
Abstract
Background
Since 2007, bacterial canker caused by Pseudomonas syringae pv. actinidiae (Psa) has become a pandemic disease leading to important economic losses in every country where kiwifruit is widely cultivated. Options for controlling this disease are very limited and rely primarily on the use of bactericidal compounds, such as copper, and resistance inducers. Among the latter, the most widely studied is acibenzolar-S-methyl. To elucidate the early molecular reaction of kiwifruit plants (Actinidia chinensis var. chinensis) to Psa infection and acibenzolar-S-methyl treatment, a RNA seq analysis was performed at different phases of the infection process, from the epiphytic phase to the endophytic invasion on acibenzolar-S-methyl treated and on non-treated plants. The infection process was monitored in vivo by confocal laser scanning microscopy.
Results
De novo assembly of kiwifruit transcriptome revealed a total of 39,607 transcripts, of which 3360 were differentially expressed during the infection process, primarily 3 h post inoculation. The study revealed the coordinated changes of important gene functional categories such as signaling, hormonal balance and transcriptional regulation. Among the transcription factor families, AP2/ERF, MYB, Myc, bHLH, GATA, NAC, WRKY and GRAS were found differentially expressed in response to Psa infection and acibenzolar-S-methyl treatment. Finally, in plants treated with acibenzolar-S-methyl, a number of gene functions related to plant resistance, such as PR proteins, were modulated, suggesting the set-up of a more effective defense response against the pathogen. Weighted-gene coexpression network analysis confirmed these results.
Conclusions
Our work provides an in-depth description of the plant molecular reactions to Psa, it highlights the metabolic pathway related to acibenzolar-S-methyl-induced resistance and it contributes to the development of effective control strategies in open field.
Similar content being viewed by others

Background
Kiwifruit is an economically important crop in several countries such as New Zealand, Italy, Chile, Iran and China, with Italy and New Zealand being the largest exporters of kiwifruit [1]. However, since 2007, kiwifruit industries suffered from a global outbreak of bacterial canker caused by the Gram-negative bacterium Pseudomonas syringae pv. actinidiae (Psa) [2, 3]. Psa enters plants through stomata or wounds and successively colonizes the vascular system, and it can spread systemically to young twigs a few minutes after penetration [4]. Molecular studies on Psa populations identified five biovars named biovar 1, 2, 3, 5 and 6 [3, 5,6,7]. Biovar 4 is now classified as a different pathovar called actinidifoliorum [8]. Biovar 3 is the biovar that caused the global outbreak [7]. Biovar 3 strains are characterized by the presence of pathogenesis-related sequences (integrative conjugative elements, ICEs), horizontally acquired from other P. syringae pathovars [9], but do not produce any known toxin. The control of this disease is primarily based on the use of bactericidal foliar copper application and on the induction of natural plant resistance [10, 11]. acibenzolar-S-methyl is among the most effective resistance inducers and it is able to activate Systemic Acquired Resistance (SAR) in kiwifruit plants [12]. Plants possess two levels of defense mechanisms against pests and pathogens: the general Pathogen associated molecular Pattern-Triggered Immunity (PTI) and the more specific Effector-Triggered Immunity (ETI), which is initiated after recognition of specific pathogen effector proteins [13]. Plants have evolved surveillance mechanisms that are activated upon recognition of “non-self” (or “damaged-self”) molecular patterns or signals by cell surface located receptors called Pattern Recognition Receptors (PRRs) [14]. Acute PRR signaling results in the accumulation of Reactive Oxygen Species (ROS), activation of ion channels and of defense-related Mitogen-Activated Protein Kinases (MAPKs), and transcriptional reprogramming [15]. Transcriptional reprogramming, driven by specific transcription factors (TFs), is a main feature of plant response to biotic stresses [16]. Specific members of the major TF families (AP2/ERF, bHLH, bZIP, MYB, NAC and WRKY) are particularly committed to regulating plant responses to pathogens. Furthermore, a cross-talk occurs among a core of TFs and genes related to phytohormone biosynthesis and signaling. Phytohormones, such as jasmonic acid (JA) ethylene (ET) and salicylic acid (SA), are signaling molecules that regulate the main part of plant response to pathogens [17] and TFs are involved in the convergence of different plant hormonal signaling pathways [18]. Often JA and ET work synergistically to trigger resistance against necrotrophic pathogens, whereas SA acts on the resistance against biotrophic ones. SA and JA/ET defense pathways are reciprocally antagonistic, therefore an elevated resistance against biotrophs is often correlated with susceptibility to necrotrophs, and vice versa [19, 20]. Many pathogens have evolved sophisticated strategies to manipulate the plant hormonal balance. The best-characterized mechanism involves the bacterial phytotoxin coronatine (COR), an analog of methyl-JA, which causes both the repression of the SA defense pathway and stomata opening, conditions which facilitate bacterial colonization of host tissues [21].
Little is known on the molecular interactions between A. chinensis var. chinensis and Psa. Up to very recently, how infection by Psa or treatment by elicitors of host resistance modifies gene expression in Actinidia sp. was still mostly unexplored. It has been demonstrated that elicitors of the SA pathway, but not of the ET/JA pathway, limit disease severity in both A. chinensis var. chinensis and A. chinensis var. deliciosa [12], and that following elicitation the genes PR1, PR8, ICS and PAL were over expressed in A. chinensis var. deliciosa and to lesser extent in A. chinensis var. chinensis [12]. Elicitation by chitosan also lead to the overexpression of the genes PR1 and PR5 [22]. The genes β 1–3 glucosidase, WRKY40, PR6 and AP2ERF2 were found overexpressed after infection by Psa [23] while analysis of the transcriptome of A. deliciosa found that several WRKY genes as well as several resistance genes (RPS2 and RPM1) were upregulated after inoculation with Psa [24]. An analysis of the WRKY genes of Actinidia species lead to the discovery of 97 genes which could be grouped in three categories [25]. After inoculation with Psa several of those WRKY genes were overexpressed (e.g. WRKY38, and WRKY 95) while some were repressed (e.g. WRKY96) [25]. It is now known that Psa infection results in massive changes of the transcriptome and that different species of Actinidia react differently to the infection [24]. Comparison of the transcriptome of A. chinensis var. chinensis, A. arguta and A. eriantha lead to the suggestion that resistance to Psa was related to the expression of a number of long non-coding RNAs that act in concert with coding genes [24]. To date, no comparison has been done of the whole transcriptome after inoculation of elicited versus non-elicited plants. This study aimed to compare gene expression in A. chinensis var. chinensis plants after inoculation with Psa, on plants which were elicited or not.
Comparative transcriptome profiling of A. chinensis var. chinensis in response to Psa was carried out to describe the molecular response of leaf cells to Psa and the molecular mechanisms underlying the increased resistance observed after the exogenous application of acibenzolar-S-methyl. The transcriptome analysis was tailored to the different steps of the infection process, from the epiphytic phase until the invasion of the host tissues. The progression of the infection was monitored in vivo by fluorescent stereomicroscopy and confocal laser scanning microscopy. Along with de novo assembly and characterization of the transcriptome, analysis of global patterns of gene expression and functional categorization was performed. Knowledge of the mechanisms involved in the acquired resistance of kiwifruit plants after the acibenzolar-S-methyl application could be useful to assist in identifying molecular markers in breeding for resistance.
Methods
Plant material
In vitro rooted plantlets of A. chinensis var. chinensis from micropropagation, 5–8 cm shoot length, were used for the experiment. The plants were grown in 1000 mL plastic jars containing 200 mL of half-strength Murashige and Skoog basal medium basal medium without sucrose [26]. The plants were left for 15 days in these jars before starting the experiments. For the whole duration of the experiment the plants were kept in a growing chamber at 22 ± 1 °C, with a 16:8 h light:dark period.
Acibenzolar-S-methyl treatment and Psa infection
The experiment was carried out with plants treated with acibenzolar-S-methyl or with water 15 days before inoculation with Psa, or mock inoculation using buffer. Acibenzolar-S-methyl-treated plants were immersed for 5 s in sterile solution 1.7 mM acibenzolar-S-methyl (Bion®50WG, Syngenta, Basel, Switzerland), whereas the untreated plants were submerged in water, 15 days before Psa inoculation [12]. The Psa strain CFBP7286-GFPuv was used for inoculation [27]; the inoculum was prepared from 48 h grown colonies resuspended in 10 mM MgSO4 to a final concentration of 108 cfu/mL. Inoculation was performed by immersing the plants in the Psa suspension for 1 min. The mock-inoculated plants were immersed in sterile 10 mM MgSO4. The Psa-inoculated plants were indicated as “Inoculated” (I), the mock inoculated plants were referred as “Healthy Control” (HC), the plants treated with acibenzolar-S-methyl only were indicated as “ASM” (ASM) and the plants treated with acibenzolar-S-methyl and then inoculated with Psa were indicated as “ASM-Inoculated” (ASM.I). Samples for RNA-seq analysis were taken 3, 24 and 48 h post-inoculation (hpi) and frozen by dipping in liquid nitrogen. The experiment was carried out with five biological replicates of five plants each: two replicates were used for transcriptome analysis according to recommended RNA-seq standards (Encode project https://genome.ucsc.edu/ENCODE/protocols/dataStandards/ENCODE_RNAseq_Standards_V1.0.pdf), one was kept till the development of the symptoms to assure the effectiveness of the inoculation, one was employed to assess Psa colonization and one was used for the confocal laser scanning microscope analysis.
Assessment of Psa colonization
Disease incidence and severity were assessed according to Cellini et al., 2014 [12]. Epiphytic and endophytic Psa populations were monitored in the first 48 h post inoculation. For this purpose, plants were washed in 10 mL of sterile 10 mM MgSO4 under gentle agitation for 15 min, then the washing solution was serially diluted and each dilution was plated in triplicate to evaluate Psa epiphytic population as cfu/mL. To assess the endophytic Psa population, the plants, washed as above, were externally sterilized by dipping each of them for 1 min in 70% ethanol, then 1 min in 1% sodium hypochlorite followed by 2 min in sterile water. After this treatment, the plants were homogenized in 10 mL of sterile 10 mM MgSO4. The homogenate was 10 folds serially diluted and each dilution was plated in triplicate as previously described. Bacterial colonies were enumerated at 0, 3, 6, 12, 24 and 48 h after inoculation and Psa population was calculated as cfu g− 1 of plant fresh weight.
Real time monitoring of the colonization of the host tissues
In order to match the transcriptome analysis to the different steps of the infection process, from the epiphytic colonization until the invasion of the host tissues, the infection was monitored in vivo by fluorescent stereomicroscopy and confocal laser scanning microscopy. At 3, 6, 12, 24 and 48 h post inoculation, leaves were initially observed with a Nikon SMZ25 fluorescence microscope (Nikon Instruments Corporation, Tokyo, JAPAN), with an optical system providing a zoom ratio of 25:1 (zoom range 0.63 x - 15.75 x) and epifluorescence filter cube selection (excitation wavelength of GFP-B: 460–500 nm, emission wavelength: 510–560 nm). Once Psa colonization was located on the leaf lamina, further observations were performed using a NIKON C1-S confocal laser scanning microscope equipped with an Argon laser. Optical sections of leaf lamina were acquired at 40, 60 and 100 x with Nikon PlanApo objectives and the BHS (GHS) filter set. Images were acquired and analysed by the NIS-Elements C Microscope Imaging Software.
RNA extraction and libraries preparation
Total RNA was extracted from 100 mg of ground tissue using Plant/Fungi Total RNA Purification kit (NorgenBiotek Corp., Canada) following manufacturer’s instructions. RNA samples were treated with RNase-free DNase I (Ambion, TX, USA) to remove contaminating DNA. Purity and concentration of the samples were estimated with a spectrophotometer. The integrity of the RNA (RIN > 8) was evaluated on an RNA 6000 Nano LabChiprun on Agilent 2100 Bioanalyzer (Agilent Technologies, Germany). Only samples with a RIN over 8 were used for the experiment. Two micrograms of total RNA were subjected to library preparation using the TruSeq RNA sample preparation v2 kit (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. Libraries were amplified by 15 cycles of PCR and then size selected for an average size of 300 bp using a 2% low range ultra-agarose gel (BIO-RAD). Library quality and size were assayed on the Agilent 2100 Bioanalyzer.
Sequencing, raw reads processing and data analysis
Libraries were paired end sequenced for 75 bp using an Illumina Genome Analyser (GAIIx) generating about 730 millions of raw reads. FastQ file generation was performed by CASAVA v1.8. Raw Illumina reads were processed using the CLC Genomics Workbench software (CLCbio, Aarhus, Denmark) to remove low-quality reads, adapters and duplicated sequences. Transcriptome de novo assembly was performed using Trinity software [28] using the following parameters: fixed default k-mer size 25, minimum contig length 200 bp and minimum k-mer coverage 3. Redundant sequences present in the assembly were removed using CD-HIT software [29] with a similarity threshold of 90%. Moreover, the contigs were blasted against Pseudomonas syringae genomes (http://www.pseudomonas-syringae.org) and the contigs showing a significant match were removed.
Contigs annotation was produced with Blast2GO [30] searching for matches against the NCBI non-redundant protein database, RefSeq protein, Swiss-Prot/Uniprot database, NCBI non-redundant nucleotide database, InterPro database and the Kiwifruit Genome database (http://bioinfo.bti.cornell.edu/cgi-bin/kiwi/download.cgi) with a E value threshold of 1e− 5 and by using the functionality of InterProScan v5.0 that allowed retrieval of domain/motif information in the InterPro as well as in other domain databases. Furthermore, local BLASTX alignments were run against the Clusters of Orthologous Groups (COGs) database and the Kiwifruit Genome database [31]. Reads were mapped to the assembled contigs and counted using the CLC software. Differentially expressed genes (DEG) were determined using the R package DESeq [32]. Gene Ontology enrichment analysis was performed using the GOseq package [33]. For a complete functional annotation of the transcriptome, a local BLASTx was performed to query the kiwifruit plant protein databases obtained from the Kiwifruit Genome database.
MapMan, KEGG tools and WGCNA
MapMan figures were obtained with the Mercator tool, using default parameters (http://mapman.gabipd.org/web/guest/mercator), to assign MapMan bins to Kiwifruits transcripts [34]. Log2 fold changes as obtained from DESeq output were used as MapMan input to represent expression changes. The KASS software was used to generate the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway picture. The KEGG database integrates genomic information with higher order functional information by collecting manually drawn pathway maps representing current knowledge on cellular processes and standardized gene annotations [35].
Gene expression was also assessed with weighted-gene co-expression network analysis (WGCNA) [36]. To analyse networks co-expression, transcripts were filtered for normalized count > 10 and the genes that have a high percentage of missing counts (> 90%) were removed, leading to a total number of 27.384 transcripts. The pickSoftThreshold was used to select the lower power for which the scale-free topology fit index curve flattened out upon reaching a high value. The weighted adjacency matrix of a signed network was obtained at power 9 (R function adjacency). Hierarchical clustering was conducted using the R package flashClust and the cuttreeDynamic function (dendro = geneTree, distM = dissTOM, method = “hybrid”, deepSplit = 2, pamRespectsDendro = F, minClusterSize = 30) were employed to identify modules. These functions have been shown to be the best approach for biologically meaningful results [37].
Validation of DEGs by qRT-PCR
The transcription of twenty DEGs was determined using quantitative real time PCR (qRT-PCR). Primers were designed using Primer3 plus Software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) and their specificity was checked by blasting their sequences against the NCBI database. The genes employed in the validation experiment and the primer information are reported in Additional file 1: Table S1.
Samples were collected from three biological replicates in an experiment conducted in the same conditions as the one used for the RNA-seq analysis. Total RNA was treated with RNase-free DNase I (Ambion) to remove the contaminating DNA and the first cDNA strand was synthesized from 1.0 μg of total RNA by reverse transcription using Superscript II (Invitrogen, Life Technologies GmbH, Darmstadt, Germany).
Real-time quantification was performed using the ABI 7300 Real Time System (Applied Biosystems). KAPA Sybr Fast qPCR kit (Resnova) master mix was used for 40 cycles with the following profile: 95 °C for 15 s, 60 °C for 20 s, 72 °C for 40 s. Melting curve analysis was performed to verify single product amplification with temperature ranging from 55 to 95 °C by increasing of 1 °C every step. All reactions were run in triplicate for each biological replicate and β-Actin (ACT1, accession number EF063572) was used as the reference gene. Primers for β-Actin (Additional file 1: Table S1) were designed by Primer 3 plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) based on complete mRNA sequence of Actinidia deliciosa ACT1 (ACT1), accession number EF063572. Transcript abundances are given as the mean ± SE of replicates. Relative transcription levels were calculated using the 2-△△CT method [38].
Results and discussion
Host tissues colonization
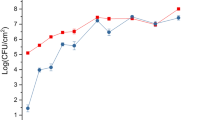
Psa epiphytic population reached their maximum concentration about 6 h post-inoculation (hpi), a result determined by the high inoculum used in this experiment and by Psa epiphytic fitness (Fig. 1a). The acibenzolar-S-methyl treatment did not influence the epiphytic growth on host leaves. From 6 hpi onwards, Psa was also found inside the host tissues (Fig. 1b), although in acibenzolar-S-methyl treated plants, Psa growth was reduced in comparison with non-treated plants. Furthermore, from 24 hpi onwards, Psa endophytic populations decreased in acibenzolar-S-methyl treated plants reaching less than 10 cfu g− 1 fresh tissues at 48 hpi (Fig. 1b).

Epiphytic (a) and endophytic (b) colonization of Actinidia chinensis var. chinensis plants by Pseudomonas syringae pv. actinidiae (Psa) strain CFBP7286-GFPuv at 3, 6, 12, 24 and 48 h after inoculation. Plants were either treated with water (Control) or acibenzolar-S-methyl application (ASM) 15 days before inoculation
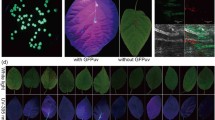
Observations under a confocal laser scanning microscopyconfirmed the dynamic of Psa colonization. At 6 hpi, Psa was localized around the stomata and on the edges of the stomata among epidermic cells (Fig. 2b). At 24 hpi, Psa was clearly observed inside the stomatal chamber and in the spongy mesophyll, indicating that the bacterium rapidly entered the host tissues via the stomata and extensively colonized the leaves (Fig. 2c). The assessment of the endophytic population indicated that Psa had already entered the leaves at 6 hpi; however, Psa was visible by confocal laser scanning micrographs only between 12 and 24 hpi, suggesting that a certain population threshold was needed to make colonization visible. At 24 hpi, colonization of secondary and tertiary leaf veins was observed (Fig. 2d), suggesting the beginning of systemic invasion of the kiwifruit vine.

Confocal laser scanning micrographs of Actinidia chinensis var. chinensis leaves inoculated with Pseudomonas syringae pv. actinidiae Psa strain CFBP7286-GFPuv. Panel a: before infection, white arrows indicate stomata; Panel b: 6 h after infection. It is possible to observe Psa cells colonizing the leaf surface and the stomata entrance (arrows); Panel c: 24 h after infection, vertical sectioning of leaf lamina (Z axis: - 20 μm) showing Psa invading the spongy mesophyll below stomata (arrows); Panel d: 24 h after infection. Psa colonizing the leaf veins (arrows)
RNA sequencing and transcriptome assembly
After quality filtering, between 11 and 46 million reads were obtained for each RNA sample (Additional file 2: Table S2 A). Pearson correlations between biological replicates were always above 0.95 and samples undergoing the same treatment clustered together. High-quality reads were used to produce the reference transcriptome of A. chinensis var. chinensis by de novo assembly. The overall transcriptome consisted of 63,943 contigs. Then, redundant contigs, as well as those belonging to P. syringae, were removed leading to 39,584 contigs, for a total assembly size of 39.95 Mbp. The sequence length ranged from 201 bp to 11,870 bp with an average size of 933 bp and a N50 of 1472 bp.
Annotation and classification of A. chinensis var. chinensis transcriptome
The main features of the annotation of the A. chinensis var. chinensis reference transcriptome are summarized in Additional file 3: Figure S1 A, while the complete information is presented in Additional file 2: Table S2 B.
The E-value distribution of the top hits for each contig in the NR protein database showed that 18,369 contigs had strong homology with an E value < 1.0 e− 5 (Additional file 3: Figure S1 B). The sequence similarity distribution of the contigs against the NR protein database shows that 24,867 contigs (85.93%) have a similarity ranging from 100 to 70% (Additional file 3: Figure S1 C). The BLASTx performed to query the Actinidia protein databases, obtained from the Kiwifruit Genome database, led to a match with 30,634 annotated contigs (77.39%).
The contigs of the reference transcriptome were classified in 13,619 Clusters of Orthologous Groups (COGs) functional annotations and grouped into 24 function categories (Additional file 3: Figure S1 D). Moreover, 21,636 contigs of the A. chinensis var. chinensis transcriptome were categorized into 101 functional groups of Groups of Orthologous (GO) slim plant (Additional file 3: Figure S1 E). As final result, a total of 34,039 contigs were annotated by integrating the BLAST results from all the database queried (Additional file 3: Figure S1 F and H, Additional file 2: Table S2 C and D).
This work identified a total of 4533 DEGs (Additional file 4: Table S3). The interaction between kiwifruit plants and Psa revealed 3360 DEGs, most of them were identified at 3 hpi (2747 DEGs). Few DEGs were highlighted at 24 hpi and at 48 hpi (272 and 341 DEGs, respectively; Table 1). The comparison between HC and Psa-inoculated plants at 3 hpi showed that 1596 DEGs were up-regulated at least two-fold, with about 37% of them being up-regulated more than five-fold and 214 more than ten-fold. About 42% of DEGs were down-regulated. At 24 hpi and 48 hpi the response was dominated by a down-regulation of gene expression with 57 and 76% of DEGs repressed, respectively. The small number of DEGs at 24 hpi and 48 hpi, and the prevalence of down-regulation may reflect the ability of Psa bacterial cells to suppress the plant defense pathways.
ASM plants reacted differently to Psa infection than non-treated ones. At 3 hpi in ASM plants, a substantially lower number of DEGs (510) were modulated in comparison to untreated plants. In contrast, at 24 hpi in ASM plants, 1374 DEGs were identified and at 48 hpi, the number of DEGs was 1252 (Table 1). Noteworthy, the treatment with acibenzolar-S-methyl sustained a much stronger molecular response to Psa infection at 24 hpi and 48 hpi, in agreement with the observed decreasing of Psa population inside the leaves (Fig. 1).
In untreated plants, out of 2747 DEGs detected at 3 hpi, only 188 were shared with 24 hpi, and this number decreased to 145 at 48 hpi. Acibenzolar-S-methyl treated and untreated plants shared 225 DEGs at 3 hpi, 41 at 24 hpi and 53 at 48 hpi (Fig. 3a).

Panel a: Venn diagrams showing the overlapping of the DEGs modulated in response to Pseudomonas syringae pv. actinidiae (Psa) infection at 3, 24 and 48 hpi in plants not treated with acibenzolar-S-methyl. Panel b: Venn diagrams showing the overlapping of the DEGs modulated in response to Psa infection at 3, 24 and 48 hpi in plants untreated and treated with acibenzolar-S-methyl. Panel c: Hierarchical clustering analysis made with Pearson correlation of Log2FC of all DEGs detected in the transcriptome analysis (4533; Additional file 4: Table S3). Each row represents a transcript; each column represents a comparison. A dendrogram of the correlation among transcripts is shown on the left of the heatmap. A clear separation between untreated and acibenzolar-S-methyl treated plants is evident from the upper cluster of the heatmap
In inoculated ASM plants, out of 510 DEGs modulated at 3 hpi, 341 were shared with 24 hpi and this number became 315 at 48 hpi (Fig. 3b). In ASM plants, regardless of inoculation status, a core group of 229 DEGs were detected at all-time points after inoculation. Finally, 151 DEGs were identified exclusively in non-inoculated ASM (Fig. 3b).
The hierarchical clustering analysis based on transcript expression levels had grouped the DEGs in two main clusters related to the acibenzolar-S-methyl treatment. This result indicated that acibenzolar-S-methyl strongly affected the plant response to Psa infection, suggesting that this compound activated defensive responses which were not induced in untreated plants (Fig. 3c). For the validation of our transcriptomic data twenty DEGs were selected for qRT-PCR analysis (Additional file 5: Figure S2). Our qRT-PCR data strongly correlated with the RNA-seq expression data.
Biochemical pathways analysis
The kiwifruit transcripts modulated in response to Psa infection were analyzed by the KEGG orthology database. Among the 4533 DEGs identified in the transcriptomic analysis, 1331 were assigned to KEGG orthologs. Among them, KEGG analyses identified several pathways, such as plant hormone signal transduction, cysteine-methionine metabolism and plant-pathogen interaction. These pathways were differentially modulated according to the progression of Psa in the tissues and to presence/absence of acibenzolar-S-methyl treatment. In addition, the visualization of the DEGs with MapMan analysis highlighted the involvement of the hormonal signaling and the modulation of the transcription factors during the progression of Psa invasion and presence/absence of acibenzolar-S-methyl treatment (Additional file 6: Figure S3).
Pathogen recognition related genes
Perception of microbe-associated molecular patterns or pathogen-associated molecular patterns (PAMPs/MAMPs) via activation of cell-surface-resident pattern recognition receptors (PRRs) initiates the PAMP-triggered immunity (PTI) [13]. In kiwifruit plants inoculated with Psa, 198 transcripts encoding putative Pathogen Recognition-Related (PRR) proteins were found to be differentially expressed (Fig. 4a, Additional file 7: Table S4). These PRRs are Receptor Like Kinases (RLK) or Leucine-rich repeat receptor-like protein kinases (LRR-RLKs).

Panel a: Heatmap of Differentially Expressed Genes (DEGs) encoding putative Pattern Recognition Receptors (PRRs). Panel b: Heatmap of DEGs involved in the defense response against Psa. Up-regulated transcripts (Log2FC ≥ 1) are in green, down-regulated transcripts (Log2FC ≤ − 1) are in red. Each row represents a transcript, each column a comparison. For the description of the gene names represented in the heatmaps refers to Additional file 4: Table S3 and Additional file 8: Table S5
The most significant up-regulation of RLK and LRR-RLK transcripts was observed in inoculated, ASM plants with an increase in the number of up-regulated transcripts from 3 to 48 hpi. On the other hand, a different set of PRR transcripts were found up-regulated almost exclusively at 3 hpi in infected untreated kiwifruit plants (Fig. 4a).
Transcripts homologous to FLAGELLING SENSING 2 (Ach_contig26439; Ach_contig38670, Ach_contig38671), the receptor kinase for bacterial flagellin (flg22), were up-regulated. Ach_contig26439 was up-regulated in untreated plants only at 3 hpi, while Ach_contig38670 and Ach_contig38671 were up-regulated in ASM plants regardless of infection. In other pathosystems, the activation of flg22 leads to the accumulation of ET and SA [39,40,41]. Other transcripts associated with bacterial recognition were up-regulated after infection by Psa. The bacterial elongation factor Tu (EF-Tu) (Ach_contig7359), was up-regulated at 3 hpi in untreated samples only. Instead, transcripts belonging to the serine/threonine receptor kinase family, containing the LysM domain [42], were up-regulated in both acibenzolar-S-methyl-treated and untreated plants at 3 hpi (Fig. 4a). LysM domains bind Peptidoglycans (PGNs) of both Gram-positive and Gram-negative bacteria and are known to mediate resistance against bacterial pathogens [43].
Oligogalacturonidases (OGs) fragments, released from plant cell walls, act as potent defense response elicitors [44]. The WALL-ASSOCIATED KINASES (WAKs) work as pathogen-related signals molecules, which are able to detect the presence of OGs [45, 46].
In Arabidopsis, the increase of expression of some WAK genes e.g. WAK2, [47] or WAKL10 and WAKL22 [48] was found related to pathogen infection. Moreover, WAK1 has been linked with both to P. syringae infection and exogenous application of SA [49]. In rice, OsWAK25 transcript accumulates after acibenzolar-S-methyl-treatment [50], and in tomato and wheat, WAK genes have been found to be related with pathogen infection [51, 52].
In the kiwifruit-Psa interaction, 10 DEGs belonging to the WAK family (Ach_contig12264; Ach_contig3903; Ach_contig11995; Ach_contig25885; Ach_contig25886; Ach_contig35387; Ach_contig37852; Ach_contig35388; Ach_contig15313) were up-regulated in untreated infected plants at 3 hpi and as well as in ASM plants regardless of sampling time (Fig. 4a and Additional file 7: Table S4).
Members of the RLK1 family are good indicators for cell wall integrity. Among them, Theseus1 (THE1) and Feronia (FER) were differentially expressed in kiwifruit-Psa interaction [53, 54]. Transcripts annotated as receptor-like kinase FERONIA were up-regulated only in ASM plants regardless of infection (Fig. 4a). Among them, Ach_contig13965 was used to confirm these data by qRT-PCR (Additional file 5: Figure S2). In response to environmental stress and SA, FER receptor kinase may negatively regulate the biosynthesis of S-adenosyl methionine (SAM) which, in turn, down-regulates the SAM-dependent pathways, such as the one leading to ET biosynthesis [55].
BRASSINOSTEROID-INSENSITIVE ASSOCIATED KINASE 1 (BAK1) is required for responses triggered by the orphan PAMPs such as Peptidoglycans (PGNs) and lipopolysaccharide (LPS) [56] Moreover, BAK1 is essential for PRRs for the responses against different bacterial and fungal pathogens [57]. Three transcripts homologs to BAK1 were found differentially expressed in kiwifruit after Psa interaction. Ach_contig24575 and Ach_contig24576 were up-regulated in infected plants at 3 hpi only, while Ach_contig29878 was up-regulated in acibenzolar-S-methyl-treated and inoculated samples, at all-time points. The expression of the BAK1-Ach_contig29878 reflects the ability of ASM plants to activate the downstream immune response (Fig. 4a). Moreover, overexpression of SUPPRESSOR OF BIR1 (SOBIR1) leads to a constitutive activation of disease-resistance responses [58, 59]. Two transcripts homologs to SOBIR1 (Ach_contig37485; Ach_contig37484) were up-regulated only in ASM plants (Fig. 4a, Additional file 7: Table S4). Transcriptome analysis of other plant-pathogen interactions revealed that the SOBIR1 gene is transcriptionally regulated by biotic stress [15]. In Sinapis alba a SOBIR1 homologue was up-regulated after infection with Alternaria brassicicola [60]. Similarly, in Malus domestica a homologue of AtSOBIR1 was up-regulated in plants resistant to Erwinia amylovora [61]. In the kiwifruit-Psa interaction, the SOBIR1 expression was related to acibenzolar-S-methyl application, suggesting a possible role for this gene in the SAR, as proposed for Arabidopsis [62].
Despite the high number of genes coding for PRR found in the A. chinensis var. chinensis genome [63] and the up-regulation of many of them at 3 hpi in untreated plants, this response is not sufficient to confer resistance to Psa. Thus, without acibenzolar-S-methyl treatment, kiwifruit is not capable of sustaining the activation of effective defensive mechanisms against the pathogen.
Defense-related genes
Transcripts encoding several classes of chitinases (Ach_contig8637, Ach_contig3262, Ach_contig27724, Ach_contig24132, Ach_contig7693, Ach_contig25203), β-1,3-glucanase (Ach_contig27175), endochitinase (Ach_contig996, Ach_contig34595; Ach_contig6256) and A. chinensis thaumatin-like proteins (Ach_contig10838, Ach_contig19122) were up-regulated in ASM plants, indicating that these transcripts are related to the enhanced resistance response. The transcripts encoding PR1 homologs (Ach_contig12206, Ach_contig14525) were up-regulated in all infected samples with a higher expression in the ASM plants. The expression profile of Ach_contig14525 was confirmed by qRT-PCR (Additional file 5: Figure S2). Transcripts putatively encoding PR10 (Ach_contig15022; Ach_contig7880; Ach_contig18265; Ach_contig18266) were found up-regulated only in untreated plants at 3 hpi. OsPR10a and Pg1, PR10 orthologs, were up-regulated in response to exogenous ethylene treatment, in rice [64] and ginseng [65]. Moreover, two alfalfa PR10 genes, MsPR10.1A and MsPR10.1B, were responsive to ethylene and abscisic acid (ABA) treatment [65].
Actinidin 2d (Ach_contig26480; Ach_contig26479) is a member of the papain-like cysteine proteases family (PLCPs). Recent evidence indicates a key role for PLCPs in plant immunity [66, 67]. In the kiwifruit-Psa interaction the transcripts homologs to Actinidin showed a strong up-regulation in ASM plants with or without Psa inoculation. On the other hand, the same transcripts were down-regulated in untreated infected plants. This finding supports the idea that the up-regulation of transcripts coding for Actinidin enzymes is associated with the increased resistance of ASM plants to Psa. Indeed, it has been suggested that the presence of basic and acidic isoforms of Actinidin are reminiscent of PR proteins, having a role in defense against pathogens [68]. In addition, about 50 transcripts putatively annotated as disease resistance genes (homologues of TIR- and CC-NBS-LRR genes) were DEGs in one or more samples (Fig. 4b, Additional file 8: Table S5). Among them, three transcripts (Ach_contig24825; Ach_contig17717; Ach_contig17718) belonging to Resistance Gene Analogs RGA-like genes, and seven (Ach_contig18842; Ach_contig13980; Ach_contig35140; Ach_contig28816; Ach_contig35282; Ach_contig27985; Ach_contig35141) belonging to cysteine-rich like protein were induced in ASM plants upon inoculation.
ROS detoxification related genes
One of the earliest responses after pathogen recognition is the oxidative burst achieved through the production of ROS caused by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and the respiratory burst due to the oxidase homologues (RBOHs) multigenic family [69]. Accordingly, the transcriptomic analysis of the kiwifruit-Psa interaction has identified many DEGs involved in ROS production and detoxification. Four transcripts belonging to NADPH-oxidase (Ach_contig28941, Ach_contig28940, Ach_contig28939, Ach_contig28930) were up-regulated at 3 hpi in untreated plants, whereas only Ach_contig37977 was up-regulated in the acibenzolar-S-methyl-treated samples (Fig. 5). Moreover, antioxidant enzymes including superoxide dismutase (Ach_contig30858), catalase (Ach_contig25559, Ach_contig25558, Ach_contig25560, Ach_contig25556, Ach_contig25561), peroxidase (Ach_contig20101, Ach_contig10320, Ach_contig6123, Ach_contig27264, Ach_contig17103, Ach_contig18018, Ach_contig7182, Ach_contig18508, Ach_contig3949, Ach_contig35730, Ach_contig10321), ascorbate-oxidases (Ach_contig15094, Ach_contig26749), ascorbate peroxidase (Ach_contig12730, Ach_contig19106) and dehydroascorbate reductase (Ach_contig20425) were differentially expressed in several conditions (Fig. 5, Additional file 9: Table S6). Differential regulation of antioxidant enzymes, partially mediated by SA, may contribute to the increases of ROS and to the activation of defenses [70].

Heatmaps of Differentially Expressed Genes (DEGs) encoding putative Reactive Oxygen Species (ROS). Up-regulated transcripts (Log2FC ≥ 1) are in green, down-regulated transcripts (Log2FC ≤ − 1) are in red. Each row represents a transcript, each column a comparison. For the description of the gene names represented in the heatmaps refer to Additional file 9: Table S6
Hypersensitive Induced Response (HIR) proteins are a group of proteins involved in Hypersensitive Response [71]. Although there are no reports about HIR genes induced during SAR, the PR-1 gene expression was elevated in transgenic Arabidopsis overexpressing the rice HR-induced gene OsHIR-1 [72]. Interestingly, in ASM plants (both infected and not infected), a robust activation of transcripts encoding a protein related to HIR-1 (Ach_contig24151, Ach_contig27830, Ach_contig24154, Ach_contig24153, Ach_contig24161, Ach_contig24158) was found. Among them, Ach_contig24153 was used to confirm these data by qRT-PCR (Additional file 5: Figure S2). Besides, four transcripts homologous to peroxidase 4 (Ach_contig14667, Ach_contig14668, Ach_contig13292, Ach_contig13293) and belonging to the PR9 protein subfamily [73] were up-regulated in ASM plants regardless Psa inoculation. Members of the PR9 family are able to catalyze the synthesis of bioactive molecules that limit bacterial pathogen spreading through the establishment of physical barriers or the generation of toxic compounds, such as ROS [74].
Photosynthesis-related genes
The infection strongly down-regulated photosynthetic related genes (Additional file 10: Table S7) as observed in other plant-pathogen interactions [75,76,77,78]. Moreover, in our experiment about 30 DEGs (Additional file 10: Table S7) were annotated as cytochrome P450 (CYP450). The cytochrome P450 (P450) superfamily is the largest family of plant metabolic enzymes and plant P450 families are highly divergent, reflecting diversification and neofunctionalization [79, 80]. CYP450s are involved in SA-dependent defense responses [81] such as lignin biosynthesis, callose deposition and cell wall reinforcement [82]. Nonetheless, P450 gene expression was not found to be consistently differentially expressed after acibenzolar-S-methyl treatment, or infection.
Hormonal-pathway-related genes
Several pathovars of P. syringae are known to produce compounds which manipulate the plant hormonal balance [83]. A well-known example of hormone manipulation by P. syringae involves the production of the phytotoxin coronatine (COR), which mimics JA functions. Other P. syringae pathovars, such as P. syringae pv. tomato (strain DC3000), modulate ABA level in order to suppresses stomata closure [84, 85]. Finally, some pathovars, such as P. savastanoi pv. glycinea and P. savastanoi pv. phaseolicola are capable of producing ET, which acts as a virulence factor by impacting ET production by the host [86]. Among the five biovars of Psa, strains of biovar 1 produce phaseolotoxin, a phytotoxin causing the halo blight disease, and strains of biovar 2 produce coronatine. Strains of biovar 3 do not produce any known toxin [7], and genes coding for ET biosynthesis, such as 2-oxoglutarate-dependent ethylene-forming enzyme (EFE), have not been found either. However, production of ethylene by some strains of Psa biovar 3 has been detected [12]. A subset of DEGs involved in ET biosynthesis and ET-signaling were found in kiwifruit plants upon Psa infection (Fig. 6a, Additional file 11: Table S8). The transcripts encoding homocysteine methyl transferase (HMT, Ach_contig26132, Ach_contig16088, Ach_contig22185), methionine synthase and S-adenosyl methionine-synthase (SAMS; Ach_contig18109, Ach_contig18113, Ach_contig17516, Ach_contig18112, Ach_contig18110, Ach_contig20053, Ach_contig18114) were up-regulated at 3 hpi in untreated plants only (Fig. 6a). The key enzymes of ET biosynthesis, 1-AMINO CYCLOPROPANE-1-CARBOXYLATE SYNTHASE (ACS) and ACC OXIDASE (ACO), were also detected following Psa infection (Fig. 6a). Two transcripts encoding ACS enzymes were found significantly modulated only in control plants at 3 hpi: Ach_contig15019, encoding ACS2, was up-regulated, while Ach_contig23058, encoding ACS5, known to be unresponsive to exogenous ET treatment [87], was down-regulated.

Heatmap describing the modulation of Differentially Expressed Genes (DEGs) involved in the hormonal pathways. a Ethylene (ET), b Jasmonic Acid (JA), c Salicylic Acid (SA) and d Abscisic acid (ABA). Up-regulated genes (Log2FC ≥ 1) are in green, whereas down-regulated ones (Log2FC ≤ − 1) are in red. The description of the genes represented in the heatmaps is reported in Additional file 11: Table S8, Additional file 12: Table S9, Additional file 13: Table S10, Additional file 14: Table S11, respectively
Two transcripts homologous to ENHANCED DISEASE RESISTANCE 1 (EDR1, Ach_contig3372; Ach_contig33724) were found up-regulated at 3 hpi in untreated plants (Fig. 6a). In Arabidopsis, EDR1 negatively regulates SA-dependent defense responses, ABA signalling, and ET-induced senescence [88]. EDR1 confers sensitivity to various pathogens such as Erwinia cichoracearum in cucurbits and P. syringae pv. tomato (strain DC3000) in tomato. EDR1 is required for resistance to some hemibiotrophic/necrotrophic fungal pathogens through the induction of plant defensin (PDF), probably interfering with MYC2 function [89]. At 3 hpi in untreated plants, transcripts (Ach_contig10181) belonging to the PDF family were found to be up-regulated (Fig. 6a). Moreover, Constitutive Triple Response1 (CTR1; Ach_contig33722, Ach_contig33724) and Ethylene Insensitive2 (EIN2; Ach_contig23335, Ach_contig23336), which are key genes of the ET signaling pathway, were found to be up-regulated. At 3 hpi, in both untreated and ASM plants, transcripts encoding EIN3 BINDING F-BOX1 (EBF1) and EBF2 (Ach_contig34711, Ach_contig34712) were up-regulated. In Arabidopsis, these genes were up-regulated even in the absence of infection, and EBF2 transcription was rapidly induced after exogenous application of ET [90].
The expression of ETHYLENE INSENSITIVE2 (EIN2) is supported by the activation of MYB44 [91] and, in turn, activates the expression of downstream transcription factors such as ETHYLENE RESPONSE FACTOR (AP2/ERF) [92]. Indeed, two MYB44 transcripts (Ach_contig27284 and Ach_contig27287) and about 25 transcripts encoding AP2/ERF transcription factors were up-regulated in response to the Psa infection in untreated plants (Fig. 6a).
Genes involved in JA-mediated responses were differentially expressed, mainly at 3 hpi in untreated plants (Fig. 6b, Additional file 12: Table S9). Transcripts corresponding to Allene Oxide Cyclase (AOC, Ach_contig13768) and OPC 8:0 CoA Ligase1 (OPCL1, Ach_contig13768) [93], two enzymes of the JA biosynthetic pathway, were all up-regulated. In addition, the expression of JASMONATE-ZIM DOMAIN (JAZ, Ach_contig31198, data confirmed by qRT-PCR, Additional file 5: Figure S2) was also found to be increased upon infection. JAZ proteins induce the expression of genes involved in the formation of the repressor complexes consisting of MYC2, a βHLH-type transcriptional regulator, NINJA adaptor-proteins and TOPLESS (TPL) co-repressors [94,95,96]. In ASM plants, none of these genes was modulated in relation to infection (Fig. 6b), thus suggesting that acibenzolar-S-methyl might minimize the hormonal imbalance caused by Psa to hijack plant defenses [12].
Few DEGs related to SA biosynthesis and signaling were detected in ASM plants regardless of Psa infection (Fig. 6c; Additional file 13: Table S10). SA biosynthesis occurs via the shikimic acid pathway, which involves the conversion of chorismate to isochorismate by Isochorismate-synthase (ICS) [91]. Control plants infected with Psa did not show modulation of ICS, while in acibenzolar-S-methyl-treated and infected plants, a transcript encoding ICS1 (Ach_contig10145) was found down-regulated at 24 hpi. In Arabidopsis, ICS1 regulation is linked to calcium signaling through the transcriptional regulation of Enhanced Disease Susceptibility-1 (EDS1) [97, 98]. In the same model plant, EDS1 works together with phytoalexin-deficient 4 (PAD4) to promote the hypersensitive response and SA accumulation [93]. In response to Psa, in kiwifruit, the transcript coding for EDS1-like lipase (Ach_contig27833) was up-regulated in all ASM samples. However, differently from Arabidopsis, PAD4 was not up-regulated in ASM kiwifruit plants. The up-regulation of PAD4 transcripts was instead detected in untreated infected plants at 3 hpi. Interestingly, in Arabidopsis, the up-regulation of PAD4 is essential for boosting ET production after pathogen infection [99,100,101]. Thus, the induction of PAD4 in kiwifruit untreated infected plants supports the role of ET as a positive regulator of host susceptibility in this pathosystem.
In ASM plants, both at 24 hpi and 48 hpi, transcripts coding for Nonexpresser of PR proteins NPR1 and NPR3 (Ach_contig14340 and Ach_contig32812, respectively) [102] were up-regulated. NPR1 and NPR3 are the main transcriptional regulators of the SA pathway [103]. The expression profile of Ach_contig14340 was confirmed by qRT-PCR (Additional file 5: Figure S2). The Arabidopsis thaliana NPR1-proteins act in concert with NIM1-INTERACTING 1 (NIMIN-1), NIMIN-2, and NIMIN-3 [104]. Accordingly, the transcripts encoding NIMIN2 (Ach_contig17288; Ach_contig17287) were also induced in ASM plants (Fig. 6c; Additional file 13: Table S10): data confirmed by qRT-PCR (Additional file 5: Figure S2). These results confirm the priming effect of acibenzolar-S-methyl on SA-dependent defensive responses. Moreover, SA-dependent responses were activated by the infection only in ASM, thus corroborating the findings that SA mediates resistance against Psa in A. chinensis [12].
One of the first line of plant defenses against bacterial pathogen is the modulation of stomata closure [105]. SA induces stomatal closure in Vicia faba [106], Phaseolus vulgaris [107] and Arabidopsis [108] when applied on leaves. However, the effect of exogenous application of SA on stomatal movements may vary according to the plant species and the mode of application [109]. In this view, ABA plays a role in early plant defenses by inducing stomata closure upon recognition of pathogen-associated patterns [82, 110]. However, in post-infection phases, ABA generally exerts a negative role on plant immunity [111,112,113]. For example, P. syringae effector AvrPtoB stimulates ABA biosynthesis to weaken plant immune responses [78, 114]. ABA effect on stomata is mediated by cell turgor and Ca2+ balance in guard cells [115]. Indeed, Ca2+-independent protein kinase SNF1 Related Kinase 2 (SNRK2) is a key regulator of the stomata closure [116]. In our experiment, two transcripts (Ach_contig20697 and Ach_contig15315) related with SNRK2 were identified as DEGs. Ach_contig20697 was down-regulated in untreated infected plants at 3 hpi, whereas Ach_contig15315 was up-regulated both in response to acibenzolar-S-methyl and to infection in treated samples at 24 hpi and 48 hpi (Fig. 6d; Additional file 14: Table S11).
In the absence of ABA, the PROTEIN PHOSPHATASE 2C (PP2C) acts as an inhibitor of the ABA signaling response by binding and blocking SNRK2 [117, 118]. In untreated plants at 3 hpi, several PP2Cs of class A [119], namely Ach_contig26540, Ach_contig9883, Ach_contig9880 and Ach_contig28395, were found to be down-regulated. Moreover, in the same plants at 3 hpi, four transcripts (Ach_contig20119, Ach_contig32298, Ach_contig20123, Ach_contig20122 and Ach_contig20120) encoding a class of intracellular ABA receptors (PYR/PYL/RCAR) [116] known to interact with PP2Cs [120], were also found to be up-regulated. In ASM plants at 24 hpi, the same transcripts were instead down-regulated (Fig. 6d). Arabidopsis plants are not able to close their stomata, even in the presence of ABA, if they are exposed to ET, confirming that ET represses ABA-dependent stomata closure [121]. In untreated plants at 3 hpi, a transcript related to ABA repressor 1 (ABR1, Ach_contig18951, Fig. 8), which is a negative regulator of ABA signaling [122], was found to be up-regulated. ABR1 upregulation was followed by down regulation at 24 hpi and 48 hpi (data confirmed by qRT-PCR; Additional file 5: Figure S2). Furthermore, in the same plants and at the same time points, transcripts encoding for the E3 ligase ARM REPEAT PROTEIN INTERACTING with ABF2-like (ARIA-like TFs; Ach_contig27093, Ach_contig27096) were found to be up-regulated. ARIA-like TFs were involved in the ubiquitination of ABF2, which controls ABA-responsive gene expression [123]. Finally, in ASM plants at 24 hpi, the bZIP transcription factor ABI5 (Ach_contig26433, Ach_contig25391, Ach_contig25394), known to be transcriptionally induced by ABA [124, 125], was up-regulated (Fig. 6d).
All these findings suggest a possible role of ET as a Psa virulence factor concurring in stomata opening by repressing ABA-mediated signaling.
Transcription factors
Transcription factors (TFs) play a key role in the regulation of plant responses to pathogens. In the kiwifruit transcriptome, 3009 transcripts belonging to 53 families of TFs and to 13 families of Chromatin Remodeling Factors were detected (Additional file 15: Figure S4; Additional file 16: Table S12). Concerning TFs, 367 transcripts were DEGs in at least in one condition (Fig. 7a, Additional file 17: Table S13). A total of 216 TFs were modulated at 3 hpi in untreated plants. Out of these, 199 TFs were modulated only in this condition (Fig. 7b) with the most represented families corresponding to APETALA2/ETHYLENE RESPONSIVE FACTOR (AP2/ERF) and WRKY. In ASM plants, 187 transcripts coding for TFs were modulated, with 11 identified either in ASM plants without Psa or in untreated plants at 3 hpi. Finally, 39 TFs were exclusively modulated at either at 24 or 48 hpi (Fig. 7c).

Panel a: Heatmap showing differentially expressed transcription factors (TFs). Each row represents a transcript; each column represents a library comparison. A dendrogram of the correlation among transcripts is shown on the left of the heatmap. The green color represents up-regulated genes (Log2FC ≥ 1) and red color represents down-regulated genes (Log2FC ≤ − 1). The description of the genes represented in the heatmap is reported in Additional file 16: Table S12; Panel b: Venn diagram showing the overlapping of differentially expressed TFs modulated in infected plants at 3, 24 and 48 hpi. Panel c: Venn diagram showing the overlapping of TFs modulated in acibenzolar-S-methyl treated plants inoculated with Psa at 3, 24 and 48 hpi
AP2/ERF is a large family of plant TFs divided into three subfamilies: AP2, RAV and ERF [126]. Individual members of the ERF family have been shown to be either positive or negative regulators of the defense response. A total of 35 AP2/ERF were modulated in the kiwifruit-Psa interaction (Fig. 8a). Among them, 23 AP2/ERF, mainly related to ERF subfamily IX [127], were up-regulated in untreated plants at 3 hpi, and only three of them were also down-regulated in ASM samples inoculated with Psa (Fig. 8a, Additional file 18: Table S14).

Heatmaps showing transcription factors (TFs) differentially expressed in the kiwifruit-Psa interaction. Panel a: Ethylene responsive Factors (ERFs); Panel b: NAC TFs; Panel c: WRKY TFs. The green color represents up-regulated genes (Log2FC ≥ 1) and red color represents down-regulated genes (Log2FC ≤ − 1). Panel d: Four-ways Venn diagram showing WRKY TFs comparison between early stages of Psa inoculation in kiwifruit untreated plants against acibenzolar-S-methyl treated ones. For the description of the gene names represented in the heatmap refer to Additional file 18: Table S14, Additional file 19: Table S15 and Additional file 20: Table S16, respectively
TFs belonging to ERF subfamily IX have been often linked with response to pathogen infection in Arabidopsis, rice and grape [126, 128]. In detail, in Arabidopsis, ERF1, belonging to subfamily IX, is involved in both JA and ET signaling. ERF1 over-expression increases resistance to necrotrophic fungi and enhances susceptibility to the hemibiotroph P. syringae pv. tomato [53]. Moreover, Ach_contig21919; Ach_contig30206; Ach_contig30205; Ach_contig21920; Ach_contig13845; Ach_contig13844, belonging to family IX, were annotated as AdERF11, AdERF12 and AdERF13. These ERF TFs of Actinidia deliciosa are known to control the expression of other ERF genes in response to ET and JA [127].
Two transcripts (Ach_contig5488; Ach_contig19387) belonging to subfamily VIII were up-regulated at 3 hpi in untreated plants (Additional file 16: Table S12). Interestingly, the expression of genes of subfamily VIII can be rapidly induced by ET and JA synergistically [128, 129]. Likewise, three transcripts (Ach_contig12383; Ach_contig23443; Ach_contig12380), up-regulated at 3 hpi in untreated plants, were identified as AdERF4 that, along with AdERF5 (Ach_contig33797), belongs to subfamily VII (Additional file 18: Table S14). The members of this subfamily have been associated with the modulation of ET during hypoxia and with grape resistance to the necrotrophic pathogen Botrytis cinerea [130]. Finally, the transcript corresponding to ABR1-like, belonging to Subfamily X, was one of the most up-regulated genes as reported in section “Hormonal Balance”.
Plants treated with acibenzolar-S-methyl showed a general down-regulation of ERF genes with the exception of transcript Ach_contig8529 which is an ortholog of Tobacco stress-induced gene 1 (Tsi1) (Fig. 8a). Tsi1, which exhibits characteristic features of group VI, is known to be responsive to SA treatment [131]. Moreover, the over-expression of Tsi1 in tobacco and pepper increased resistance to viral, bacterial and oomycete pathogens [131, 132]. Therefore, Tsi1 could be one of the main candidate genes involved in increased defenses against Psa in ASM kiwifruit plants.
Additionally, to AP2/ERF-related transcripts, 20 contigs belonging to the NAC TF family were identified (Fig. 8b). The NAC TF family, which include three subfamilies, namely NAM, ATAF and CUC, regulates a variety of plant processes, including the response to biotic and abiotic stresses [133, 134]. Within this family, in Arabidopsis, ATAF1/NAC2 is known to be a negative regulator of ABA signalling and is related to ET signaling [135, 136]. In our experiments, in untreated infected plants at 3 hpi, an up-regulation of transcripts homologous to ATAF1/NAC2 (Ach_contig25527; Ach_contig25525) was recorded. Moreover, others transcripts (Ach_contig26632; Ach_contig19753; Ach_contig31507; Ach_contig32980; Ach_contig25010; Ach_contig25004; Ach_contig25007), homologous to NAC TFs, were also up-regulated (Fig. 8b, Additional file 19: Table S15).
Acibenzolar-S-methyl modulated a number of NAC TFs (Fig. 8b). Some NAC TFs are positive regulators of SA-mediated responses, such-as NAC090 [137] whose transcript (Ach_contig20506) was up-regulated in all ASM samples. The transcript Ach_contig20762, a homolog to ANAC100 (Arabidopsis NAC domain containing protein 100), was down-regulated in all ASM samples. Close homologs of ANAC100 are involved in ET signaling and biotic stress response [138]. Therefore, the down-regulation of this transcript in ASM plants may indicate a regulatory SA-dependent mechanism.
Finally, acibenzolar-S-methyl treatments also up-regulated a transcript homologous to ANAC042, a TF involved in the induction of camalexin biosynthesis, in Arabidopsis. Camalexin, a phytoalexin, is accumulated in response to pathogen infection through complex signaling networks, including SA-, JA-, and ET-dependent pathways, as well as glutathione status and ROS generation [139]. Phytoalexins are known to have an antimicrobial activity [140, 141].
A total of 53 WRKY TFs [142] were listed as DEGs, and among them, 23 were exclusively modulated in untreated plants at 3 hpi (Fig. 8c and d), highlighting the role of these TFs in the transcriptional reprogramming of the kiwifruit plant during Psa infection (Fig. 8c, Additional file 20: Table S16). Transcripts Ach_contig37648, Ach_contig20598, Ach_contig20597, Ach_contig37652, Ach_contig15612 and Ach_contig17244, related to the rapidly pathogen induced AtWRKY33, were up-regulated in untreated plants at 3 hpi. AtWRKY33 acts as a positive regulator of resistance toward the necrotrophic fungi Alternaria brassicicola and Botrytis cinerea [143]. In addition, its expression does not require SA-dependent signaling, but it is dependent on PAD4 [144, 145].
After P. syringae infection in Arabidopsis, WRKY33 is released in the nucleus where it induces JA\ET-related defense genes, repressing at the same time the genes related to SA-dependent defense [146]. Ach_contig20597, belonging to WRKY33, was used to confirm these data by qRT-PCR (Additional file 5: Figure S2). Moreover, WRKY33 and WRKY22 are known to activate the PAMP signaling cascade through MAPK cascade signal transduction pathways. WRKY22 activation is driven by the MAPK3/MAPK6 signal cascade and requires the cooperation of other WRKY TFs for the induction of resistance to bacterial and fungal pathogens [147]. In our experiments, Ach_contig16581 and Ach_contig14586 were annotated as WRKY22. In addition, Ach_contig22394, Ach_contig26220 Ach_contig26219, annotated as MAPK3 and MAPK6 related to MAPK3/MAPK6, were up-regulated at 3 hpi in untreated plants (Additional file 21: Table S17).
AtWRKY18 is closely related to AtWRKY40 and AtWRKY60. These three WRKY TFs interact in a complex manner to regulate plant defensive responses [148]. A transcript related to AtWRKY18 (Ach_contig11697) was identified as a DEG in ASM plants at 24 hpi and 48 hpi. Moreover, eight transcripts putatively encoding AtWRKY40 (Ach_contig15900; Ach_contig15903; Ach_contig6471; Ach_contig6470; Ach_contig7044; Ach_contig5585; Ach_contig10300; Ach_contig19510) were up-regulated at several time points following Psa inoculation in both control and ASM plants. In transgenic Arabidopsis, where AtWRKY18 was constitutively expressed, an increased resistance to P. syringae was observed. On the other hand, its co-expression with WRKY40 and WRKY60 has a redundant function and negatively regulates the resistance to P. syringae [149].
The acibenzolar-S-methyl treatment acts on a class of WRKY TFs more related to SAR response, modulating distinct transcripts from those observed in infected untreated plants. In detail, five transcripts (Ach_contig20526; Ach_contig20525; Ach_contig22553; Ach_contig22554; Ach_contig16016), related to AtWRKY70 (Additional file 5: Figure S2 and Additional file 20: Table S16), were reported as DEGs and up-regulated specifically after acibenzolar-S-methyl treatment (Fig. 8c). Among them, Ach_contig20526 was used to confirm these data by qRT-PCR (Additional file 5: Figure S2). AtWRKY70, similarly to WRKY51 and WRKY50, acts as a positive regulator of SA-dependent responses and as a negative regulator of JA-dependent responses [150, 151]. Moreover, AtWRKY70, together with its closely related AtWRKY53, plays a key role in the regulation of SAR and innate immunity in Arabidopsis [137]. Recent evidence suggests that AtWRKY70, and other WRKY TFs, are the targets of Nonexpresser of PR proteins NPR1, a key regulator of SA-dependent defenses and SAR [152]. In the kiwifruit plant-Psa interaction, three transcripts (Ach_contig16947; Ach_contig20013; Ach_contig20015), related to WRKY50 and WRKY51, were observed as DEGs and their expressions were preferentially linked to acibenzolar-S-methyl treatment (Fig. 8c). In this view, our results suggest that WRKY TF may be involved in the defense response to Psa and may be associated with ET and JA signaling in kiwifruit plants.
Network analysis WGCNA
Another perspective on this transcriptomic analysis was given by weighted gene co-expression network analysis (WGCNA). WGCNA allows to get a better understanding of which genes within this plant-pathogen interaction signalling networks, were the most connected hubs. Twenty-one modules were detected, assigned colour names and correlated to Psa inoculation and ASM treatment effects over the time points.
The kME (module eigengene-based connectivity) measure was calculated for each gene to every twenty-one modules with the score ranging between 1 (perfectly positively correlated) to − 1 (perfectly negatively correlated). kME scores were computed for each module in order to detect genes which can act as a hub in more than one network. The most interesting module detected in our analysis was “darkorange2” which is highly correlated (0.98; Pval 5e-12) with the ASM treatment (Additional file 22: Figure S5). Moreover, “darkorange2” shown positive correlation along the time course of inoculated ASM plants but negative correlation with Psa inoculated untreated plants.
The top hub of “darkorange2” network was an inactive leucine-rich repeat receptor-like protein kinase; several other leucine-rich repeat receptor-like were detected in this module indicating the importance of these classes of genes in the defense response [153]. About 222 hubs transcripts involved in the defense response with a kME score greater than 0.90 were detected in this module (Additional file 23: Table S18). About 179 of them were identified as DEGs in several of the comparisons discussed above. Among them, Ach_contig19122, which encoded thaumatin-like protein, was differentially expressed in ASM plants along the time course and was positively correlated with “darkorange2”. In contrast, Ach_contig19122 was negatively correlated with the “skyblue2” network which represents a module preferentially associated with the Psa infection in untreated plants (Additional file 22: Figure S5). Also Ach_contig26479, encoding actinidin act2d, and Ach_contig7693, encoding a class I chitinase, corroborated this observation (Additional file 23: Table S18). WGCNA also confirmed a clear separation by treatment and infection (Additional file 22: Figure S5).
Conclusions
In this study, we characterized the transcriptome of A. chinensis var. chinensis and identified genes differentially expressed after infection with Psa in plants treated and not treated with acibenzolar-S-methyl. Inoculation by Psa led to a large plant response in the initial phase with 1596 genes being over expressed and 1152 being repressed at 3 hpi. Nevertheless, this response was much reduced at 24 hpi and 48 hpi when the number of DEGs was only 272 and 341 respectively. Thus, while endophytic populations of Psa increased rapidly, the plants’ reactions were relatively muted. It is interesting to speculate whether this limited reaction to Psa infection is a consequence or a cause of Psa invading the plant tissues.
Some clues for answering this question might be found comparing gene expression in plants treated or not treated with acibenzolar-S-methyl prior to inoculation. In non-infected plants acibenzolar-S-methyl modulated the expression of 475 genes including an up-regulation of several PRRs, defense-related genes (e.g. NBS-LRR genes and Actinidin) and genes involved in the SA pathway (e.g. NIMIN2 and EDS1). The expression of genes involved in the JA/ET pathways was mostly unchanged: these results were expected since acibenzolar-S-methyl is known to elicit the SA pathway. In ASM plants the initial reaction (3 hpi) was not as pronounced as in the acibenzolar-S-methyl-untreated ones but it grew stronger with time (from 510 DEGs at 3 hpi to 1374 and 1252 DEGs at 24 hpi and 48 hpi, respectively). Notwithstanding, at 24 hpi or 48 hpi the DEGs in the ASM plants were largely different from those found in the acibenzolar-S-methyl-untreated ones with only 46 DEGs in common between acibenzolar-S-methyl-untreated (24 hpi or 48 hpi) and -treated without inoculation. A larger overlap was found in ASM plants before and after inoculation (acibenzolar-S-methyl-treated vs. -treated and inoculated at 24 hpi or 48 hpi 282 DEGs). Among the genes induced by acibenzolar-S-methyl, we found PRR genes and those involved in ROS detoxification (e.g. catalase and superoxide dismutase) or in the SA elicitation pathway (e.g. EDR1 and AP2/ERF). In addition, none of the genes involved in JA (e.g. AOC, JAZ proteins) or ET (e.g. EDR1, ACS3, SAMS, HMT) elicitation pathways that were up-regulated at 3 hpi in acibenzolar-S-methyl-untreated plants, were found to be DEGs in acibenzolar-S-methyl-treated samples.
A number of the genes up-regulated after acibenzolar-S-methyl treatment, that were involved in or contributed to the SA elicitation pathway, were further up-regulated after inoculation (e.g. EDS1, NIMIN2). A similar profile was found for several TFs such as WRKY40 and WRKY70. These findings suggest that the expression of these genes is primed by the acibenzolar-S-methyl treatment.
Genes involved in the ET pathway (e.g. AdERF11–14, HMT and SAMS) as well as genes involved in the JA pathway (e.g. AOC and OPCL1) were up-regulated in acibenzolar-S-methyl-untreated plants at 3 hpi. Since the JA and the ET pathways are antagonistic to the SA pathway, which is known to limit Psa infection [12], the induction of SA by acibenzolar-S-methyl treatment might contribute to the resistant phenotype. Inoculation of untreated plants also leads to increased expression of ERF genes also involved in the ET pathway. In contrast, inoculation of ASM plants leads to over-expression of genes involved in the SA pathway but not those involved in the ET or JA pathway.
The consistent picture emerging from this study suggests that the host response is partly dictated by the pathogen, which reduces the defense capacity of the plant, as long as within 24 hpi the affected cells are unable to prevent bacterial multiplication and therefore disease. In contrast, in ASM plants a different molecular response primed by acibenzolar-S-methyl treatment blocks Psa multiplication, thus preventing the disease symptoms. Moreover, these results elucidate and confirm the mechanisms of Psa control strategies in open field based on acibenzolar-S-methyl.
Some cultivars of A. chinensis var. deliciosa, e.g. ‘Hayward’, are reported to show a stronger reaction to acibenzolar-S-methyl treatment than the cultivar used in this study [12]; it would be interesting to compare gene expression of such cultivar with that presented in this study.
This study not only gave us a better understanding of the early interaction between Psa and its host, but it also indicates new avenues for the selection of novel elicitors and selection of kiwifruit genotypes which will respond well to those elicitors or selection of kiwifruit genotypes which will not respond to Psa manipulation.
Abbreviations
- ABA:
-
Abscisic acid
- ABI:
-
Abscisic acid insensitive
- ABRE:
-
ABA-responsive element
- ACO:
-
1-amino cyclopropane-1-carboxylate oxidase
- AOS:
-
Allene oxide synthase
- AP2/ERF:
-
APETALA 2/ethylene-responsive element binding factor
- ASM:
-
Plants treated with acibenzolar-S-methyl not inoculated with Psa
- ASM.I3, ASM.I24, ASM.I48:
-
Plants treated with acibenzolar-S-methyl and then inoculated with Psa at 3, 24, 48 h post inoculation
- CaM:
-
Calmodulin
- CLSM:
-
Confocal laser scanning micrographs
- COI:
-
Coronatine insensitive
- DEGs:
-
Differentially expressed genes
- EDS:
-
Enhanced disease susceptibility
- EFE:
-
2-oxoglutarate-dependent ethylene-forming enzyme
- EIL:
-
Ethylene insensitive like
- EIN:
-
Ethylene insensitive
- ET:
-
Ethylene
- ETI:
-
Effector-triggered immunity
- FDR:
-
False discovery rate
- FMO1:
-
Flavin-contain monooxigenase1
- HC:
-
Healthy Control plants
- hpi:
-
Hour post inoculation
- HR:
-
Hypersensitive response
- I3, I24, I48:
-
Inoculated plants, 3, 24, 48 h post-inoculation
- JA:
-
Jasmonic acid
- JAZ:
-
Jasmonate-zim domain
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- kME:
-
Module eigengene-based connectivity
- log2fc:
-
Log2 fold change
- LOX:
-
Lipoxygenase
- LRR:
-
Leucine rich-repeat
- LRR-RLKs:
-
Leucine-rich repeat receptor-like protein kinases
- MAMP/PAMP:
-
Microbe/pathogen-associated molecular pattern
- MAPK:
-
Mitogen-activated protein kinase
- MeJA:
-
Methyl jasmonic acid
- NAC:
-
No apical meristem, ATAF1/2, CUP-SHAPED COTYLEDON 2
- NBS-LRR:
-
Nucleotide-binding site leucine-rich repeat
- NCED:
-
9-cis-epoxycarotenoid dioxygenase
- NPR:
-
Nonexpresser of PR genes
- PAD:
-
Phytoalexin deficient
- PDF:
-
Plant defensin
- PP2C:
-
Type 2C protein phosphatase
- PR:
-
Pathogenesis related
- Psa:
-
Pseudomonas syringae pv. actinidiae
- PTI :
-
PAMP-triggered immunity
- PYR/PYL/RCAR:
-
Pyrabactin resistance/PY- like/regulatory component of ABA receptor
- qRT-PCR:
-
quantitative reverse transcription PCR
- R-genes:
-
Resistance genes
- RLK:
-
Receptor Like Kinases
- RNAseq:
-
RNA-Sequencing
- ROS:
-
Reactive oxygen species
- SA:
-
Salicylic acid
- SAR:
-
Systemic acquired resistance
- SnRK:
-
SNF1-related protein kinase
- TFs:
-
Transcription factors
- TLPs:
-
Thaumatin-like proteins
- WGCNA:
-
Weighted gene coexpression network analysis
- WRKY:
-
Transcription factor containing a highly conserved WRKY domain
References
Ferguson AR. Kiwifruit in the world – 2014. Acta Hortic. 2015;1096:33–46.
Balestra GM, Mazzaglia A, Quattrucci A, Renzi M, Rossetti A. Current status of bacterial canker spread on kiwifruit in Italy. Aust Plant Dis Notes. 2009;4(1):34–6.
Vanneste JL, Yu J, Cornish DA, Tanner DJ, Windner R, Chapman JR, et al. Identification, virulence and distribution of two biovars of Pseudomonas syringae pv. actinidiae in New Zealand. Plant Dis. 2013;97:708–19.
Gao QM, Zhu S, Kachroo P, Kachroo A. Signal regulators of systemic acquired resistance. Front Plant Sci. 2015;6:228.
Fujikawa T, Sawada H. Genome analysis of the kiwifruit canker pathogen Pseudomonas syringae pv. actinidiae biovar 5. Sci Rep. 2016;6:21399.
Sawada H, Kondo K, Nakaune R. Novel biovar (biovar 6) of Pseudomonas syringae pv. actinidiae causing bacterial canker of kiwifruit (Actinidia deliciosa) in Japan. Jpn J Physiol. 2016;82:101–15.
Vanneste JL. The scientific, economic, and social impacts of the New Zealand outbreak of bacterial canker of kiwifruit (Pseudomonas syringae pv. actinidiae). Annu Rev Phytopathol. 2017;55(1):377–99.
Cunty A, Poliakoff F, Rivoal C, Cesbron S, Fischer-Le Saux M, Lemaire C, et al. Characterization of Pseudomonas syringae pv. actinidiae (Psa) isolated from France and assignment of Psa biovar 4 to a de novo pathovar: Pseudomonas syringae pv. actinidifoliorum pv. nov. Plant Pathol. 2015;64:582–96.
Butler MI, Stockwell PA, Black MA, Day RC, Lamont IL, Poulter RTM. Pseudomonas syringae pv. actinidiae from recent outbreaks of kiwifruit bacterial canker belong to different clones that originated in China. PLoS ONE. 2013;8(2):e57464.
Donati I, Buriani G, Cellini A, Mauri S, Costa G, Spinelli F. New insights on the bacterial canker of kiwifruit (Pseudomonas syringae pv. actinidiae). J Berry Res. 2014;4:53–67.
Collina M, Donati I, Bertacchini E, Brunelli A, Spinelli F. Greenhouse assays on the control of the bacterial canker of kiwifruit (Pseudomonas syringae pv. Actinidiae). J Berry Res. 2016;6(4):407–15.
Cellini A, Fiorentini L, Buriani G, Yu J, Donati I, Cornish DA, et al. Elicitors of the salicylic acid pathway reduce incidence of bacterial canker of kiwifruit caused by Pseudomonas syringae pv.actinidae. Ann Appl Biol. 2014;165:441–53.
Li J, Brader G, Palva ET. The WRKY70 transcription factor: a node of convergence for jasmonate-mediated and salicylate-mediated signals in plant defense. Plant Cell. 2004;16(2):319–31.
Couto D, Zipfel C. Regulation of pattern recognition receptor signalling in plants. Nat Rev Immunol. 2016;16:537.
Macho A, Zipfel C: Plant PRRs and the activation of innate immune signaling. vol. 54; 2014.
Moore JW, Loake GJ, Spoel SH. Transcription dynamics in plant immunity. Plant Cell Online. 2011;23(8):2809–20.
Kazan K, Lyons R. Intervention of phytohormone pathways by pathogen effectors. Plant Cell. 2014;26(6):2285–309.
Pieterse CM, Van der Does D, Zamioudis C, Leon-Reyes A, Van Wees SC. Hormonal modulation of plant immunity. Annu Rev Cell Dev Biol. 2012;28:489–521.
Shigenaga AM, Berens ML, Tsuda K, Argueso CT. Towards engineering of hormonal crosstalk in plant immunity. Curr Opin Plant Biol. 2017;38:38164–72.
Koornneef A, Pieterse CM. Cross talk in defense signaling. Plant Physiol. 2008;146(3):839–44.
Uppalapati SR, Ishiga Y, Wangdi T, Kunkel BN, Anand A, Mysore KS, et al. The phytotoxin coronatine contributes to pathogen fitness and is required for suppression of salicylic acid accumulation in tomato inoculated with Pseudomonas syringae pv. tomato DC3000. Mol Plant-Microbe Interact. 2007;20(8):955–65.
Corsi B, Forni C, Riccioni L, Linthorst JMH. Enhancement of PR1 and PR5 gene expressions by chitosan treatment in kiwifruit plants inoculated with Pseudomonas syringae pv. actinidiae. Eur J Plant Pathol. 2017;147:471–89.
Wurms KV, Gould E, Ah Chee A, Taylor J, Curran B, Reglinski T. Elicitor induction of defence genes and reduction of bacterial canker in kiwifruit. New Zealand Plant Protection. 2017;70:272–84.
Wang T, Wang G, Jia ZH, Pan DL, Zhang JY, Guo ZR. Transcriptome analysis of kiwifruit in response to Pseudomonas syringae pv. actinidiae infection. Int J Mol Sci. 2018;19(2)
Jing Z, Liu Z. Genome-wide identification of WRKY transcription factors in kiwifruit (Actinidia spp.) and analysis of WRKY expression in responses to biotic and abiotic stresses. Genes Genomics. 2018;40(4):429-46. https://doi.org/10.1007/s13258-017-0645-1. Epub 2018 Jan 6.
Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant. 1962;15(3):473–97.
Spinelli F, Speakman J-B, Rademacher W, Halbwirth H, Stich K, Costa G. Luteoforol, a flavan 4-ol, is induced in pome fruits by prohexadione-calciumand shows phytoalexin-like properties against Erwinia amylovora and other plant pathogens. Eur J Plant Pathol. 2005;112(2):133–42.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–9.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4(1):41.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14.
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, et al. Mapman: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37(6):914–39.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559.
Song L, Langfelder P, Horvath S. Comparison of co-expression measures: mutual information, correlation, and model based indices. BMC Bioinf. 2012;13:328.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–8.
Boller T, Felix G. A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu Rev Plant Biol. 2009;60:379–406.
Tsuda K, Sato M, Glazebrook J, Cohen JD, Katagiri F. Interplay between MAMP-triggered and SA-mediated defense responses. Plant J. 2008;53(5):763–75.
Mishina TE, Zeier J. Pathogen-associated molecular pattern recognition rather than development of tissue necrosis contributes to bacterial induction of systemic acquired resistance in Arabidopsis. Plant J. 2007;50(3):500–13.
Willmann R, Lajunen HM, Erbs G, Newman M-A, Kolb D, Tsuda K, et al. Arabidopsis lysin-motif proteins LYM1 LYM3 CERK1 mediate bacterial peptidoglycan sensing and immunity to bacterial infection. Proc Natl Acad Sci. 2011;108(49):19824–9.
Shinya T, Motoyama N, Ikeda A, Wada M, Kamiya K, Hayafune M, et al. Functional characterization of CEBiP and CERK1 homologs in Arabidopsis and rice reveals the presence of different chitin receptor systems in plants. Plant Cell Physiol. 2012;53(10):1696–706.
Galletti R, De Lorenzo G, Ferrari S. Host-derived signals activate plant innate immunity. Plant Signal Behav. 2009;4(1):33–4.
Brutus A, Sicilia F, Macone A, Cervone F, De Lorenzo G. A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides. Proc Natl Acad Sci. 2010;107(20):9452–7.
Ferrari S, Savatin D, Sicilia F, Gramegna G, Cervone F, De Lorenzo G. Oligogalacturonides: plant damage-associated molecular patterns and regulators of growth and development. Front Plant Sci. 2013;4:49.
Kohorn B, Kohorn S. The cell wall-associated kinases, WAKs, as pectin receptors. Front Plant Sci. 2012;3:88.
Meier S, Ruzvidzo O, Morse M, Donaldson L, Kwezi L, Gehring C. The Arabidopsis wall associated kinase-like 10 gene encodes a functional guanylyl cyclase and is co-expressed with pathogen defense related genes. PLoS One. 2010;5(1):e8904.
He ZH, He D, Kohorn BD. Requirement for the induced expression of a cell wall associated receptor kinase for survival during the pathogen response. Plant J. 1998;14(1):55–63.
Harkenrider M, Sharma R, De Vleesschauwer D, Tsao L, Zhang X, Chern M, et al. Overexpression of rice wall-associated kinase 25 (OsWAK25) alters resistance to bacterial and fungal pathogens. PLoS One. 2016;11(1):e0147310.
Rosli HG, Zheng Y, Pombo MA, Zhong S, Bombarely A, Fei Z, et al. Transcriptomics-based screen for genes induced by flagellin and repressed by pathogen effectors identifies a cell wall-associated kinase involved in plant immunity. Genome Biol. 2013;14(12):R139.
Yang K, Qi L, Zhang Z. Isolation and characterization of a novel wall-associated kinase gene TaWAK5 in wheat (Triticum aestivum). The Crop Journal. 2014;2(5):255–66.
Kessler SA, Shimosato-Asano H, Keinath NF, Wuest SE, Ingram G, Panstruga R, et al. Conserved molecular components for pollen tube reception and fungal invasion. Science. 2010;330(6006):968–71.
Keinath NF, Kierszniowska S, Lorek J, Bourdais G, Kessler SA, Shimosato-Asano H, et al. PAMP (pathogen-associated molecular pattern)-induced changes in plasma membrane compartmentalization reveal novel components of plant immunity. J Biol Chem. 2010;285(50):39140–9.
Mao D, Yu F, Li J, BVd P, Tan D, Li J, et al. FERONIA receptor kinase interacts with S-adenosylmethionine synthetase and suppresses S-adenosylmethionine production and ethylene biosynthesis in Arabidopsis. Plant Cell Environ. 2015;38(12):2566–74.
Heese A, Hann DR, Gimenez-Ibanez S, Jones AM, He K, Li J, et al. The receptor-like kinase SERK3/BAK1 is a central regulator of innate immunity in plants. Proc Natl Acad Sci U S A. 2007;104(29):12217–22.
Chinchilla D, Zipfel C, Robatzek S, Kemmerling B, Nurnberger T, Jones JD, et al. A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature. 2007;448(7152):497–500.
Gao QM, Venugopal S, Navarre D, Kachroo A. Low oleic acid-derived repression of jasmonic acid-inducible defense responses requires the WRKY50 and WRKY51 proteins. Plant Physiol. 2011;155(1):464–76.
Liebrand TW, van den Burg HA, Joosten MH. Two for all: receptor-associated kinases SOBIR1 and BAK1. Trends Plant Sci. 2014;19(2):123–32.
Ghose K, Dey S, Barton H, Loake GJ, Basu D. Differential profiling of selected defence-related genes induced on challenge with Alternaria brassicicola in resistant white mustard and their comparative expression pattern in susceptible India mustard. Mol Plant Pathol. 2008;9(6):763–75.
Jensen PJ, Halbrendt N, Fazio G, Makalowska I, Altman N, Praul C, et al. Rootstock-regulated gene expression patterns associated with fire blight resistance in apple. BMC Genomics. 2012;13(1):9.
Pan Y, Pylatuik JD, Ouyang J, Famili AF, Fobert PR. Discovery of functional genes for systemic acquired resistance in Arabidopsis thaliana throught integrated data mining. J Bioinf Comput Biol. 2004;02(04):639–55.
Huang S, Ding J, Deng D, Tang W, Sun H, Liu D, et al. Draft genome of the kiwifruit Actinidia chinensis. Nat Commun. 2013;4:2640.
Jwa NS, Kumar Agrawal G, Rakwal R, Park CH, Prasad Agrawal V. Molecular cloning and characterization of a novel Jasmonate inducible pathogenesis-related class 10 protein gene, JIOsPR10, from rice (Oryza sativa L.) seedling leaves. Biochem Biophys Res Commun. 2001;286(5):973–83.
Yasnetskaya EG, Bulgakov VP, Gorbach VI, Shevchenko NM, Fedoreyeva LI, Zhuravlev YN, et al. Ethephon- and jasmonate-elicited pathogenesis-related ribonucleases in cultured ginseng cells. Russ J Plant Physiol. 2003;50(4):492–7.
Bahramnejad B, Goodwin PH, Zhang J, Atnaseo C, Erickson LR. A comparison of two class 10 pathogenesis-related genes from alfalfa and their activation by multiple stresses and stress-related signaling molecules. Plant Cell Rep. 2010;29(11):1235–50.
Misas-Villamil Johana C, Hoorn Renier AL, Doehlemann G. Papain-like cysteine proteases as hubs in plant immunity. New Phytol. 2016;212(4):902–7.
Gavrović-Jankulović M, Ćirković T, Vučković O, Atanasković-Marković M, Petersen A, Gojgić G, et al. Isolation and biochemical characterization of a thaumatin-like kiwi allergen. J Allergy Clin Immunol. 2002;110(5):805–10.
Suzuki N, Miller G, Morales J, Shulaev V, Torres MA, Mittler R. Respiratory burst oxidases: the engines of ROS signaling. Curr Opin Plant Biol. 2011;14(6):691–9.
Mittler R, Herr EH, Orvar BL, van Camp W, Willekens H, Inzé D, et al. Transgenic tobacco plants with reduced capability to detoxify reactive oxygen intermediates are hyperresponsive to pathogen infection. Proc Natl Acad Sci. 1999;96(24):14165–70.
Nadimpalli R, Yalpani N, Johal GS, Simmons CR. Prohibitins, stomatins, and plant disease response genes compose a protein superfamily that controls cell proliferation, ion channel regulation, and death. J Biol Chem. 2000;275(38):29579–86.
Zhou L, Cheung M-Y, Li M-W, Fu Y, Sun Z, Sun S-M, et al. Rice hypersensitive induced reaction protein 1 (OsHIR1) associates with plasma membrane and triggers hypersensitive cell death. BMC Plant Biol. 2010;10(1):290.
van Loon LC, Rep M, Pieterse CMJ. Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol. 2006;44(1):135–62.
Passardi F, Penel C, Dunand C. Performing the paradoxical: how plant peroxidases modify the cell wall. Trends Plant Sci. 2004;9(11):534–40.
Bilgin Damla D, Zavala Jorge A, Zhu JIN, Clough Steven J, Ort Donald R, De LEH. Biotic stress globally downregulates photosynthesis genes. Plant Cell Environ. 2010;33(10):1597–613.
Denoux C, Galletti R, Mammarella N, Gopalan S, Werck D, De Lorenzo G, et al. Activation of defense response pathways by OGs and Flg22 elicitors in Arabidopsis seedlings. Mol Plant. 2008;1(3):423–45.
Kerchev Pavel I, Fenton B, Foyer Christine H, Hancock RD. Plant responses to insect herbivory: interactions between photosynthesis, reactive oxygen species and hormonal signalling pathways. Plant Cell Environ. 2012;35(2):441–53.
Sugano S, Jiang CJ, Miyazawa S, Masumoto C, Yazawa K, Hayashi N, et al. Role of OsNPR1 in rice defense program as revealed by genome-wide expression analysis. Plant Mol Biol. 2010;74(6):549–62.
Dixon RA. Natural products and plant disease resistance. Nature. 2001;411:843.
Geisler K, Hughes RK, Sainsbury F, Lomonossoff GP, Rejzek M, Fairhurst S, et al. Biochemical analysis of a multifunctional cytochrome P450 (CYP51) enzyme required for synthesis of antimicrobial triterpenes in plants. Proc Natl Acad Sci. 2013;110(35):E3360–E7.
Zhao Y, Wei T, Yin KQ, Chen Z, Gu H, Qu LJ, et al. Arabidopsis RAP2.2 plays an important role in plant resistance to Botrytis cinerea and ethylene responses. New Phytol. 2012;195(2):450–60.
Glazebrook J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol. 2005;43:205–27.
Zheng Z, Qamar Synan A, Chen Z, Mengiste T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2006;48(4):592–605.
Melotto M, Mecey C, Niu Y, Chung HS, Katsir L, Yao J, et al. A critical role of two positively charged amino acids in the Jas motif of Arabidopsis JAZ proteins in mediating coronatine- and jasmonoyl isoleucine-dependent interactions with the COI1 F-box protein. Plant J. 2008;55(6):979–88.
de Torres M, Mansfield JW, Grabov N, Brown IR, Ammouneh H, Tsiamis G, et al. Pseudomonas syringae effector AvrPtoB suppresses basal defence in Arabidopsis. Plant J. 2006;47(3):368–82.
Weingart H, Ullrich H, Geider K, Volksch B. The role of ethylene production in virulence of Pseudomonas syringae pvs. Glycinea and phaseolicola. Phytopathology. 2001;91(5):511–8.
McAtee PA, Richardson AC, Nieuwenhuizen NJ, Gunaseelan K, Hoong L, Chen X, et al. The hybrid non-ethylene and ethylene ripening response in kiwifruit (Actinidia chinensis) is associated with differential regulation of MADS-box transcription factors. BMC Plant Biol. 2015;15:304.
Frye CA, Tang D, Innes RW. Negative regulation of defense responses in plants by a conserved MAPKK kinase. Proc Natl Acad Sci U S A. 2001;98(1):373–8.
Hiruma K, Nishiuchi T, Kato T, Bednarek P, Okuno T, Schulze-Lefert P et al: Arabidopsis ENHANCED DISEASE RESISTANCE 1 is required for pathogen-induced expression of plant defensins in nonhost resistance, and acts through interference of MYC2-mediated repressor function. vol. 67; 2011.
Binder BM, Walker JM, Gagne JM, Emborg TJ, Hemmann G, Bleecker AB, et al. The Arabidopsis EIN3 binding F-box proteins EBF1 and EBF2 have distinct but overlapping roles in ethylene signaling. Plant Cell. 2007;19(2):509–23.
Liu R, Chen L, Jia Z, Lü B, Shi H, Shao W, et al. Transcription factor AtMYB44 regulates induced expression of the ETHYLENE INSENSITIVE2 gene in Arabidopsis responding to a harpin protein. Bull Am Phys Soc. 2011;24(3):377–99.
Alonso JM, Hirayama T, Roman G, Nourizadeh S, Ecker JR. EIN2, a bifunctional transducer of ethylene and stress responses in Arabidopsis. Science. 1999;284(5423):2148–52.
Wasternack C, Song S. Jasmonates: biosynthesis, metabolism, and signaling by proteins activating and repressing transcription. J Exp Bot. 2017;68(6):1303–21.
Niu Y, Figueroa P, Browse J. Characterization of JAZ-interacting bHLH transcription factors that regulate jasmonate responses in Arabidopsis. J Exp Bot. 2011;62(6):2143–54.
Qi T, Song S, Ren Q, Wu D, Huang H, Chen Y, et al. The Jasmonate-ZIM-domain proteins interact with the WD-repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell. 2011;23(5):1795–814.
Song S, Huang H, Gao H, Wang J, Wu D, Liu X, et al. Interaction between MYC2 and ETHYLENE INSENSITIVE3 modulates antagonism between jasmonate and ethylene signaling in Arabidopsis. Plant Cell. 2014;26(1):263–79.
Gao M, Wang X, Wang D, Xu F, Ding X, Zhang Z, et al. Regulation of cell death and innate immunity by two receptor-like kinases in Arabidopsis. Cell Host Microbe. 2009;6(1):34–44.
Seyfferth C, Tsuda K. Salicylic acid signal transduction: the initiation of biosynthesis, perception and transcriptional reprogramming. Front Plant Sci. 2014;5:697.
Feys BJ, Moisan LJ, Newman M-A, Parker JE. Direct interaction between the Arabidopsis disease resistance signaling proteins, EDS1 and PAD4. EMBO J. 2001;20(19):5400–11.
Groen SC, Whiteman NK, Bahrami AK, Wilczek AM, Cui J, Russell JA, et al. Pathogen-triggered ethylene signaling mediates systemic-induced susceptibility to herbivory in Arabidopsis. Plant Cell. 2013;25(11):4755–66.
Heck S, Grau T, Buchala A, Métraux JP, Nawrath C. Genetic evidence that expression of NahG modifies defence pathways independent of salicylic acid biosynthesis in the Arabidopsis–Pseudomonas syringae pv. Tomato interaction. Plant J. 2003;36(3):342–52.
Fu ZQ, Yan S, Saleh A, Wang W, Ruble J, Oka N, et al. NPR3 and NPR4 are receptors for the immune signal salicylic acid in plants. Nature. 2012;486(7402):228–32.
Moreau M, Tian M, Klessig DF. Salicylic acid binds NPR3 and NPR4 to regulate NPR1-dependent defense responses. Cell Res. 2012;22(12):1631–3.
Weigel RR, Pfitzner UM, Gatz C. Interaction of NIMIN1 with NPR1 modulates PR gene expression in Arabidopsis. Plant Cell. 2005;17(4):1279–91.
Montillet JL, Leonhardt N, Mondy S, Tranchimand S, Rumeau D, Boudsocq M, et al. An abscisic acid-independent oxylipin pathway controls stomatal closure and immune defense in Arabidopsis. PLoS Biol. 2013;11(3):e1001513.
Mori IC, Pinontoan R, Kawano T, Muto S. Involvement of superoxide generation in salicylic acid-induced stomatal closure in Vicia faba. Plant Cell Physiol. 2001;42(12):1383–8.
LarquÉ-Saavedra A. The antiranspirant effect of acetylsalcylic acid on Phaseolus vulgaris. Physiol Plant. 1978;43(2):126–8.
Khokon Md. Atiqur R, Okuma E, Hossain Mohammad A, Munemasa S, Uraji M, Nakamura Y, et al. Involvement of extracellular oxidative burst in salicylic acid-induced stomatal closure in Arabidopsis. Plant, Cell Environ. 2011;34(3):434–43.
Hao YJ, Wei W, Song QX, Chen HW, Zhang YQ, Wang F, et al. Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. 2011;68(2):302–13.
Ton J, Flors V, Mauch-Mani B. The multifaceted role of ABA in disease resistance. Trends Plant Sci. 2009;14(6):310–7.
Mohr PG, Cahill DM. Suppression by ABA of salicylic acid and lignin accumulation and the expression of multiple genes, in Arabidopsis infected with Pseudomonas syringae pv. tomato. Funct Integr Genomics. 2007;7(3):181–91.
Thaler JS, Bostock RM. Interactions between abscisic-acid-mediated responses and plant resistance to pathogens and insects. Ecology. 2004;85(1):48–58.
Yasuda M, Ishikawa A, Jikumaru Y, Seki M, Umezawa T, Asami T, et al. Antagonistic interaction between systemic acquired resistance and the abscisic acid-mediated abiotic stress response in Arabidopsis. Plant Cell. 2008;20(6):1678–92.
De Torres ZM, Bennett Mark H, Truman William H, Grant MR. Antagonism between salicylic and abscisic acid reflects early host–pathogen conflict and moulds plant defence responses. Plant J. 2009;59(3):375–86.
Munemasa S, Hauser F, Park J, Waadt R, Brandt B, Schroeder JI. Mechanisms of abscisic acid-mediated control of stomatal aperture. Curr Opin Plant Biol. 2015;28:154–62.
Weiner JJ, Peterson FC, Volkman BF, Cutler SR. Structural and functional insights into core ABA signaling. Curr Opin Plant Biol. 2010;13(5):495–502.
Miyakawa T, Fujita Y, Yamaguchi-Shinozaki K, Tanokura M. Structure and function of abscisic acid receptors. Trends Plant Sci. 2013;18(5):259–66.
Park SY, Fung P, Nishimura N, Jensen DR, Fujii H, Zhao Y, et al. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science. 2009;324(5930):1068–71.
Ma Y, Szostkiewicz I, Korte A, Moes D, Yang Y, Christmann A, et al. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science. 2009;324(5930):1064–8.
Nishimura N, Sarkeshik A, Nito K, Park SY, Wang A, Carvalho PC, et al. PYR/PYL/RCAR family members are major in-vivo ABI1 protein phosphatase 2C-interacting proteins in Arabidopsis. Plant J. 2010;61(2):290–9.
Tanaka Y, Sano T, Tamaoki M, Nakajima N, Kondo N, Hasezawa S. Ethylene inhibits abscisic acid-induced stomatal closure in Arabidopsis. Plant Physiol. 2005;138(4):2337–43.
Pandey GK, Grant JJ, Cheong YH, Kim BG, Li L, Luan S. ABR1, an APETALA2-domain transcription factor that functions as a repressor of ABA response in Arabidopsis. Plant Physiol. 2005;139(3):1185–93.
Kim S, Choi HI, Ryu HJ, Park JH, Kim MD, Kim SY. ARIA, an Arabidopsis arm repeat protein interacting with a transcriptional regulator of abscisic acid-responsive gene expression, is a novel abscisic acid signaling component. Plant Physiol. 2004;136(3):3639–48.
Casaretto J. The transcription factors HvABI5 and HvVP1 are required for the abscisic acid induction of gene expression in barley aleurone cells. Plant Cell Online. 2002;15(1):271–84.
Lopez-Molina L, Mongrand S, Chua NH. A postgermination developmental arrest checkpoint is mediated by abscisic acid and requires the ABI5 transcription factor in Arabidopsis. Proc Natl Acad Sci U S A. 2001;98(8):4782–7.
Licausi F, Ohme-Takagi M, Perata P. APETALA2/ethylene responsive factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs. New Phytol. 2013;199(3):639–49.
X-r Y, Allan AC, Xu Q, Burdon J, Dejnoprat S, Chen K-s, et al. Differential expression of kiwifruit ERF genes in response to postharvest abiotic stress. Postharvest Biol Technol. 2012;66:1–7.
Nakano T, Suzuki K, Ohtsuki N, Tsujimoto Y, Fujimura T, Shinshi H. Identification of genes of the plant-specific transcription-factor families cooperatively regulated by ethylene and jasmonate in Arabidopsis thaliana. J Plant Res. 2006;119(4):407–13.
Cheng MC, Liao PM, Kuo WW, Lin TP. The Arabidopsis ETHYLENE RESPONSE FACTOR1 regulates abiotic stress-responsive gene expression by binding to different cis-acting elements in response to different stress signals. Plant Physiol. 2013;162(3):1566–82.
Zhang X, Dong J, Liu H, Wang J, Qi Y, Liang Z. Transcriptome sequencing in response to salicylic acid in Salvia miltiorrhiza. PLoS One. 2016;11(1):e0147849.
Park JM, Park C-J, Lee S-B, Ham B-K, Shin R, Paek K-H. Overexpression of the tobacco Tsi1 gene encoding an EREBP/AP2–type transcription factor enhances resistance against pathogen attack and osmotic stress in tobacco. Plant Cell. 2001;13(5):1035–46.
Shin R, Park JM, An J-M, Paek K-H. Ectopic expression of Tsi1 in transgenic hot pepper plants enhances host resistance to viral, bacterial, and oomycete pathogens. Mol Plant-Microbe Interact. 2002;15(10):983–9.
Nakashima K, Yamaguchi-Shinozaki K, Shinozaki K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front Plant Sci. 2014;5:170.
Nuruzzaman M, Sharoni AM, Kikuchi S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front Microbiol. 2013;4:248.
Wang X, Basnayake BMVS, Zhang H, Li G, Li W, Virk N, et al. The Arabidopsis ATAF1, a NAC transcription factor, is a negative regulator of defense responses against necrotrophic fungal and bacterial pathogens. Mol Plant-Microbe Interact. 2009;22(10):1227–38.
Jensen Michael K, Hagedorn Peter H, De Torres-Zabala M, Grant Murray R, Rung Jesper H, Collinge David B, et al. Transcriptional regulation by an NAC (NAM–ATAF1,2–CUC2) transcription factor attenuates ABA signalling for efficient basal defence towards Blumeria graminis f. Sp. hordei in Arabidopsis. Plant J. 2008;56(6):867–80.
Wang D, Amornsiripanitch N, Dong X. A genomic approach to identify regulatory nodes in the transcriptional network of systemic acquired resistance in plants. PLoS Pathog. 2006;2(11):e123.
Voitsik AM, Muench S, Deising HB, Voll LM. Two recently duplicated maize NAC transcription factor paralogs are induced in response to Colletotrichum graminicola infection. BMC Plant Biol. 2013;13:85.
Glawischnig E. The role of cytochrome P450 enzymes in the biosynthesis of camalexin. Biochem Soc Trans. 2006;34(6):1206–8.
Saga H, Ogawa T, Kai K, Suzuki H, Ogata Y, Sakurai N, et al. Identification and characterization of ANAC042, a transcription factor family gene involved in the regulation of camalexin biosynthesis in Arabidopsis. Mol Plant-Microbe Interact. 2012;25(5):684–96.
Spinelli F, Donati I, Vanneste JL, Costa M, Costa G. Real time monitoring of the interactions between Pseudomonas syringae pv. actinidiae and Actinidia species. Acta Hortic. 2011;913:461–6.
Eulgem T, Somssich IE. Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol. 2007;10(4):366–71.
Zheng XY, Spivey NW, Zeng W, Liu PP, Fu ZQ, Klessig DF, et al. Coronatine promotes Pseudomonas syringae virulence in plants by activating a signaling cascade that inhibits salicylic acid accumulation. Cell Host Microbe. 2012;11(6):587–96.
Birkenbihl RP, Kracher B, Somssich IE. Induced genome-wide binding of three Arabidopsis WRKY transcription factors during early MAMP-triggered immunity. Plant Cell. 2017;29(1):20–38.
Lippok B, Birkenbihl RP, Rivory G, Brümmer J, Schmelzer E, Logemann E, et al. Expression of AtWRKY33 encoding a pathogen- or PAMP-responsive WRKY transcription factor is regulated by a composite DNA motif containing W box elements. Mol Plant-Microbe Interact. 2007;20(4):420–9.
Qiu JL, Fiil BK, Petersen K, Nielsen HB, Botanga CJ, Thorgrimsen S, et al. Arabidopsis MAP kinase 4 regulates gene expression through transcription factor release in the nucleus. EMBO J. 2008;27(16):2214–21.
Asai T, Tena G, Plotnikova J, Willmann MR, Chiu W-L, Gomez-Gomez L, et al. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature. 2002;415:977.
Birkenbihl RP, Diezel C, Somssich IE. Arabidopsis WRKY33 is a key transcriptional regulator of hormonal and metabolic responses toward Botrytis cinerea infection. Plant Physiol. 2012;159(1):266–85.
Xu X, Chen C, Fan B, Chen Z. Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell. 2006;18(5):1310–26.
Li B, Meng X, Shan L, He P. Transcriptional regulation of pattern-triggered immunity in plants. Cell Host Microbe. 2016;19(5):641–50.
Gao X, Huang Q, Zhao Z, Han Q, Ke X, Qin H, et al. Studies on the infection, colonization, and movement of Pseudomonas syringae pv. actinidiae in kiwifruit tissues using a GFPuv-labeled strain. PLoS ONE. 2016;11(3):e0151169.
Ishihama N, Yoshioka H. Post-translational regulation of WRKY transcription factors in plant immunity. Curr Opin Plant Biol. 2012;15(4):431–7.
Marone D, Russo MA, Laidò G, De Leonardis AM, Mastrangelo AM. Plant nucleotide binding site-leucine-rich repeat (NBS-LRR) genes: active guardians in host defense responses. Int J Mol Sci. 2013;14(4):7302–26.
Acknowledgements
We thank Lorenzo Tosi (Agrea Centro Studi, Verona, Italy), Graziano Vittone (Agrion, Cuneo, Italy), Loredana Antoniacci (Phytosanitary Service Emilia-Romagna Region, Italy) and Sauro Graziani (Agrintesa, Ravenna, Italy) for the discussion about Psa control strategies in open field.
Funding
The study was supported by Italian MIPAAF projects INTERACT (D.M. 26045–5/12/2011) and ARDICA (D.M. 27234/7303/2011) for the experiments set up, plants treatments, mRNA extraction and part of related personnel cost; EU project DROPSA (FP7-KBBE-2013-7, number 613678) for CLSM analysis, mRNA sequencing, data analysis and interpretation, experiments for qPCR validations, writing the manuscript and part of related personnel cost.
Availability of data and materials
RNA-seq data of this study have been deposited at the NCBI under the BioProject ID PRJNA436459 and Biosample: SAMN08625669, SAMN08625670, SAMN08625671, SAMN08625672, SAMN08625673, SAMN08625674, SAMN08625675, SAMN08625676, SAMN08625677, SAMN08625678, SAMN08625679, SAMN08625680, SAMN08625681, SAMN08625682, SAMN08625683, SAMN08625684. Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GGMO00000000. The version described in this paper is the first version, GGMO01000000.
Author information
Authors and Affiliations
Contributions
GT, FS, VM, JLV conceived and designed the experiments; FS, GB, AC, provided all the genetic materials. GB, ID, GT carried out the artificial inoculations. VM performed the RNA extraction and library preparations. AL, VM, LO performed bioinformatics analyses. LO carried out the sequencing. VM carried out data analysis, qRT-PCR analysis and WGCNA, and drafted the manuscript. FS carried out CLSM. FS, GT supervised the experiments. FS, GT, LC, JLV, VM revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The biological material was provided by the Department of Agricultural Sciences Alma Mater Studiorum -University of Bologna according to national (D.L. 206/2001; D.L. 224/2003) and European (98/81/CE) regulation. The biological material don’t belong to species at risk of extinction or wild fauna and flora.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential competing of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. List of primers used for qRT-PCR of selected differentially expressed genes (DEGs). (XLSX 13 kb)
Additional file 2:
Table S2. Summary of annotations of the Actinidia chinensis var. chinensis transcriptome. (DOC 66 kb)
Additional file 3:
Figure S1. Main characteristics of the annotation of the Actinidia chinensis var. chinensis reference transcriptome. Panel A: contig distribution by length. Panel B: contigs distribution by E-value. Panel C: contig distribution by similarity. Panel D The contigs of the A. chinensis var. chinensis reference transcriptome have been classified in Clusters of Orthologous Groups (COGs) functional annotations and organized into 24 function categories. Panel E Most relevant functional categories of the Groups of Orthologs obtained with the A. chinensis var. chinensis transcriptome. Venn diagrams illustrating the distribution of similarity search results made with the A. chinensis var. chinensis transcriptome. In panel F, the BLAST results against NR protein, NR nucleotide, RefSeq and SwissProt databases are reported. The total number of annotations obtained from these databases was 29,583 (74.73% of total contigs). In panel G are shown the annotation results obtained from COGs, InterProScan and KEGG. The number of contigs showing a hit against these databases was 26,353 (66.57% of total contigs). Panel H reports the intersection among the annotations presented in A and B with the results obtained from the BLASTx search against the Kiwifruit Genome protein database. Summarizing the results obtained from all the queried databases, a total of 34,039 contigs (85.99% of total contigs) were annotated. (TIF 2577 kb)
Additional file 4:
Table S3. List of all differentially expressed genes (DEGs) of Actinidia chinensis var. chinensis modulated in response to Pseudomonas syringae pv. actinidiae inoculation both in acibenzolar-S-methyl treated and untreated plants. (XLSX 1256 kb)
Additional file 5:
Figure S2. qRT-PCR analysis was employed to validate the expression of twenty differentially expressed genes (DEGs). Ach_contigs and primers used are reported in Additional file: Table ST1, the PCR conditions are described in Material and Methods. Gene expression expressed as fold change and time course is indicated in the X axis. Standard errors are indicated. Results of quantitative PCRs were in agreement with RNA-seq experiment. (TIF 1088 kb)
Additional file 6:
Figure S3. The MapMan figure of the “Biotic stress” was obtained by running the Mercator tool (http://mapman.gabipd.org/web/guest/mercator) with default parameters to assign MapMan bins to Actinidia chinensis var. chinensis transcripts. (TIF 3354 kb)
Additional file 7:
Table S4. List of subset of differentially expressed genes (DEGs) related with PRRs genes. (XLSX 505 kb)
Additional file 8:
Table S5. List of subset of differentially expressed genes (DEGs) related with defense genes. (XLSX 484 kb)
Additional file 9:
Table S6. List of subset of differentially expressed genes (DEGs) related to ROS detoxification. (XLSX 470 kb)
Additional file 10:
Table S7. List of subset of differentially expressed genes (DEGs) related to photosynthesis. (XLSX 43 kb)
Additional file 11:
Table S8. List of subset of differentially expressed genes (DEGs) involved in ethylene biosynthesis and signalling. (XLSX 467 kb)
Additional file 12:
Table S9. List of subset of differentially expressed genes (DEGs) involved in jasmonic acid-mediated response. (XLSX 462 kb)
Additional file 13:
Table S10. List of subset of differentially expressed genes (DEGs) involved in salicylic acid biosynthesis and signalling. (XLSX 464 kb)
Additional file 14:
Table S11. List of subset of differentially expressed genes (DEGs) involved in abscisic acid biosynthesis and signalling. (XLSX 463 kb)
Additional file 15:
Figure S4. Distribution of transcription factors (TFs) in Actinidia chinensis var. chinensis transcriptome based on BLASTx against plantTFDBcat (http://plantgrn.noble.org/PlantTFcat). Chromatin remodelling factors and families of TF with less than 5 transcripts are not shown. C2C2 family harbours: CO-like Dof, GATA, LSD and YABBY TFs. AP2/ERF includes the AP2, ERF and RAV classes of TFs. ARF and B3 classes belong to B3 superfamily of TFs. HD-ZIP, TALE, WOX, HB-PHD, and HB-other were grouped in the HB class. GARB family of TFs consists of ARR and G2-like classes. Others category consists of FHA-SMAD, GAGA-Binding-like, GeBP, HSF-type-DNA-binding, Nin-like, PAZ-Argonaute, PLATZ, Znf-B, Znf-LSD, SAP, SBP, TCP, TUBBY, FAR, CG1-CAMTA, E2F-DP, STY-LRP1, CW-Zn. Numbers of transcripts for each family were summarized in the figure and are detailed in Additional file: Table S12. (TIF 750 kb)
Additional file 16:
Table S12. Distribution of transcription factors (TFs) in Actinidia chinensis var. chinensis transcriptome. (XLSX 357 kb)
Additional file 17:
Table S13. List of subset of differentially expressed genes (DEGs) related to transcription factors. (XLSX 545 kb)
Additional file 18:
Table S14. List of subset of differentially expressed genes (DEGs) related to AP2/ERF transcription factors. (XLSX 463 kb)
Additional file 19:
Table S15. List of subset of differentially expressed genes (DEGs) related to NAC transcription factors. (XLSX 458 kb)
Additional file 20:
Table S16. List of subset of differentially expressed genes (DEGs) related to WRKY transcription factors. (XLSX 463 kb)
Additional file 21:
Table S17. List of subset of differentially expressed genes (DEGs) related to MAPK cascade signal transduction. (XLSX 458 kb)
Additional file 22:
Figure S5. Heatmap of the correlation of WGCNA modules with traits (ASM treatment and Psa inoculation). 21 modules were detected and named with colour names. The grey category is not a real module: it collect all the leftover genes not enough correlated with one of the other significant coloured modules. In each square the upper value is kME (module eigengene-based connectivity) while the lower value is the P-value of the correlation. (TIF 1752 kb)
Additional file 23:
Table S18. List of the genes obtained by WGCNA and the kMEs for each module. (XLSX 9185 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Michelotti, V., Lamontanara, A., Buriani, G. et al. Comparative transcriptome analysis of the interaction between Actinidia chinensis var. chinensis and Pseudomonas syringae pv. actinidiae in absence and presence of acibenzolar-S-methyl. BMC Genomics 19, 585 (2018). https://doi.org/10.1186/s12864-018-4967-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4967-4