2, nad3, nad4, nad4L, nad5 and nad6 :
-
Mitochondrial genes for NADH dehydrogenase subunits 1–6 and 4 L
- ORF :
-
open reading frame
- rnS and rnL :
-
Genes for small and large subunits of ribosomal RNA
- rRNA :
-
Ribosomal RNA
- trnA or A :
-
tRNA gene for alanine
- tRNA :
-
Transfer RNA
- trnC or C :
-
tRNA gene for cysteine
- trnD or D :
-
tRNA gene for aspartic acid
- trnE or E :
-
tRNA gene for glutamic acid
- trnF or F :
-
tRNA gene for phenylalanine
- trnG or G :
-
tRNA gene for glycine
- trnH or H :
-
tRNA gene for histidine
- trnI or I :
-
tRNA gene for isoleucine
- trnK or K :
-
tRNA gene for lysine
- trnL 1 or L 1 :
-
tRNA gene for leucine (anticodon NAG)
- trnL 2 or L 2 :
-
tRNA gene for leucine (anticodon YAA)
- trnM or M :
-
tRNA gene for methionine
- trnN or N :
-
tRNA gene for asparagine
- trnP or P :
-
tRNA gene for proline
- trnQ or Q :
-
tRNA gene for glutamine
- trnR or R :
-
tRNA gene for arginine
- trnS 2 or S 2 :
-
tRNA gene for serine (anticodon NGA)
- trnS 1 or S 1 :
-
tRNA gene for serine (anticodon NCU)
- trnT or T :
-
tRNA gene for threonine
- trnV or V :
-
tRNA gene for valine
- trnW or W :
-
tRNA gene for tryptophan
- trnY or Y :
-
tRNA gene for tyrosine
- μl:
-
Microliter
References
Ross-Davis A, Stewart J, Hanna J, Kim M, Knaus B, Cronn R, et al. Transcriptome of an Armillaria root disease pathogen reveals candidate genes involved in host substrate utilization at the host-pathogen interface. For Pathol. 2013;43(6):468–77.
Hicke J, Allen C, Desai A, Dietze M, Hall R, Ted Hogg E, et al. Effects of biotic disturbances on forest carbon cycling in the United States and Canada. Glob Chang Biol. 2011;18(1):7–34.
Baumgartner K, Coetzee M, Hoffmeister D. Secrets of the subterranean pathosystem of Armillaria. Mol Plant Pathol. 2011;12(6):515–34.
Smith M, Bruhn J, Anderson J. The fungus Armillaria bulbosa is among the largest and oldest living organisms. Nature. 1992;356(6368):428–31.
Shaw CG, Roth LF. Persistence and distribution of a clone of Armillaria mellea in a ponderosa pine forest. Phytopathology. 1976;66:1210–3.
Anderson J, Ullrich R, Roth L, Filip G. Genetic identification of clones of Armillaria mellea in coniferous forests in Washington. Phytopathology. 1979;69:1109–11.
Ferguson B, Dreisbach T, Parks C, Filip G, Schmitt C. Coarse-scale population structure of pathogenic Armillaria species in a mixed-conifer forest in the Blue Mountains of Northeast Oregon. Can J For Res. 2003;33:612–23.
Smith M, Anderson J. Mitochondrial DNAs of the fungus Armillaria ostoyae: restriction map and length variation. Curr Genet. 1994;25(6):545–53.
Smith ML, Duchesne LC, Bruhn JN, Anderson JB. Mitochondrial genetics in a natural population of the plant pathogen Armillaria. Genetics. 1990;126(3):575–82.
Saville BJ, Kohli Y, Anderson JB. mtDNA recombination in a natural population. Proc Natl Acad Sci U S A. 1998;95(3):1331–5.
Basse C. Mitochondrial inheritance in fungi. Curr Opin Microbiol. 2010;13(6):712–9.
Adams K. Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol. 2003;29(3):380–95.
John P, Whatley F. Paracoccus denitrificans and the evolutionary origin of the mitochondrion. Nature. 1975;254(5500):495–8.
Herrmann J. Converting bacteria to organelles: evolution of mitochondrial protein sorting. Trends Microbiol. 2003;11(2):74–9.
Thrash J, Boyd A, Huggett M, Grote J, Carini P, Yoder R, et al. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Sci Rep. 2011;1:13.
Fitzpatrick D, Creevey C, McInerney J. Genome phylogenies indicate a meaningful α-proteobacterial phylogeny and support a grouping of the mitochondria with the Rickettsiales. Mol Biol Evol. 2005;23(1):74–85.
Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Pontén T. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396(6707):133.
Bullerwell CE, Lang BF. Fungal evolution: the case of the vanishing mitochondrion. Curr Opin Microbiol. 2005;8(4):362–9.
Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283(5407):1476–81.
Pramateftaki PV, Kouvelis VN, Lanaridis P, Typas MA. The mitochondrial genome of the wine yeast Hanseniaspora uvarum: a unique genome organization among yeast/fungal counterparts. FEMS Yeast Res. 2005;6(1):77–90.
Losada L, Pakala S, Fedorova N, Joardar V, Shabalina S, Hostetler J, et al. Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol Lett. 2014;352(2):165–73.
Burger G, Gray M, Franz Lang B. Mitochondrial genomes: anything goes. Trends Genet. 2003;19(12):709–16.
Salavirta H, Oksanen I, Kuuskeri J, Mäkelä M, Laine P, Paulin L, et al. Mitochondrial genome of Phlebia radiata is the second largest (156 kbp) among fungi and features signs of genome flexibility and recent recombination events. PLoS One. 2014;9(5):e97141.
Stone CL, Buitrago MLP, Boore JL, Frederick RD. Analysis of the complete mitochondrial genome sequences of the soybean rust pathogens Phakopsora pachyrhizi and P. meibomiae. Mycologia. 2010;102:887–97.
Zhang Y, Zhang S, Zhang G, Liu X, Wang C, Xu J. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet Biol. 2015;77:95–107.
Formey D, Molès M, Haouy A, Savelli B, Bouchez O, Bécard G, et al. Comparative analysis of mitochondrial genomes of Rhizophagus irregularis –syn. Glomus irregular – reveals a polymorphism induced by variability generating elements. New Phytol. 2012;196(4):1217–27.
Sethuraman J, Majer A, Friedrich NC, Edgell DR, Hausner G. Genes within genes: multiple LAGLIDADG homing endonucleases target the ribosomal protein S3 gene encoded within an rnl group I intron of Ophiostoma and related taxa. Mol Biol Evol. 2009;26(10):2299–315.
Monteiro-Vitorello CB, Hausner G, Searles DB, Gibb EA, Fulbright DW, Bertrand H. The Cryphonectria parasitica mitochondrial rns gene: plasmid-like elements, introns and homing endonucleases. Fungal Genet Biol. 2009;46(11):837–48.
Mardanov AV, Beletsky AV, Kadnikov VV, Ignatov AN, Ravin NV. The 203 kbp mitochondrial genome of the phytopathogenic fungus Sclerotinia borealis reveals multiple invasions of introns and genomic duplications. PLoS One. 2014;9(9):e107536.
Hausner G. Introns, mobile elements, and plasmids. In: Bullerwell C, editor. Organelle Genetics. Berlin, Heidelberg: Springer; 2011. p. 329–57.
Beaudet D, Nadimi M, Iffis B, Hijri M. Rapid mitochondrial genome evolution through invasion of mobile elements in two closely related species of arbuscular mycorrhizal fungi. PLoS One. 2013;8(4):e60768.
Férandon C, Moukha S, Callac P, Benedetto JP, Castroviejo M, Barroso G. The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS One. 2010;5(11):e14048.
Kanzi AM, Wingfield BD, Steenkamp ET, Naidoo S, van der Merwe NA. Intron derived size polymorphism in the mitochondrial genomes of closely related Chrysoporthe species. PLoS One. 2016;11(6):e0156104.
Joardar V, Abrams NF, Hostetler J, Paukstelis PJ, Pakala S, Pakala SB, et al. Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics. 2012;13(1):698.
Vaughn JC, Mason MT, Sper-Whitis GL, Kuhlman P, Palmer JD. Fungal origin by horizontal transfer of a plant mitochondrial group I intron in the chimeric coxI gene of Peperomia. J Mol Evol. 1995;41(5):563–72.
Gonzalez P, Barroso G, Labarère J. Molecular analysis of the split cox1 gene from the Basidiomycota Agrocybe aegerita: relationship of its introns with homologous Ascomycota introns and divergence levels from common ancestral copies. Gene. 1998;220(1):45–53.
Férandon C, Xu J, Barroso G. The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet Biol. 2013;55:85–91.
Goddard MR, Burt A. Recurrent invasion and extinction of a selfish gene. Proceedings of the National Academy of Sciences USA. 1999;96(24):13880–5.
Cusimano N, Zhang LB, Renner SS. Reevaluation of the cox1 group I intron in Araceae and angiosperms indicates a history dominated by loss rather than horizontal transfer. Mol Biol Evol. 2007;25(2):265–76.
Sanchez-Puerta MV, Cho Y, Mower JP, Alverson AJ, Palmer JD. Frequent, phylogenetically local horizontal transfer of the cox1 group I intron in flowering plant mitochondria. Mol Biol Evol. 2008;25(8):1762–77.
Duò A, Bruggmann R, Zoller S, Bernt M, Grünig CR. Mitochondrial genome evolution in species belonging to the Phialocephala fortinii sl-Acephala applanata species complex. BMC Genomics. 2012;13(1):166.
Torriani SF, Penselin D, Knogge W, Felder M, Taudien S, Platzer M, et al. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet Biol. 2014;62:34–42.
Liang X, Tian X, Liu W, Wei T, Wang W, Dong Q. Comparative analysis of the mitochondrial genomes of Colletotrichum gloeosporioides sensu lato: insights into the evolution of a fungal species complex interacting with diverse plants. BMC Genomics. 2017;18(1):171.
Li Y, Hu XD, Yang R-H, Hsiang T, Wang K, Liang D-Q, Liang F, Cao D-M, Zhou F, Wen G, et al. Complete mitochondrial genome of the medicinal fungus Ophiocordyceps sinensis. Sci Rep. 2015;5:13892.
Zhang S, Hao AJ, Zhao YX, Zhang XY, Zhang YJ. Comparative mitochondrial genomics toward exploring molecular markers in the medicinal fungus Cordyceps militaris. Sci Rep. 2017;7:40219.
Shen XY, Li T, Chen S, Fan L, Gao J, Hou CL. Characterization and phylogenetic analysis of the mitochondrial genome of Shiraia bambusicola reveals special features in the order of Pleosporales. PLoS One. 2015;10(3):e0116466.
Yoon H, You Y-H, Woo J-R, Park Y-J, Kong W-S, Lee B-M, et al. The mitochondrial genome of the white-rot fungus Flammulina velutipes. J Gen Appl Microbiol. 2012;58(4):331–7.
Koch RA, Wilson AW, Séné O, Henkel TW, Aime MC. Resolved phylogeny and biogeography of the root pathogen Armillaria and its gasteroid relative, Guyanagaster. BMC Evol Biol. 2017;17(1):33.
Coetzee M, Wingfield B, Wingfield M. Armillaria root-rot pathogens: species boundaries and global distribution. Pathogens. 2018;7(4):83.
Bullerwell CE, Gray MW. Evolution of the mitochondrial genome: protist connections to animals, fungi and plants. Curr Opin Microbiol. 2004;7(5):528–34.
Fourie G, Van der Merwe NA, Wingfield BD, Bogale M, Tudzynski B, Wingfield M, et al. Evidence for inter-specific recombination among the mitochondrial genomes of Fusarium species in the Gibberella fujikuroi complex. BMC Genomics. 2013;14(1):605.
Beaudet D, Terrat Y, Halary S, de la Providencia IE, Hijri M. Mitochondrial genome rearrangements in Glomus species triggered by homologous recombination between distinct mtDNA haplotypes. Genome Biol Evol. 2013;5(9):1628–43.
Lang BF, Laforest M-J, Burger G. Mitochondrial introns: a critical view. Trends Genet. 2007;23:119–25.
Haugen P, Simon DM, Bhattacharya D. The natural history of group I introns. Trends Genet. 2005;21:111–9.
Wu Y, Yang J, Yang F, Liu T, Leng W, Chu Y, et al. Recent dermatophyte divergence revealed by comparative and phylogenetic analysis of mitochondrial genomes. BMC Genomics. 2009;10(1):238.
Al-Reedy RM, Malireddy R, Dillman CB, Kennell JC. Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genet Biol. 2012;49(1):2–14.
Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure. 2011;19(1):7–15.
Franco MEE, López SMY, Medina R, Lucentini CG, Troncozo MI, Pastorino GN, et al. The mitochondrial genome of the plant-pathogenic fungus Stemphylium lycopersici uncovers a dynamic structure due to repetitive and mobile elements. PLoS One. 2017;12(10):e0185545.
Cahan P, Kennell JC. Identification and distribution of sequences having similarity to mitochondrial plasmids in mitochondrial genomes of filamentous fungi. Mol Gen Genomics. 2005;273(6):462–73.
Brugger K, Torarinsson E, Redder P, Chen L, Garrett RA. Shuffling of Sulfolobus genomes by autonomous and non-autonomous mobile elements. Biochem Soc Trans. 2004;32:179–83.
Griffiths AJ. Natural plasmids of filamentous fungi. Microbiol Mole Biol Rev. 1995;59(4):673–85.
Mouhamadou B, Barroso G, Labarere J. Molecular evolution of a mitochondrial polB gene, encoding a family B DNA polymerase, towards the elimination from Agrocybe mitochondrial genomes. Mol Gen Genomics. 2004;272(3):257–63.
Wang Y, Zeng F, Hon CC, Zhang Y, Leung FCC. The mitochondrial genome of the basidiomycete fungus Pleurotus ostreatus (oyster mushroom). FEMS Microbiol Lett. 2008;280(1):34–41.
Guo T, Wang HC, Xue WQ, Zhao J, Yang ZL. Phylogenetic analyses of Armillaria reveal at least 15 phylogenetic lineages in China, seven of which are associated with cultivated Gastrodia elata. PLoS One. 2016;11(5):e0154794.
Saville BJ, Yoell H, Anderson JB. Genetic exchange and recombination in populations of the root-infecting fungus Armillaria gallica. Mol Ecol. 1996;5:485–97.
Devey ME, Bell JC, Smith DN, Neale DB, Moran GF. A genetic linkage map for Pinus radiata based on RFLP, RAPD, and microsatellite markers. Theor Appl Genet. 1996;92(6):673–9.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Fernandes F, Pereira L, Freitas AT. CSA: an efficient algorithm to improve circular DNA multiple alignment. BMC Bioinformatics. 2009;10(1):230.
Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32(1):11–6.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64.
Kofler R, Schlotterer C, Lelley T. SciRoKo: a new tool for whole genome microsatellite search and investigation. Bioinformatics. 2007;23(13):1683–5.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80.
Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–403.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32(1):268–74.
Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27(2):221–4.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–52.
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2016;34(3):772–3.
Le SQ, Gascuel O. LG: an improved, general amino-acid replacement matrix. Mol Biol Evol. 2008;25(7):1307–20.
Gu X, Fu YX, Li WH. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol Biol Evol. 1995;12(4):546–57.
Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 1994;39(3):306–14.
Acknowledgments
Authors are thankful to the editor and two anonymous reviewers for their suggestions that helped us improve the manuscript.
Funding
This study was funded by the Research Grant No. 14.Y26.31.0004 from the Government of the Russian Federation. The funding body did not contribute in the design of the study, collection, analysis, interpretation of data, or writing the manuscript.
Availability of data and materials
All sequences described in this study are available in GenBank under the accession numbers MH282847 (A. sinapina), MH407470 (A. borealis), MH660713 (A. solidipes) and MH878687 (A. gallica).
Author information
Authors and Affiliations
Contributions
KVK, YAP, NVO, AIK & INP designed the study. KVK, NVO & YAP administered the project. NVO & EPS carried out most of the sequencing. AIK, YAP, VVB, VVS, DAK & EPS carried out bioinformatics analysis. VVS & DAK provided computer support. INP provided fungal material. AIK, EPS, JBA, INP & KVK drafted the manuscript. EPS, VVS, DAK & JBA contributed to analysis and interpretation of data and revised the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Tandem repeats detected in four Armillaria mitogenomes using Tandem Repeats Finder. (XLSX 14 kb)
Additional file 2:
Table S2. Microsatellite or simples equence repeat (SSR) loci in four Armillaria mitogenomes. (XLSX 11 kb)
Additional file 3:
Table S3. Comparison of codon usage of 14 oxidative phosphorylation genes and the codon-anticodon recognition pattern of tRNA genes identified in A. borealis, A. gallica, A. sinapina, and A. solidipes. The number of plus signs indicates the presence and numbers of the respective tRNA gene. (XLSX 17 kb)
Additional file 4:
Table S4. Classification of introns in mitogenomes of four Armillaria species. (XLSX 9 kb)
Additional file 5:
Table S5. Mapping Armillaria transcriptome reads on mitogenomes. (DOCX 14 kb)
Additional file 6:
Table S6. Names and accession numbers for 21 Basidiomycota species downloaded from GenBank and used for phylogenetic analysis. (XLSX 9 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kolesnikova, A.I., Putintseva, Y.A., Simonov, E.P. et al. Mobile genetic elements explain size variation in the mitochondrial genomes of four closely-related Armillaria species. BMC Genomics 20, 351 (2019). https://doi.org/10.1186/s12864-019-5732-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5732-z