
Abstract
Background
Avocado (Persea americana Mill.) is a basal angiosperm from the Lauraceae family. This species has a diploid genome with an approximated size of ~ 920 Mbp and produces a climacteric, fleshy and oily fruit. The flowering and fruit set are particularly prolonged processes, lasting between one to three months, generating important differences in physiological ages of the fruit within the same tree. So far there is no detailed genomic information regarding this species, being the cultivar ‘Hass’ especially important for avocado growers worldwide. With the aim to explore the fruit avocado transcriptome and to identify candidate biomarkers to monitore fruit development, we carried out an RNA-Seq approach during 4 stages of ‘Hass’ fruit development: 150 days after fruit set (DAFS), 240 DAFS, 300 DAFS (harvest) and 390 DAFS (late-harvest).
Results
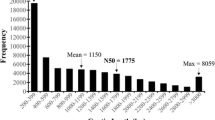
The ‘Hass’ de novo transcriptome contains 62,203 contigs (x̅=988 bp, N50 = 1050 bp). We found approximately an 85 and 99% of complete ultra-conserved genes in eukaryote and plantae database using BUSCO (Benchmarking Universal Single-Copy Orthologs) and CEGMA (Core Eukaryotic Gene Mapping Approach), respectively. Annotation was performed with BLASTx, resulting in a 58% of annotated contigs (90% of differentially expressed genes were annotated). Differentially expressed genes analysis (DEG; with False Discovery Rate ≤ 0.01) found 8672 genes considering all developmental stages. From this analysis, genes were clustered according to their expression pattern and 1209 genes show correlation with the four developmental stages.
Conclusions
Candidate genes are proposed as possible biomarkers for monitoring the development of the ‘Hass’ avocado fruit associated with lipid metabolism, ethylene signaling pathway, auxin signaling pathway, and components of the cell wall.
Similar content being viewed by others

Background
Avocado (Persea americana Mill.) belongs to the Lauraceae family, one of the oldest and largest flowering plant families that includes over 50 genera [1]. Due to its nutritional properties, avocado is one of the major fruit crops worldwide and its commercial production is based on selections within three races and hybrids between them: the Mexican race P. americana var. drymifolia, the Guatemalan race P. americana var. guatemalensis and the West Indian race P. americana var. americana [2]. Depending on the race and variety, the development of the avocado fruit may vary, but generally is characterized by a biphasic behavior: the first phase is fruit maturation and occur while the fruit is attached to the tree, this stage is pre-climacteric (on-tree storage) and is associated to low ethylene levels and respiration rate. Later, a second phase or climacteric peak begins after the detachment of the fruit and it is characterized by an increase in ethylene levels and respiration rate, resulting in ripening and softening [2,3,4,5].
Fruit development and ripening of avocado are unique, with a constant cell division and cell growth during development. While for other angiosperms, the cell division ceases followed by cell growth [6]. During development, one of the main characteristics of avocado is the large accumulation of oil fraction that is used as harvest and quality indexes, which can be up to 9–15% of oil in fresh weight, levels that are dependent on maturity stage and growing conditions in ‘Hass’ avocado [7,8,9,10]. Additionally, the mesocarp also accumulates high nutrient levels, which includes 6% of carbohydrates, 2% of proteins and vitamins E, C, B2, B12, B1, K and D [6, 11].
Despite the economic and nutritional importance of avocado fruit, genomic data and resources are scarce. In last years, the development of new sequencing platforms and bioinformatic tools has been critical to study non-model organisms that lack a reference genome at a low cost [12,13,14]. One of these platforms, RNA sequencing (RNA-seq), is useful to find functional elements of the genome and to understand different developmental stages and tissues. Thus, the transcriptomic analysis allows us to identify candidate genes that could be associated with phenotype and biological processes [15, 16].
Transcriptomic changes during ripening stages (pre-climacteric; climacteric and post-climacteric) have been previously studied in the Mexican avocado var. drymifolia [17]. Regarding to ‘Biological process’GO terms, the authors reported significant changes in transcripts related to major biological changes like: ‘monosaccharide metabolic process’, ‘carbohydrate catabolic process’, ‘cell wall macromolecule catabolic process’, ‘cellular lipid metabolic process’ and ‘secondary metabolic process’, indicating that the most important changes associated to ripening include color (loss of green), firmness (cell wall degrading activities), taste (increase in sugars) and flavor (increase of volatile compounds) [17]. Furthermore, Kilaru and colleagues [18] studied the mesocarp tissue of avocado fruit at five stages to generate temporal transcriptomic data in order to associate expression patterns of lipid biosynthesis genes with oil accumulation and fatty acid content. However, molecular markers or biomarkers associated with fruit development and/or harvest date for marker-assisted selection are not yet available.
To further elucidate some of the molecular factors involved in the regulation of fruit development of the economically important ‘Hass’ avocado variety, we conducted a de novo assembly of the transcriptome from the developing mesocarp at 150, 240, 300 (first harvest) and 390 (second harvest) days after fruit set (DAFS) using HiSeq 2500 (Illumina) platform. From this data, our aim was to explore the fruit avocado transcriptome during development in order to identify candidate genes that could be further characterized as potential biomarkers to monitor fruit development. Finally, to confirm RNA-seq data and de novo assembly, genes related to lipid metabolism were analyzed through quantitative Real Time PCR.
Results
Illumina sequencing and assembly of the avocado de novo transcriptome
In order to obtain insights of the dynamics of the ‘Hass’ avocado transcriptome during fruit development, RNA libraries were constructed and sequenced at four developmental stages (150, 240, 300 and 390 DAFS) using a HiSeq 2500 (Illumina) sequencer. Sequencing yielded a total of 261,409,226 pair-end reads (2 × 150 bp long) considering three biological replicates for each developmental stage (12 libraries). Raw data were submitted and they are available at the NCBI database (BioProject: PRJNA483022). The reads were trimmed to remove low quality reads, obtaining a total of 260,628,748 processed reads that were used as input to assemble the transcriptome with Trinity software (see Additional file 1). From the processed reads, a total of 62,203 contigs were assembled with an average size of 987 bp an N50 of 1050 bp and 62,167 predicted genes (Table 1). From CEGMA, a 99% of complete genes were found, meantime for BUSCO a total of 972 complete and single copy genes (84.1%) were found (Table 1).
In order to estimate the variability of gene global expression among fruit development stages and their biological replicates, the PCA explained up to 36.4% of the variability in PC1 and PC2 (Fig. 1a), while in PC2 and PC3 explained 22.90%. The PCA graph results displayed consistency among biological replicate samples and stages of fruit development. The first component explained 23.18% of variance and separates 150 DAFS from 390 DAFS. On the other hand, PC3 (9.72%) achieve the separation of 240 DAFS from 300 DAFS. In addition, Pearson correlation matrix shows high similarity among biological replicates, which is consistent with PCA (Fig. 1b). In both cases (PCA analysis and correlation matrix), homogeneity is shown between the biological replicates and, on the other hand, there are significant changes in the transcriptome during fruit development. To compare transcriptomic data among samples, normalization of the reads through transcripts per million (TPM) was performed. In relation with the overrall gene expression, we obtained an homogeneous level of gene expression (1,000,000 – 1,100,000 TPM) among the four developmental stages and biological replicates (Fig. 1c).

Overall gene expression during fruit developmental in P. americana by RNA-seq strategy. a PCA of the genes global expression from four development stages (150, 240, 300 and 390 days after fruit set or DAFS) and their three biological replicates (R1, R2 and R3). b Similarity heatmap based on the Pearson correlation among fruit developmental stage and biological replicates (c) Total TPM-TMM normalized expression for each stages and biological replicates
Functional annotation and global GO terms of the de novo transcriptome
To annotate the 62,203 contigs, we searched for reference sequences using BLASTx (https://blast.ncbi.nlm.nih.gov/Blast.cgi), against the NCBI database. Among the total assembled contigs 36,749 transcripts (57.5%) had a significant hit (Fig. 2). From these, a 32.4% were annotated using BLASTx obtaining as best hits Nelumbo nucifera, followed by Vitis vinifera (10.0%), Elaeis guineensis (6.8%) and Phoenix dactylifera (6.3%) (see Additional file 2). The 36,749 expressed genes were also used for Gene Ontology (GO) analysis and associated to different GO terms (Fig. 2). The GO terms were classified, and transcripts were assigned to biological process (GO:0008150), molecular function (GO:0003674) and cellular component (GO:0005575) categories. In the biological process category, the two largest groups were ‘Metabolic process’ and ‘Cellular lipid metabolic process’; meanwhile ‘Cell’ and ‘Intracellular’ are the largest groups for the cellular component category. For the molecular function category, ‘Binding’ and ‘Catalytic activity’ accounted for most of the annotated genes (Fig. 2).

Annotation and relevant GO terms of the de novo transcriptome assembled. 36,749 of 62,203 contigs were annotated with BLASTx. The top hit species were Nelumbo nucifera (32.4%), Vitis vinifera (10.0%), Elaeis guineensis (6.8%) and Phoenix dactylifera (6.3%). Top 10 relevant level 1 GO categories from unigenes are displayed for biological process (BP) on green, cellular component (CC) on blue and molecular function (MF) on purple
GO classification of DEGs and global differential expression analysis during development
In order to globally understand the development process, differentially expressed genes were used to perform GO analyzes, using the comparisons of 150 DAFS vs 240 DAFS, 240 DAFS vs 300 DAFS and 300 DAFS vs 390 DAFS. For the first comparison (150 DAFS vs 240 DAFS), we obtained a significant enrichment (from 35.71 to 45.83%) of transcripts involved in the processes of ‘lipid localization’ (GO:0010876), ‘tubulin complex’ (GO:0045298), ‘fatty-acid synthase activity’ (GO:0004312) and ‘acyl carrier activity’ (GO:0000036) (Additional file 3). When comparing 240 DAFS and 300 DAFS (first harvest), we can observe the overexpression (from 7.06 to 9.43%) of genes related to ‘response to hydrogen peroxide’ (GO:0042542) and ‘response to reactive oxygen species’ (GO:0000302) (Additional file 3). Finally, for the third comparison (300 DAFS vs 390 DAFS), genes related to ‘post-embryonic morphogenesis’ (GO:0009886) were overexpressed with a 14.29% of frequency (Additional file 3). Additionally, we identified transcription factors among the DEG for all the conditions during fruit development (Additional file 4).
On the other hand, in order to identify candidate genes involved in the development of avocado fruit, we compared samples from 150, 240, 300 and 390 DAFS considering three biological replicates using differential expression analysis. We obtained 8672 differentially expressed genes [FDR ≤0.01 and Fold Change (FC) ≥1] that revealed significant differences and were clustered according to their regulation pattern (Fig. 3a). Fifteen subclusters were obtained (K = 15), where subclusters 4, 7 and 11 contain a total of 1209 genes that show a correlation with the four developmental stages (Fig. 3b; see Additional file 5 and Additional file 6). Among the 1209 genes, 382 transcripts belonging to subcluster 4 increase their abundance between 150 DAFS and 390 DAFS. On the contrary, transcripts of subcluster 7 (724 genes) and 11 (103 genes), strongly decrease their abundance starting from 150 DAFS (Fig. 3b). Functional annotation of these transcripts was performed using the KEGG automatic annotation server, where pathways of ‘carotenoid biosynthesis’, ‘fatty acid biosynthesis’, ‘starch and sucrose metabolism’ and ‘phenylpropanoid biosynthesis’ were identified (Additional file 7). Furthermore, genes from each subcluster were selected as candidates for further analysis as possible biomarkers for fruit development and harvest date in avocado. In subcluster 4, selected genes that constantly increase their expression were related to lipid storage, carbohydrate metabolism, phenylpropanoid biosynthesis, intracellular signal transduction, metabolic process, ethylene signaling pathway, auxin signaling pathway and cytokinin metabolic process (Table 2). In subcluster 7, genes that constantly decrease their expression were associated to sugar transport, cell signaling, lipid catabolism, lipid metabolic process, pigment biosynthetic process and cell signaling (Table 2). For subcluster 11, genes that constantly decrease their expression were related to the fatty acid biosynthesis pathway and components of cell wall (Table 2). On the other hand, we validated one gene selected from each subcluster (4, 7 and 11) by qPCR as biological validation. Concomitant with the subclusters profile, we obtained the same pattern in the qPCR analysis (Fig. 3c).

Differentially Expressed Genes analsyis. Profiling expression of DEG in P. americana cv. ‘Hass’(a) Heatmap of DEGs (FC ≥ 1 and FDR ≤ 0.01) across all four developmental stages, resulting in 8672 differentially expressed genes (DEGs). Y-axis corresponds to the different genes used as input information, X-axis shows the different conditions with their respective biological replicates. The color key represents the median centered log2 TMM-normalized TPM values. b Subcluster Plots, three out of fifteen (K = 15) subclusters were used to find candidate genes because of their expression profiles. The X-axis shows the four developmental stages and the Y-axis is the median centered log2 expression. c Validation of gene expression by qPCR for selected genes from subclusters 4, 7 and 11: CBV_PAM10189 (subcluster 4), CBV_PAM17119 (subcluster 7) and CBV_PAM1355 (subcluster 11). The Y-axis displays relative expression levels to PamActin gene, while the X-axis indicates the development stage
Validation of DEGs by quantitative real-time PCR (qRT-PCR)
To validate the de novo assembly and transcriptomic data, genes related to lipid metabolism, fatty acid biosynthesis and storage were used to analyze through qRT-PCR (Fig. 4). Therefore, the expression analysis for Stearoyl-(ACP) 9-Desaturase 6, Beta Ketoacyl-(ACP) Synthase III, Glycoside Hydrolase Isoform 2, 3-Oxoacyl-(ACP) Synthase I, Lipid-Phosphate Phospatase 2 and Oleosin were consistent with the transcriptomic data (Fig. 4). The Pearson correlation (r > 0.85) showed that there is a positive correlation between the two methods, validating the transcriptome analysis (Fig. 4; see Additional file 8).

Correlation between qRT-PCR and in silico gene expression. The figure shows six transcripts analyzed this study: (a) Stearoyl-(ACP) 9-desaturase 6, (b) Beta-ketoacyl-ACP Synthase III, (c) Glycoside Hydrolase Isoform 2, (d) 3-Oxoacyl-(ACP) Synthase I, (e) Lipid Phosphate Phosphatase 2 and (f) Oleosin. The left Y axis displays the normalized relative expression levels to PamActin gene, while the right Y axis shows the FC values from the differential expression analysis, X axis indicates the development stage. R indicates the spearman correlation coefficient between in vitro and in silico expression values
Discussion
The de novo assembly and functional annotation of the avocado transcriptome
In this study we report the de novo assembly of the ‘Hass’ avocado transcriptome to identify candidate genes that correlate their RNA levels with different developmental stages (150, 240, 300 and 390 DAFS), allowing their future study as potential biomarkers for monitoring developmental stage and harvest date. In the last years, the development of new sequencing technologies and informatic tools, together with the available data of avocado form previous studies, has helped to improve assemblies and therefore, to better understand biological and molecular processes. In this work, from a short-read assembly we could obtain 62,203 contigs and 62,167 genes using Trinity (Table 1), while a previous study of the ‘Hass’ transcriptome reported a total of 134,329 contigs with a short-read assembly and another study of Mexican avocado obtained 83,650 contigs using a hybrid assembly (long reads + short reads) [17, 18]. Thus, in the present study we obtained an improved short-read assembly (62,203 contigs) with 85–99% of complete genes, providing with additional data that can contribute to the characterization of avocado development.
Transcriptomic analysis has been fundamental for the characterization and discovery of new molecular insights for biological processes like fruit development and ripening. In the plant model tomato, a spatiotemporal resolved transcriptome of tomato fruit revealed that the ripening program comprises gradients of gene expression from internal tissues to outward as maturation progressed [16, 19]. On the other hand, for non-model crops, like hami melon (Cucumis melo), sweet orange (Citrus sinensis), eggplant (Solanum melongena) and sweet cherry (Prunus avium), this platform has been critical to better understand the development process without a reference genome [20,21,22,23]. For P. americana, the normalized transcriptomic data was classified according to the transcript profile across the different developmental stages (Fig. 1). In this case, the PC could group the three biological replicates together and separate 150 DAFS from 390 DAFS and 240 DAFS from 300 DAFS (Fig. 1a). Together with this, the correlation analysis shows a decreasing similarity between 150 DAFS and 240, and 300 and 390 DAFS (Fig. 1b), reflecting the results obtained by PCA and indicating changes in gene expression during fruit development.
To annotate and to identify similarities between avocado transcripts and known plant proteins, we used BLASTx against the non-redundant NCBI database and obtained that 57.5% of total transcripts (36,749 out of 62,203 contigs) have similarity with annotated proteins (Fig. 2; Additional file 2). These genes were associated to GO terms, where the most represented groups from the biological process category corresponded to ‘metabolic’ and ‘cellular lipid’ processes, coinciding with a constant increase in oil content (from 2.8% at 150 DAFS to 11.5% at 390 DAFS) and fruit weight (Additional file 9). These results are related to the maturation process, which occur while the fruit is still in the tree, characterized by an active cell growth and cell division until the last stage of maturation [24]. Moreover, during this process of maturation and not later in ripening, the biosynthesis of fatty acids and triacylglycerols (TAGs) is being carried out [6, 18, 25].
Gene ontology (GO) classification of DEGs during development
In order to globally comprehend the development process, differentially expressed genes were used to perform GO analyzes, using the comparisons of 150 DAFS vs 240 DAFS, 240 DAFS vs 300 DAFS and 300 DAFS vs 390DAFS. For the first comparison (150 DAFS vs 240 DAFS), we obtained an enrichment of transcripts involved in the processes of ‘lipid localization’ (GO:0010876), ‘tubulin complex’ (GO:0045298), ‘fatty-acid synthase activity’ (GO:0004312) and ‘acyl carrier activity’ (GO:0000036), involved in the fatty-acid biosynthesis (Additional file 3). In these stages, the oil content increases from 2.8 to 6% (Additional file 9), which is stored as TAG and involves the synthesis of fatty-acids (FA) in the plastid. Initially acetyl-CoA is carboxilated to form malonyl-CoA, followed by its repeated condensation with a growing acyl-carrier protein to finally obtain de novo synthesized FA. Finally, these FA are incorportated into glycerol backbones to generate TAG in the endoplasmic reticulum [26]. As previously reported in avocado, the oil content and transcript levels of acyl-carrier proteins increase representing about 24% of the total fatty-acid gene expression during maturation of the mesocarp [18].
Later in development, when comparing 240 DAFS and 300 DAFS (first harvest), we observed the overexpression of genes related to ‘response to hydrogen peroxide’ (GO:0042542) and ‘response to reactive oxygen species’ (GO:0000302), suggesting that reactive oxygen species (ROS) generated earlier in cell metabolism are being regulated during maturation (Additional file 3). Furthermore, it is known that during fruit ripening in tomato and muskmelon, there has to be a balance between the production of ROS and its removal by the action of antioxidant mechanisms [27, 28]. From these mechanisms, ascorbic acid is a powerfull antioxidant involved in the removal of ROS during cell growth, cell division, cell wall expansion and organogenesis [28,29,30,31]. In the case of avocado, this process might be occurring before fruit ripening since its development is characterized by constant cell division and cell growth.
For the third comparison during late development (300 DAFS vs 390 DAFS), genes related to ‘post-embryonic morphogenesis’ (GO:0009886) and ‘response to virus’ (GO:0009615) were overexpressed (Additional file 3). These results are associated to a full development of the seed together with an increase in susceptibility to biotic stress during ripening, since the unripe mesocarp of avocado is more tolerant to pathogen attack [6]. For example, changes related to susceptibility to pathogen infection were previously observed in tomato, were the presence of the vacuolar protease SlVPE3 is necessary for resistance against the fungal pathogen Botrytis cinerea during ripening [32].
Differentially expressed genes during the fruit developmental process
Among the four developmental stages, we obtained 8672 differentially expressed genes, which were clustered in 15 groups according to their regulation pattern (Fig. 3a). In order to obtain candidate genes as potential biomarkers for the monitoring of fruit development and/or harvest date, transcripts from subcluster 4, 7 and 11 were selected due to their expression changes during the four stages of development and functional annotation was performed with BLASTx against the KEGG GENES database to identify associated pathways (Fig. 3b; Additional file 7). For these subclusters, transcripts that increase (subcluster 4) or decrease (subcluster 7 and 11) their levels were related to different processes like carotenoid biosynthesis, which is involved in the flesh color that in ‘Hass’ depends on the ripe state, changing from light green to dark green after harvest as observed in Additional file 9 [33]. The pathways of fatty acid biosynthesis and starch and sucrose metabolism were also observed, where sugars are important to promote cell growth and to control fruit metabolism during development [34]. Additionally, the phenylpropanoid biosynthesis pathway was also represented, where the secondary metabolites act as antioxidants and in tomato fruit are involved in pigmentation and aroma [35].
To obtain candidate genes for fruit development, we selected transcripts from subcluster 4 that constantly increase their expression and genes from subcluster 7 and 11 that constantly decrease their expression. For subcluster 4, we identified a homolog with similarity to oleosin (CBV_PAM10189), which usually is involved in the storage of oil bodies in seeds and pollens, however, it has also been reported its presence in avocado mesocarp, suggesting an additional role for these lipid droplet proteins during avocado fruit development [17, 36]. The homolog to beta-glucosidase 22 (CBV_PAM50790) was also present in this group and is involved in many processes of plant metabolism like the formation of intermediates for cell wall lignification and plant defense against biotic stress [37]. This is concomitant with the lignification of the endocarp for seed protection and the increasing susceptibility to pathogen infection during development [32, 38]. Additionally, the homolog of cinnamoyl-CoA reductase (CBV_PAM74617) is also associated to the lignin biosynthesis pathway. Lignin is synthesized from the phenylpropanoid pathway, where cinnamoyl-CoA reductase is a key enzyme for this process [39].
On the other side, the homolog of UDP-glucosyltransferase 85A4 (CBV_PAM23929) is involved in the transfer of glycosyl residues from nucleotide sugars to acceptor molecules and can regulate many properties of plant hormone and secondary metabolites like their bioactivity, solubility and their transport properties within the cell and throughout the plant, while zeatin O-glucosyltransferase-like (CBV_PAM38155) has a role in maintaining cytokinin homeostasis during development [40, 41]. Meantime, chitinases are pathogenesis-related proteins that are constitutively present in plants, however they can be developmentally upregulated by a growth regulator such as ethylene [42, 43]. In our case, the increase in the expression of the putative chitinase 1 (CBV_PAM2452), together with the ethylene-responsive transcription factor RAP2–4-like (CBV_PAM3819) could indicate the increase of ethylene biosynthesis, which regulates the ripening of climacteric fruit such as avocado [44]. In the case of auxins, they play a critical role during fruit development since they regulate fruit set, fruit growth and ripening, causing the increase of auxin-induced proteins like 5NG4 (CBV_PAM11370) and some ABC transporters (CBV_PAM11012) [45, 46]. Together with ethylene and auxin, transmembrane transport of gibberellin (GA) is required during plant growth and development. The coordinated work of phytohormones is crucial for fruit development, being GA an essential hormone that promotes fruit growth [47, 48].
It was previously described in Arabidopsis thaliana that SWEET proteins are involved in the transport of GA, indicating that the decrease of transcripts with homology to a bidirectional sugar transporter SWEET14 (CBV_PAM37547) in subcluster 7 could be associated to changes in GA transport in fruit maturity [49]. Additionally, a decrease of expression in the homolog of a Rapid Alkalinization Factor (RALF) (CBV_PAM51147) is also related to plant development and growth (Additional file 5 and Additional file 6: Table S2). RALFs are cysteine-rich peptides that allows for the communication of signaling networks across the organism, controlling processes like stem cell division, differentiation and cell expansion [50, 51]. Other transcripts that decrease their levels are ceramides, which are a family of lipid molecules that are present within the cell membrane in higher plants and their homeostasis is maintained by ceramidases [52]. In the case of avocado, as lipid metabolism is active during development, the balance of these ceramides through the sphingolipid pathway could be associated to changes in the transcript levels of neutral-ceramidases (CBV_PAM51593, CBV_PAM52108) during fruit maturation. In addition, transcripts with similarity to polyphenol oxidases (PPO) (CBV_PAM17119, CBV_PAM55044), are also downregulated during the different developmental stages since the presence of phenolic compounds are in part responsible for color, astringency, bitterness, flavor and nutritional quality in fruits [53, 54]. In relation to cell growth and division, which are constantly occurring in avocado, the coordination and regulation of these processes are important to achieve a specific shape or size. This is in part regulated by cell surface receptors known as receptor-like protein kinases, which are also decreasing their expression while the fruit is developing (CBV_PAM38418 and CBV_PAM25422) [55].
For subcluster 11, genes that constantly decrease their expression are mainly part of the cell wall structure and lipid metabolism. From these, four genes are homologs to proline-rich proteins (PRPs) (CBV_PAM10195, CBV_PAM10256, CBV_PAM1216, CBV_PAM51992), which are a principal constituent of the cell wall (Additional file 5 and Additional file 6: Table S2). The cell wall is a dynamic and complex structure that differentiates cell types during plant growth and development [56]. Our results coincide with previous reports in tomato and a higher plant like watermelon (Citrullus lanatus), where PRPs are highly expressed during an immature stage of fruit development that requires cell wall synthesis for cell division and cell growth, also suggesting that ethylene mediated pathways can downregulate these genes [57, 58]. From the lipid metabolism, the decreasing expression of fatty acid desaturases in avocado mesocarp was previously observed and this also agrees with the changing fatty acid composition during fruit development before harvest (pre-climacteric stage). Since no significant changes in fatty acid composition were detected during ripening, some authors hypothesize that the increase of ethylene might halt lipid biosynthesis [17, 59].
Gene expression of genes related to fatty acid and TAG biosynthesis
Avocado fruit accumulate TAGs synthesized mainly from oleic acid, palmitic and linoleic acid [60, 61]. There are key enzymes involved in the TAG biosynthesis pathway, like acetyl-CoA carboxylase, diacylglycerol acyltransferase, several fatty acid desaturases and thioestherases [62,63,64,65,66,67,68]. Thus, we decided to validate the transcriptomic data using transcripts related to the TAG biosynthesis pathway (Fig. 4). Transcripts that were strongly expressed either at the start of the developmental process (150 DAFS) or at the end of the process (390 DAFS) were searched. When comparing the TPM-normalized counts with the relative abundance of transcript with qRT-PCR, the overall spearman correlation coefficient resulted over r = 0.85, which validates the in-silico assembly. We found that transcripts related to overall fatty acids and TAGs biosynthesis were upregulated at 150 DAFS and 240 DAFS, whereas downregulated in the stages of 300 DAFS and 390 DAFS. This agrees with the idea that the fruit produces the majority of its lipid content during the first half of fruit development, then those processes are not ceased but reduced [59]. In addition, oleosin was differentially expressed in mesocarp tissue between 240 DAFS to 390 DAFS, which is consistent with another study where oleosin helps to stabilize lipid storage [18].
Conclusions
In this work, the transcript changes of the mesocarp of ‘Hass’ avocado was examined focusing in fruit development in the search for potential biomarkers for this process. Based on the annotation, it was possible to detect transcripts specifically related to lipid biosynthesis and fruit development. This transcriptomic data is consistent with previous transcriptome data generated in avocado and thus, contributes to increase the coverage data available for this economically important species. In addition, candidate genes related to lipid metabolism, ethylene signaling pathway, auxin signaling pathway, cell signaling, and components of the cell wall were identified due to their differential expression during the developing fruit, providing a foundation to further characterize these genes in the search for molecular applications. On the other hand, the main future implications of these results from an agronomical point of view, is the identification of candidate biomarkers for monitoring fruit development and harvest index, which should be validated in future work.
Methods
Plant material and phenotyping
‘Hass’ is the most important variety in the world representing 95% of the cultivated area, characterized by te accumulation of high levels of oil content and a quintessentially fairly thick skin that turns near black when fully ripe. From the commercial orchard located on Panquehue Valley (32°48′37.2″S; 70°50′16.7″W)″, three independent fruits (biological replicates) were sampled at 150 (2.8% oil), 240 (6% oil), 300 (7.5% oil) and 390 (11.5% oil) days after fruit set (DAFS) (Additional file 9). The oil content for each developmental stage was measured in the mesocarp by quantifying the dry matter content, considering a ~ 9% of oil content for harvest according to [69]. We determined ethylene by enclosing individual fruits in a 5000 mL jar and then sampling the air in the container. We measured ethylene by injecting 1 mL headspace sample into a gas chromatograph (GC-2014, Shimadzu, Japan) equipped with a flame ionization detector and an alumina column (Additional file 9) [70].
RNA-Seq and de novo transcriptome assembly
RNA was extracted using RNeasy mini kit (QIAGEN, Germantown, MD, USA) from a homogenous sample of the fruit mesocarp following the manufacturer’s instruction. To verify sample integrity, total RNA was evaluated on Fragment Analyzer™ Automated CE System (Advanced Analytical Technologies, Ames, IA, USA) and quantified using Qubit® RNA BR Assay kit (Thermo Fisher Scientific). One microgram of total RNA from each sample was used as input for the Illumina® TruSeq RNA HT Sample preparation commercial kit, according with the manufacturer’s instructions. Twelve libraries (4 development stages × 3 biological replicates) were sequenced on one lane of HiSeq 2500 platform (Illumina) with reads of 2x100bp (paired-end mode).
Total reads quality parameters were analyzed (pre and post trimming) with FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) software and the trimming process was performed using FLEXBAR software (https://github.com/seqan/flexbar) in order to filter low quality reads (phred score less than 25) and remove residual adapter sequences. Remaining reads after trimming were used as input for transcriptome assembly using Trinity software [71] with “--KMER_SIZE 20 --min_contig_length 400” [72]. After assembly, we used the CD-HIT software (http://weizhongli-lab.org/cd-hit/) to remove duplicated contigs and CORSET software (https://github.com/Oshlack/Corset/) to filter out contigs with less than 36 reads.
Quality check and annotation of the novo transcriptome
Quality assessment was performed with two approaches: (i) a biological approach that used two ultra-conserved protein gene finder software: Core Eukaryotic Gene Mapping Approach (CEGMA) and Benchmarking Universal Single-Copy Orthologs (BUSCO) and (ii) an informatic approach, DETONATE and TransRate to assess the architecture and quality of the contigs. To perform the in-silico validation of de novo transcriptome, we used, which search ultra-conserved genes by sequence similarity on trinity genes. Annotation was performed with BLAST in BLASTx mode against the NCBI “non-redundant” protein database, reporting hits with minimum e-value of 10− 3 [72,73,74,75].
Principal component analysis and differential expression analysis
A principal component analysis (PCA) and heatmap were conducted to determine grouping and similarity of each sample (biological replicates) using Trinity analysis utilities. Raw counts of each contig were obtained with RSEM using Bowtie2 aligner to align the input reads to the de novo assembled transcriptome [76, 77]. From the raw counts matrix was calculated a TPM-TMM normalized matrix used for the study alongside the raw counts matrix. Differential expression analysis was performed by using R’s package edgeR from Bioconductor. Differentially expressed genes (DEGs) were used for further comparisons and downstream analyses (False Discovery Rate; FDR < 0.01).
Gene ontology (GO), subclustering and KEGG analysis from DEGs
DEGs were used for Singular enrichment analysis (SEA) with AgriGO v2.0 web software (http://bioinfo.cau.edu.cn/agriGO/analysis.php) and Arabidopsis thaliana TAIR10 as background specie [78]. DEGs-SEA data was exported to REVIGO web software (http://revigo.irb.hr/) to reduce the redundancy of GO terms (FDR < 0.05) [79].
From DEGs with at least four fold-change (log2(4)) and with an FDR less than 0.01, we normalized expression values and grouped them in similar expression patterns. Thus, we generated a heatmap using the ggplot2 package, which shows the relationships between genes and samples. Then, to generate the subclusters we extracted the hierarchical grouping obtained earlier considering K equal to 15 using the MeV software. From the clustering data, FASTA nucleotide sequences were obtained from the transcripts present in subclusters 4, 7 and 11 and loaded into KAAS (KEGG Automatic Annotation Server; https://www.genome.jp/kegg/kaas/) to provide functional annotation of genes that constantly increase/decrease their transcript levels.
RNA-seq validation through qRT-PCR analysis
In order to perform a technical validation of DEG analysis, we designed primers using the software Primer 3 Plus (http://www.bioin formatics.nl/cgi-bin/primer3plus/primer3plus.cgi) based on the sequences generated by the de novo assembly. First, from the same input used to construct the libraries, total RNA was treated with DNase I (Fermentas, Thermo Fisher Scientific, Waltham, MA, USA) according to the standard protocol. The first strand cDNA was obtained by reverse transcription using the system MMLV-RT reverse transcriptase (Promega, Madison, WI, USA) and oligo dT primers. The cDNA concentration was obtained by measuring absorbance at 260 nm. Each cDNA sample was diluted to 50 ng*μL− 1 before being used in qRT-PCR assays. The qRT-PCR assay was performed using a LightCycler® 96 Real-Time PCR kit (Roche Diagnostics, Mannheim, Germany) with LC-FastStart DNA Master SYBR Green I to measure the DNA product derived from RNA, as previously described by García-Rojas and colleages [80]. Additionally, to validate the gene expression profiles in the selected subclusters, qRT-PCR of one gene belonging to each subcluster (4, 7 and 11) was performed using total RNA from five independent fruits at 150, 240, 300 and 390 DAFS. All qRT-PCR were performed using five biological replicates and the gene expression values were normalized relative to the PamActin gene (GenBank JN786942).
Abbreviations
- BLAST:
-
Basic Local Alignment Search Tool
- BUSCO:
-
Benchmarking Universal Single-Copy Orthologs
- CEGMA:
-
Core Eukaryotic Genes Mapping Approach
- DAFS:
-
Days After Fruit Set
- DEGs:
-
Differentially Expressed Genes
- DETONATE:
-
DE novo TranscriptOme rNa-seq Assembly
- FC:
-
Fold Change
- FDR:
-
False Discovery Rate
- GA:
-
Gibberellin
- GO:
-
Gene Ontology
- MAS:
-
Marker-Assisted Selection
- MMLV-RT:
-
Moloney Murine Leukemia Virus Reverse Transcriptase
- NGS:
-
Next-Generation Sequencing
- PCA:
-
Principal Component Analysis
- PPO:
-
Polyphenol Oxidases
- PRPs:
-
Proline-Rich Proteins
- RALF:
-
Rapid Alkalinization Factor
- REVIGO:
-
Reduce + Visualize Gene Ontology
- ROS:
-
Reactive Oxygen Species
- SEA:
-
Singular Enrichment Analysis
- TAGs:
-
Triacylglycerols
- TAIR:
-
The Arabidopsis Information Resource
- TMM:
-
Trimmed Mean of M-values
- TPM:
-
Transcripts Per Million
References
Rohwer JG. Lauraceae. In: Kubitzki K, Rohwer JG, Bittrich V, editors. Flowering Plants ·Dicotyledons. Berlin: Springer Berlin Heidelberg; 1993. p. 366–391. doi.org/https://doi.org/10.1007/978-3-662-02899-5_46.
Chanderbali AS, Albert VA, Ashworth VETM, Clegg MT, Litz RE, Soltis DE, et al. Persea americana (avocado): bringing ancient flowers to fruit in the genomics era. BioEssays. 2008;30:386–96. https://doi.org/10.1002/bies.20721.
Sitrit Y, Riov J, Blumenfeld A. Regulation of ethylene biosynthesis in avocado fruit during ripening. Plant Physiol. 1986;81:130–5.
Starret AA, Laties GG. Ethylene and wound-induced gene expression in the preclimateric phase of ripening avocado fruit and mesocarp disc. Plant Physiol. 1993;103:227–34.
García-Rojas M, Morgan A, Gudenschwager O, Zamudio S, Campos-Vargas R, González-Agüero M. Biosynthesis of fatty acids-derived volatiles in ‘Hass’ avocado is modulated by ethylene and storage conditions during ripening. Sci Hortic. 2016;202:91–8.
Lewis CE. The maturity of avocados-a general review. J Sci Fd Agric. 1978;29:857–66. https://doi.org/10.1002/jsfa.2740291007.
Davenport JB, Ellis SC. Chemical changes during growth and storage of the avocado fruit. Austral J Biol Sci. 1959;12:445–54.
Biale JB, Young RE. The avocado pear. In: Hulme AC, editor. The biochemistry of fruits and their products. London: Academic Press; 1971. p. 2–63.
Whiley AW, Schaffer B. Avocado. In: Schaffer B, Andersen PC, editors. Handbook of environmental physiology of fruit crops. Boca Raton: CRC Press; 1994. p. 1–35.
Ferreyra R, Sellés G, Saavedra J, Ortiz J, Zúñiga C, Troncoso C, et al. Identification of pre-harvest factors that affect fatty acid profiles of avocado fruit (Persea americana mill) cv. ‘Hass’ at harvest. S Africa J Bot. 2016;104:15–20. https://doi.org/10.1016/j.sajb.2015.10.006.
Knight RJ. History, distribution and uses. In: Whiley AW, Schaffer B, Wolstenholme BN, editors. The avocado: botany, production and uses. Wallingford: CAB International; 2002. p. 1–14.
Schliesky S, Gowik U, Weber AP, Bräutigam A. RNA-Seq assembly-are we there yet? Front Plant Sci. 2012;3:220–30. https://doi.org/10.3389/fpls.2012.00220.
Johnson MTJ, Carpenter EJ, Tian Z, Bruskiewich R, Burrls JN, Carrigan CT, et al. Evaluating methods for isolating total RNA and predicting the success of sequencing phylogenetically diverse plant transcriptomes. PLoS One. 2012;7:e50226. https://doi.org/10.1371/journail.pone.0050226.
Xiao M, Zhang Y, Chen X, Lee E, Barber CJS, Chakrabarty R, et al. Transcriptome analysis based on next-generation sequencing of non-model plants producing specialized metabolites of biotechnological interest. J Biotechnol. 2013;166:122–34. https://doi.org/10.1016/j.jbiotec.2013.04.004.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. https://doi.org/10.1038/nrg2484.
Shinozaki Y, Nicolas P, Fernandez-Pozo N, Ma Q, Evanich DJ, Shi Y, et al. High-resolution spatiotemporal transcriptome mapping of tomato fruit development and ripening. Nat Commun. 2018;9:364. https://doi.org/10.1038/s41467-017-02782-9.
Ibarra-Laclette E, Méndez-Bravo A, Pérez-Torres CA, Albert VA, Mockaitis K, Kilaru A, et al. Deep sequencing of the Mexican avocado transcriptome, an ancient angiosperm with a high content of fatty acid. BMC Genomics. 2015;16:599. https://doi.org/10.1186/s12864-015-1775-y.
Kilaru A, Cao X, Dabbs PB, Sung H, Rahman MM, Thrower N, et al. Oil biosynthesis in a basal angiosperm: transcriptome analysis of Persea Americana mesocarp. BMC Plant Biol. 2015;15:203. https://doi.org/10.1186/s12870-015-0586-2.
Ye J, Hu T, Yang C, Li H, Yang M, Ijaz R, et al. Transcriptome profiling of tomato fruit development reveals transcription factors associated with ascorbic acid, carotenoid and flavonoid biosynthesis. PLoS One. 2015;10:e0130885. https://doi.org/10.1371/journal.pone.0130885.
Yu K, Xu Q, Da X, Guo F, Ding Y, Deng X. Transcriptome changes during fruit development and ripening of sweet orange (Citrus sinensis). BMC Genomics. 2012;13:10. https://doi.org/10.1186/1471-2164-13-10.
Alkio M, Jonas U, Declercq M, Van Nocker S, Knoche M. Transcriptional dynamics of the developing sweet cherry (Prunus avium L.) fruit: sequencing, annotation and expression profiling of exocarp-associated genes. Hortic Res. 2014;1:11. https://doi.org/10.1038/hortres.2014.11.
Zhang H, Wang H, Yi H, Zhai W, Wang G, Fu Q. Transcriptome profiling of Cucumis melo fruit development and ripening. Hortic Res. 2016;3:16014. https://doi.org/10.1038/hortres.2016.14.
Chen X, Zhang M, Tan J, Huang S, Wang C, Zhang H, et al. Comparative transcriptome analysis provides insights into molecular mechanisms for parthenocarpic fruit development in eggplant (Solanum melongena L.). PLoS One. 2017;12:e0179491. https://doi.org/10.1371/journal.pone.0179491.
Cowan AK, Moore-Gordon CS, Bertling I, Wolstenholme BN. Metabolic control of avocado fruit growth – isoprenoid growth regulators and the reaction catalyzed by 3-Hydroxy-3-Methylglutaryl coenzyme a reductase. Plant Physiol. 1997;114:511–8.
Luza JG, Lizana LA, Masson L. Comparative lipids evolution during cold storage of three avocado cultivars. Acta Hortic. 1990;(269):153–60. https://doi.org/10.17660/ActaHortic.1990.269.20.
Lung SC, Weselake RJ. Diacylglycerol acyltransferase: a key mediator of plant triacylglycerol synthesis. Lipids. 2006;41:1073–88. https://doi.org/10.1007/s11745-006-5057-y.
Lacan D, Baccou JC. High levels of antioxidant enzymes correlate with delayed senescence in nonnetted muskmelon fruits. Planta. 1998;204:377–82. https://doi.org/10.2307/23385214.
Jimenez A, Creissen G, Kular B, Firmin J, Robinson S, Verhoeyen M, et al. Changes in oxidative processes and components of the antioxidant system during tomato ripening. Planta. 2002;214:751–8.
Navas P, Gomez-Diaz C. Ascorbate free radical and it role in growth control. Protoplasma. 1995;184:8–13.
Takahama U, Oniki T. Effects of ascorbate on the oxidation of derivatives of hydroxycinnamic acid and the mechanism of oxidation of sinapic acid by cell wall-bound peroxidases. Plant Cell Physiol. 1994;35:593–600.
Joy RW, Patel KR, Thorpe TA. AsA enchancement of organogenesis in tobacco callus. Plant Cell Tissue Organ Culture. 1988;13:219–28.
Wang W, Cai J, Wang P, Tian S, Qin G. Post-transcriptional regulation of fruit ripening and disease resistance in tomato by the vacuolar protease SlVPE3. Genome Biol. 2017;18:–47. https://doi.org/10.1186/s13059-017-1178-2.
Lu QY, Zhang Y, Wang Y, Wang D, Lee RP, Gao K, et al. California ‘Hass’ avocado: profiling of carotenoids, tocopherol, fatty acid, and fat content during maturation and from different growing areas. J Agric Food Chem. 2009;57:10408–13. https://doi.org/10.1021/jf901839h.
Kanayama Y. Sugar metabolism and fruit development in the tomato. Hort J. 2017;86:417–25. https://doi.org/10.2503/hortj.OKD-IR01.
Rigano MM, Raiola A, Docimo T, Ruggieri V, Calafiore R, Vitaglione P, et al. Metabolic and molecular changes of the phenylpropanoid pathway in tomato (Solanum lycopersicum) lines carrying different Solanum pennellii wild chromosomal regions. Front Plant Sci. 2016;7:1484. https://doi.org/10.3389/fpls.2016.01484.
Huang AHC. Oleosins and oil bodies in seeds and other organs. Plant Physiol. 1996;110:1055–61.
Escamilla-trevino LL, Chen W, Card ML, Shih MC, Cheng CL, Poulton JE. Arabidopsis thaliana beta-glucosidases BGLU45 and BGLU46 hydrolyse monoligol glucosides. Phytochemistry. 2006;67:1651–60.
Dardick CD, Callahan AM, Chiozzotto R, Schaffer RJ, Piagnani MC, Scorza R. Stone formation in peach fruit exhibits spatial coordination of the lignin and flavonoid pathways and similarity to Arabidopsis dehiscence. BMC Biol. 2010;8:13. https://doi.org/10.1186/1741-7007-8-13.
Kawasaki T, Koita H, Nakatsubo T, Hasegawa K, Wakabayashi K, Takahashi H, et al. Cinnamoyl-CoA reductase, a key enzyme in lignin biosynthesis, is an effector of small GTPase Rac in defense signaling in rice. Proc Natl Acad Sci U S A. 2006;103:230–5.
Ross J, Li Y, Lim E, Bowles DJ. Higher plant glycosyltransferases. Genome Biol. 2001;2:REVIEWS3004.
Martin RC, Machteld CM, Habben JE, Mok DWS. A maize cytokinin gene encoding an O-glucosyltransferase specific to cis-zeatin. Proc Natl Acad Sci U S A. 2001;98:5922–6.
Roby D, Broglie K, Gaynor J, Broglie R. Regulation of a chitinase gene promoter by ethylene and elicitors in bean protoplast. Plant Physiol. 1991;97:433–9.
Hamid R, Khan MA, Ahmad M, Ahmad MM, Abdin MZ, Musarrat J, et al. Chitinases: an update. J Pharm Bioallied Sci. 2013;5:21–9. https://doi.org/10.4103/0975-7406.106559.
Van de Poel B, Bulens I, Hertog MLATM, Nicolai BM, Geeraerd AH. A transcriptomics-based kinetic model for ethylene biosynthesis in tomato (Solanum lycopersicum) fruit: development, validation and exploration of novel regulatory mechanism. New Phytol. 2014;202:952–63.
Kang J, Park J, Choi H, Burla B, Kretzschmar T, Lee Y, et al. Plant ABC transporters. Arabidopsis Book. 2011;9:e0153. https://doi.org/10.1199/tab.0153.
Pattison RJ, Csukasi F, Catalá C. Mechanisms regulating auxin action during fruit development. Physiol Plant. 2014;151:62–72. https://doi.org/10.1111/ppl.12142.
El-Sharkawy I, Sherif S, El Kayal W, Mahboob A, Abubaker K, Ravindran P, et al. Characterization of gibberellin-signalling elements during plum fruit ontogeny defines the essentiality of gibberellin in fruit development. Plant Mol Biol. 2014;84:399–413. https://doi.org/10.1007/s11103-013-0139-8.
El-Sharkawy I, Sherif S, Mahboob A, Subramanian J. Plum fruit development occurs via gibberellin-sensitive and-insensitive DELLA repressors. PLoS One. 2017;12:e0169440. https://doi.org/10.1371/journal.pone.0169440.
Kanno Y, Oikawa T, Chiba Y, Ishimaru Y, Shimizu T, Sano N, et al. AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat Com. 2016;7:13245. https://doi.org/10.1038/ncomms13245.
Czyzewicz N, Yue K, Beeckman T, De Smet I. Message in a bottle: small signaling peptide outputs during growth and development. J Exp Bot. 2013;64:5281–96. https://doi.org/10.1093/jxb/ert283.
Campbell L, Turner SR. A comprehensive analysis of RALF proteins in green plants suggests there are two distinct functional groups. Front Plant Sci. 2017;8:37. https://doi.org/10.3389/fpls.2017.00037.
Michaelson LV, Napier JA, Molino D, Faure JD. Plant sphingolipids: their importance in cellular organization and adaptation. Biochim Biophys Acta. 1861;2016:1329–35. https://doi.org/10.1016/j.bbalip.2016.04.003.
Macheix JJ, Fleuriet A, Billiot J. Changes and metabolism of phenolic compounds in fruits. In: Macheix JJ, Fleuriet A, Boca Raton BJ, editors. Fruit Phenolics. Boca Raton: CRC Press; 1990. p. 149–221.
Holderbaum DF, Kon T, Kudo T, Guerra MP. Enzymatic browning, polyphenol oxidase activity, and polyphenols in four apple cultivars: dynamics during fruit development. Hortscience. 2010;8:1150–4.
Lease KA, Lau NY, Schister RA, Torii KU, Walker JC. Receptor serine/threonine protein kinases in signaling: analysis of the erecta receptor-like kinase of Arabidopsis thaliana. New Phytol. 2001;151:133–43. https://doi.org/10.1046/j.1469-8137.2001.00150.x.
Fowler TJ, Bernhardt C, Tierney M. Characterization and expression of four proline-rich cell wall protein genes in Arabidopsis encoding two distinct subsets of multiple domain proteins. Plant Physiol. 1999;121:1081–91.
Santino CG, Stanford GL, Connor TW. Developmental and transgenic analysis of two tomato fruit enhanced genes. Plant Mol Biol. 1997;33:405–16.
Guo S, Liu J, Zheng Y, Huang M, Zhang H, Gong G, et al. Characterization of transcriptome dynamics during watermelon fruit development: sequencing, assembly, annotation and gene expression profiles. BMC Genomics. 2011;12:454. https://doi.org/10.1186/1471-2164-12-454.
Ozdemir F, Topuz A. Changes in dry matter, oil content and fatty acids composition of avocado during harvesting time and post-harvesting ripenning period. Food Chem. 2004;86:79–83. https://doi.org/10.1016/j.foodchem.2003.08.012.
Vrinten P, Hu Z, Munchinsky MA, Rowland G, Qiu X. Two FAD3 desaturase genes control the level of linolenic acid in flax seed. Plant Physiol. 2005;139:79–87. https://doi.org/10.1111/nph.12685.
Shanklin J, Somerville C. Stearoyl-acyl-carrier-protein desaturase from higher plants is structurally unrelated to the animal and fungal homologs. Proc Natl Acad Sci U S A. 1991;88:2510–4.
Cran DG, Possingham JV. The fine structure of avocado plastids. Ann Bot. 1973;37:993–7. https://doi.org/10.2307/42752180.
Guerra DJ, Ohlrogge JB, Frentzen M. Activity of acyl carrier protein isoforms in reactions of plant fatty acid metabolism. Plant Physiol. 1986;82:448–53. https://doi.org/10.1104/pp.82.2.448.
Okuley J, Lightner J, Feldmann K, Yadav N, Lark E, Browse J. Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid synthesis. Plant Cell. 1994;6:147–58.
Chapman KD, Ohlrogge JB. Compartmentation of triacylglycerol accumulation in plants. J Biol Chem. 2012;287:2288–94. https://doi.org/10.1074/jbc.R111.290072.
Horn PJ, James CN, Gidda SK, Kilaru A, Dyer JM, Mullen RT, et al. Identification of a new class of lipid droplet-associated proteins in plants. Plant Physiol. 2013;162:1926–36. https://doi.org/10.1104/pp.113.222455.
Gidda SK, Watt S, Collins-Silva J, Kilaru A, Arondel V, Yurchenko O, et al. Lipid droplet-associated proteins (LDAPs) are involved in the compartmentalization of lipophilic compounds in plant cells. Plant Signal Behav. 2013;8:e27141. https://doi.org/10.4161/psb.27141.
Huang MD, Huang AHC. Subcellular lipid droplets in vanilla leaf epidermis and avocado mesocarp are coated with oleosins of distinct phylogenic lineages. Plant Physiol. 2016;171:1867–78. https://doi.org/10.1104/pp.16.00322.
Lee SK, Young RE, Schiffman PM, Coggins CW. Maturity studies of avocado fruit based on picking dates and dry weigth. J Amer Soc Hort Sci. 1983;108:390–4.
Sanhueza D, Vizoso P, Balic I, Campos-Vargas R, Meneses C. Transcriptomic análisis of fruit stored under cold conditions using controlled atmosphere in Prunus persica cv. “Red pearl”. Front Plant Sci. 2015;6:788. https://doi.org/10.3389/fpls.2015.00788.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512. https://doi.org/10.1038/nprot.2013.084.
Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7.
Li B, Fillmore N, Bai Y, Collins M, Thomson JA, Stewart R, et al. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 2014;15:553. https://doi.org/10.1186/s13059-014-0553-5.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2. https://doi.org/10.1093/bioinformatics/btv351.
Smith-Unna R, Boursnell C, Patro R, Hibberd JM, Kelly S. TransRate: reference free quality assessment of de-novo transcriptome assemblies. Genome Res. 2016;26:1134–44. https://doi.org/10.1101/gr.196469.115.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. https://doi.org/10.1186/1471-2105-12-323.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–9. https://doi.org/10.1038/nmeth.1923.
Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, et al. AgriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;45:W122–9. https://doi.org/10.1093/nar/gkx382.
Supek F, Bošnjak M, Škunca N, Šmuc T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. https://doi.org/10.1371/journal.pone.0021800.
García-Rojas M, Gudenschwager O, Defilippi BG, González-Agüero M. Identification of genes possibly related to loss of quality in late-season ‘Hass’ avocados in Chile. Postharvest Biol Technol. 2012;73:1–7.
Acknowledgements
The authors would like to thank Centro de Biotecnología Vegetal at Universidad Andrés Bello and Instituto Nacional de Investigación Agropecuaria INIA-La Platina. Also, we would like to thank the reviewers for valuable comments.
Funding
This work was supported by FONDECYT project number 1170501 (BGD), FONDECYT project number 1160584 (CM), Fondo de Financiamiento de Centros de Investigación en Áreas Prioritarias CRG 15090007 (CM) and CONICYT scholarship number 21170365 (KR).
Availability of data and materials
The datasets generated during the current study are available in the NCBI repository; https://www.ncbi.nlm.nih.gov/biosample/?term=srs3599252.
Author information
Authors and Affiliations
Contributions
CVP performed the data bioinformatic analysis including de novo transcriptome assembly, DEG analysis and GO analysis. KR contributed with formal analysis and writing the manuscript. MGA. carried out the genes validation by RT-qPCR. RP and RC collaborated to writing-review and editing of manuscript. BGD contributed to the design and implementation of the research, he performed funding acquisition and reviewed and edited manuscript. CM supervised the bioinformatic analysis and he worked on writing-original draft. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Sequencing metrics for 12 cDNA libraries, which were sequenced on one lane of HiSeq2000 (2x100bp). We showed number of raw and processed reads, quality reads and mapped reads. (DOCX 14 kb)
Additional file 2:
Functional annotation of the whole de novo transcriptome of Persea Americana cv. Hass during fruit developmental stages. Trinity contigs were annotated against the nr database using Blastx (10− 3). (XLSX 4991 kb)
Additional file 3:
Gene Ontology (GO) term comparisons among fruit developmental stages (150, 240, 300 and 390 days after fruit set; DAFS). GO analysis was performed using Differentally Expressed Genes (DEG; FDR < 0.01) from comparisons among 150 DAFS vs 240 DAFS, 240 DAFS vs 300 DAFS and 300 DAFS vs 390 DAFS by AgriGO v2.0 web software. (XLSX 36 kb)
Additional file 4:
List of transcription factors differentally expressed during fruit development in Persea Americana cv. Hass. Transcription factors were selected using Differentally Expressed Genes (DEG; FDR < 0.01) from comparisons among 150 DAFS vs 240 DAFS, 240 DAFS vs 300 DAFS and 300 DAFS vs 390 DAFS. (XLSX 23 kb)
Additional file 5:
Expression subcluster matrix. Values of Trinity gene expression (TPM-TMM) for 15 subclusters identified by gene expression cluster analysis during fruit developmental stages (150, 240, 300 and 390 days after fruit set; DAFS) in Persea americana cv. Hass. (XLSX 713 kb)
Additional file 6:
Expression subcluster plots. Analysis of gene expression cluster during fruit developmental stages (150, 240, 300 and 390 days after fruit set; DAFS) in Persea americana cv. Hass. (PDF 762 kb)
Additional file 7:
KEGG pathway analysis from selected subclusters 4, 7 and 11 from gene expression cluster analysis. Using Differentally Expressed Genes (DEG; FDR < 0.01) from comparisons among 150 DAFS (Days After Fruit Set) vs 240 DAFS, 240 DAFS vs 300 DAFS and 300 DAFS vs 390 DAFS. (DOCX 290 kb)
Additional file 8:
List of the primers used for validated gene expression levels by qRT-PCR. Trinity genes were validated by qRT-PCR assays and the correlation between gene expression level obtained by qRT-PCR and RNA-Seq data are shown. (DOCX 15 kb)
Additional file 9:
Fruit developmental stages phenotyping of Persea americana cv. ‘Hass’. we phenotyped fruit weight (g), ethylene production (uL C2H4kg− 1 h− 1) and oil content (5) for fruit developmental stages during 150, 240, 300 and 390 days after fruit set. (DOCX 1082 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Vergara-Pulgar, C., Rothkegel, K., González-Agüero, M. et al. De novo assembly of Persea americana cv. ‘Hass’ transcriptome during fruit development. BMC Genomics 20, 108 (2019). https://doi.org/10.1186/s12864-019-5486-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5486-7