
Abstract
Background
To continue to meet the increasing demands of soybean worldwide, it is crucial to identify key genes regulating flowering and maturity to expand the cultivated regions into short season areas. Although four soybean genes have been successfully utilized in early maturity breeding programs, new genes governing maturity are continuously being identified suggesting that there remains as yet undiscovered loci governing agronomic traits of interest. The objective of this study was to identify novel loci and genes involved in a diverse set of early soybean maturity using genome-wide association (GWA) analyses to identify loci governing days to maturity (DTM), flowering (DTF) and pod filling (DTPF), as well as yield and 100 seed weight in Canadian environments. To do so, soybean plant introduction lines varying significantly for maturity, but classified as early varieties, were used. Plants were phenotyped for the five agronomic traits for five site-years and GWA approaches used to identify candidate loci and genes affecting each trait.
Results
Genotyping using genotyping-by-sequencing and microarray methods identified 67,594 single nucleotide polymorphisms, of which 31,283 had a linkage disequilibrium < 1 and minor allele frequency > 0.05 and were used for GWA analyses. A total of 9, 6, 4, 5 and 2 loci were detected for GWA analyses for DTM, DTF, DTPF, 100 seed weight and yield, respectively. Regions of interest, including a region surrounding the E1 gene for flowering and maturity, and several novel loci, were identified, with several loci having pleiotropic effects. Novel loci affecting maturity were identified on chromosomes five and 13 and reduced maturity by 7.2 and 3.3 days, respectively. Novel loci for maturity and flowering contained genes orthologous to known Arabidopsis flowering genes, while loci affecting yield and 100 seed weight contained genes known to cause dwarfism.
Conclusions
This study demonstrated substantial variation in soybean agronomic traits of interest, including maturity and flowering dates as well as yield, and the utility of GWA analyses in identifying novel genetic factors underlying important agronomic traits. The loci and candidate genes identified serve as promising targets for future studies examining the mechanisms underlying the related soybean traits.
Similar content being viewed by others

Background
Due to their sedentary life style and somewhat limited means of dispersal, plants have become highly attuned to their surrounding environment. Photoperiod responses are well known to impose limitations on plants such as the transition from the vegetative to flowering stage [1]. As such, plants have been grouped into short day, long day and day neutral plants depending on the number of daylight hours required to induce flowering. In addition to photoperiodism, transition from the vegetative to flowering stage is also affected by temperature (vernalisation), gibberellin hormones, as well as other factors that are not yet fully understood (example abiotic stresses) making this transition a complicated event [1]. In order to expand the growing range of short day crops, such as soybean, to far north and south regions, agricultural breeding programs have aimed to alter photoperiod responses to obtain day neutral responses.
Soybean, Glycine max (L.) Merr., is a short day flowering crop originating from Asia. Its grain is used worldwide as a human and animal food source, and for the production of oils and plastics. World soybean production was 320.2 M metric tons in 2015, which represented a 47% increase worldwide since 2005, and accounted for 29.0% of the world’s vegetable oil consumption and 70.9% of the protein meal consumption in 2015 (www.soystats.com). The expansion of soybean cultivation has been challenged by long days and short growing seasons of far northern/southern climates and cultivars that have been bred are often limited to a very narrow range of latitudes [2]. In order to continue to meet the increasing demands of soybean worldwide, it is crucial to identify key genes regulating flowering and maturity to expand the area of cultivated regions.
Several natural genetic variants controlling flowering and maturity time have been identified and heavily used in soybean breeding programs for adaptation to long day environments. These include the soybean E genes E1 to E10 [3,4,5,6,7,8,9,10,11,12], the JUVENILE (J) gene [13], and the FLOWEING LOCUS T (FT) genes [14]. Among the E genes, E1 to E4, E9 and E10 have been identified by various fine-mapping and candidate gene identification approaches [6, 7, 9,10,11,12]. E1 contains a putative bipartite nuclear localization signal and a B3-related domain [10] and acts as a floral repressor by down-regulating GmFT2a and GmFT5a [15]. The E2 gene is an orthologue to the Arabidopsis GIGANTEA (GI) gene [9]; however, unlike in Arabidopsis, E2 delays flowering under long days by inhibiting expression of GmFT2a, but not GmFT5a [9]. E3 and E4 encode the PHYTOCHROME A (PHYA) genes [6, 7], GmPHYA3 and GmPHY2, and control flowering under high and low red to far-red ratios, respectively [16]. Loss-of-function alleles exist for E1 to E4 and lead to photoperiod insensitivity by allowing higher expression levels of the florigen FT genes and promoting flowering under long day conditions. Recently, E10 was identified as FT4, which has been shown to be up-regulated by and act down-stream of E1 [11]. Samanfar et al. [11] demonstrated that the e10 haplotype promotes early flowering; however, this haplotype appears to be rare and the mechanisms by which it promotes flowering are not yet known. The remaining E genes result in early flowering under long day conditions and the genes encoding them have yet to be identified with the exception of E9, which was recently identified as the GmFT2a gene [12]. E9 and GmFT5a have been shown to have redundant roles in soybean and control flowering by inducing the expression of flower-initiating genes [14].
Despite the increasing knowledge and continuous identification of genes involved in soybean flowering, there remains a plethora of missing links as well as a need for identification of new soybean genes for flowering and maturity. Soybean flowering and maturity are often positively correlated with seed yield [8], but are not correlated with 100 seed weight [8, 17, 18], two agronomical traits that are important for meeting soybean demands and maintaining quality standards for soy food products. As such, it is important to select early maturing varieties that minimally affect yield and seed weight. Genome-wide association (GWA) analyses enable the detection of genetic differences accounting for the observed variation in phenotypes. Studies have demonstrated that GWA studies containing few individuals (< 100) can successfully identify single nucleotide polymorphisms (SNPs) affecting phenotypes if the population is sufficiently genetically diverse and population structure is accounted for [19]. This study aimed to identify new loci governing early flowering, maturity and pod filling time, as well as seed weight and yield in Canadian environments using genotyping-by-sequencing (GBS), microarray and GWA analysis approaches in a diverse set of 86 soybean genotypes belonging to early maturity groups 00 to 000. Although previous studies have reported on SNPs affecting these traits in early maturity groups 0 and 00 [20], none have been reported on markers affecting these phenotypes in Canadian environments. This study identified major markers affecting phenotypic traits of interest by identifying over 67,000 SNPs that were subsequently used in GWA analysis approaches. The successful identification of novel loci, some of which contain genes known to govern the traits of interest in other model plant species, but not yet reported in soybean were identified.
Methods
Plant materials and phenotyping
A total of 86 soybean plant introduction (PI) lines belonging to early maturity groups 000 to 00 and having diverse geographical origins (Europe, China, Japan, North Korea, Russia, and North America) were obtained from the Germplasm Resources Information Network (www.ars-grin.gov) and were used for phenotypic and genetic evaluation (Additional file 1). Soybean lines were phenotyped for days to flowering (DTF), days to pod filling (DTPF), days to maturity (DTM), yield (kg/ha) and 100 seed weight (SW) for two (2012 and 2013) and 3 years (2011 to 2013) at sites located in Québec City and Saint-Mathieu-de-Beloeil, Québec, Canada, respectively, for a total of five site-years. At each site, soybean lines were planted in single row (2011) or two row (2012 and 2013) plots using a Modified Augmented Design [21, 22]. Phenotypes for each line were calculated as follows: DTF as the day of planting to the day at which 75% of the genotype was flowering; DTM as the day of planting to the day at which 95% of the pods within the genotype were at physiological maturity; DTPF as the number of days from DTF to DTM; yield as the grain mass per plot adjusted for population and converted to kg ha− 1; and 100 seed weight was taken as the average of two measurements per plot. Phenotypic data distribution and Spearman’s pairwise correlation coefficients were calculated for all trait comparisons using R version 3.3.1 [23]. All trait data were normally distributed.
Genotyping
Sample preparation for GBS was performed as described in Tardivel et al. [24]. Briefly, soybean DNA was extracted using the Qiagen DNeasy 96 Plant Kit (Qiagen, Toronto, Canada) from 100 mg (wet weight) of soybean tissue obtained from a unique plant for each line following the manufacturer’s protocols. Libraries were prepared at the Plate-Forme d’Analyses Génomiques (Université Laval, Québec City, Canada) as described in Elshire et al. [25] using the ApeKI restriction enzyme. Sequencing was performed as single-end 100 bp reads on an Illumina HiSeq2000 System at the Genome Québec Innovation Centre (McGill University, Montreal, Canada), as part of a larger project, with 96 genotypes per sequencing lane.
Illumina sequence read data were processed using multiple publically available software tools in an in-house script (unpublished) similar to that reported by Torkamaneh and Belzile [26]. Briefly, adapters and barcodes were removed from reads using Trimmomatic v. 0.33 using ILLUMINACLIP <adapter file>:2:30:15, LEADING and TRAILING removal of three, SLIDINGWINDOW:3:20, and finally MINLEN:32. Sequences were then aligned to the soybean reference genome version 2 (NCBI assembly #GCA_000004515.3) using Burrows-Wheeler Alignment (BWA version 0.7.12-r1039) with the options –a –M –R followed by variant calling with SAMtools version 1.2.1 [27], sambamba version 0.6.4 [28] and bcftools version 1.2.1 [29]. BAM files were then pooled and variants filtered with vcftools version 0.1.15 [30] to maintain only biallelic sites with an overall mapping quality > 30, a read depth > 2, and present in a minimum of 20% of the genotypes. Missing genotypes were then self-imputed with Beagle version 4.1 [31, 32] using 10 iterations. In addition to performing GBS, data for the SoySNP50K microarray were obtained from the SoyBase database (https://www.soybase.org) [33] for each genotype. Microarray and GBS data were merged using an in house developed script in order to maximize the number of SNPs in the data set, with priority given to the microarray genotypes for common SNPs as this data was not imputed.
Genetic analyses
Population structure was modeled using fastStructure version 1.0 [34] with a simple prior and 1000 iterations for cross-validation of population structure (K) ranging from one to ten. To diminish the effect of high admixture within the population, structure analysis was performed on genotypes with linkage disequilibrium (LD) < 0.9 and minor allele frequency (MAF) > 0.05 [35]. The optimal range of K was determined based on model complexity using the marginal likelihood method using the fastStructure script chooseK.py, as well as on visualization of the log marginal likelihood, cross-validation error and population visualization using Distruct version 1.1 [36]. Genetic relationships were investigated using unrooted Neighbour Joining (NJ) tree construction implemented in TASSEL software version 5.2.17 [37].
Linkage disequilibrium (LD), estimated as the r2 between SNPs, was calculated for each chromosome based on the entire collection for 10 Mb windows using Plink version 1.90b3z [38]. LD was visualized using the mean r2 within bin sizes of 1000 SNPs for each chromosome. LD decay was calculated as the point at which the chromosomes reached 50% of their original LD value.
SNP distributions for the GBS, microarray and merged data sets were visualized using Circos version 0.67-7 [39] ideogram and karyotype options. The effects of SNPs within the genome were calculated using SnpEff version 4.3i [40]. Locations of genes were obtained from SoyBase GFF3 files for each chromosome, converted to a GTF file using the Cufflinks version 2.1.1 command gffread [41], and a database for version 2 of the soybean genome and transcriptome built using the SnpEff build –gtf22 command.
Genome-wide association analyses
Genome-wide association analyses were performed using TASSEL software version 5.2.17 [37]. General linear models (GLM) were performed with or without covariates from principle component analyses (PCA) or population structure covariates (Q = 3) obtained from fastStructure. A range of principle components (P) were used to reflect the chosen population structure (P = 3) or to represent approximately 50% of the total variation within the data set (P = 10). A kinship matrix was calculated for each data set using the scaled identity-by-state (IBS) method [42] implemented in TASSEL version 5.2.17 to determine the relatedness among the individuals. Compressed mixed linear models (CMLM) incorporating the kinship matrix as a random effect, along with the abovementioned principle components or population structure were tested. All GWA analyses were performed with MAF ≥0.05 and LD < 1, as well as with year and site as fixed effects. The fit of the different models for each trait was assessed by comparing the expected versus obtained –Log10 P-values (i.e. QQ-plots) with graphs produced in R [23]. SNPs were identified as significant in the best-fit model using Bonferroni multiple comparisons correction and visualized using the QQman package [43] in R software version 3.3.1 [23]. Significant markers within the same genomic region and in high LD were viewed as a quantitative trait locus (QTL). Candidate genes associated with SNPs were reported by calling genes within 500 kb of significant SNPs [44]. Statistical validation of SNP markers identified in the GWA analysis was done using Tukey’s HSD tests.
Results
Distribution and correlation of phenotypic traits
Phenotyping was conducted at two different sites for two or 3 years to study the distribution and correlation among the different traits of interest. Differences among sites and years were observed among traits, and as such sites and years were used as fixed effects for all linear models (Additional file 2). Normal distributions without any significant skewness were observed for all traits (Fig. 1 and Additional file 3). Coefficients of variation ranged from 10 to 18% for all traits except yield, which was highly variable among years and environments with a coefficient of variation of 47%.

Frequency distribution of soybean traits of interest. Frequency distributions are based on the average phenotype value of each soybean line across different environments and years
Significant correlations were observed between traits (Table 1). Highly significant correlations between DTM and DTF (r = 0.78) and DTM and DTPF (r = 0.75) were observed suggesting that later maturity may be partially due to later flowering and longer pod filling times. Similarly, 100 seed weight had a moderate negative correlation with DTF suggesting possible interdependence, while a low negative correlation was observed with DTM.
Distribution of SNPs and comparison of different genotyping methods
Sequencing of the GBS libraries resulted in approximately 202.2 M clean reads with an average of 2.35 M clean reads per soybean genotype with an average depth of coverage of 4.4 ± 2.2. By applying several in house filtering parameters, a total of 33,575 SNPs and 3236 INDELs were obtained. SNPs and INDELs were located throughout the genome with 69 SNP intervals over 500 kb (Additional file 4). An additional four SNPs were located in plastid DNA, with 18 SNPs and 12 INDELs in mitochondrial DNA.
Compared to the SoySNP50K microarray SNP calls, which contained a total of 34,556 SNPs, the distribution of SNPs within the GBS data set was more uniform across chromosomes, whereas SNPs within the SoySNP50K microarray data set were denser around chromosome ends (Fig. 2a). Merging of the two data sets showed that only 471 SNPs were found in both the GBS and SoySNP50K microarray data sets resulting in a total of 67,571 SNPs. Of the overlapping SNPs, a concordance rate of 98.8 ± 0.3% was observed, suggesting high reproducibility between the two data sets for common SNPs. Of the non-concordant SNPs, 62.6% were observed to be heterozygous in one of the two data sets, but homozygous in the other. Merging of the two data sets provided greater genome coverage with SNPs being relatively evenly distributed throughout the entire genome (Fig. 2a). Merging of the data sets resulted in an average SNP spacing of 13,249 bp compared to one every 28,099 bp in the GBS data set. A total of 41 gaps over 500 kb (Additional file 5) was observed in the merged data set. All gaps over 500 kb were found in centromeric or pericentromeric regions (Additional file 5). As such, this data set was used for all further analyses.

SNP distributions across the soybean genome (v2) and SNP effects within the population of plant introduction genotypes. a Gene and SNP distributions used for genotyping across the soybean chromosomes. From the outer to inner circle: Soybean chromosomes 1 to 20; gene locations on the positive and negative chromosome strands; and GBS, SoySNP50K microarray and the merged data set SNP locations. b Distribution of SNPs based on genomic region within the merged data set. c Predicted SNP effects based on degree of impact within the merged data set. d Predicted SNP effects based on function class for SNPs located within coding regions within the merged data set
Analysis with SnpEff showed that SNPs in the merged data set were found primarily outside of coding regions, while 30.6% of SNP effects were found within genes (Fig. 2b). A total of 605 SNPs were predicted to have a high impact on gene products (Fig. 2c), while 54.5%, 1.4% and 44.1% of SNP effects within coding regions resulted in missense, nonsense and silent mutations, respectively (Fig. 2d). An overall transition to transversion ratio of 2.63 was observed.
Population structure and linkage disequilibrium
Population structure analysis with fastStructure suggested between one and nine populations among the 86 plant introduction genotypes used in this study. Further analysis of the data revealed that plateauing of the marginal likelihood values started occurring at three populations (Fig. 3a, b), which is in accordance with analyses using Structure (data not shown). Principle component analysis confirmed that high similarity existed within the three populations (Fig. 3c). Neighbour joining tree analysis further confirmed the presence of three main populations and the nine subpopulations (Fig. 3d).

Genetic diversity and population structure of the 86 soybean genotypes. a Estimated log marginal likelihood (LML) calculated for populations (K) ranging from 2 to 10 using fastStructure. b Population structure of the soybean lines, where each vertical line represents a cultivar and each colour a separate population. c PCA plot of the first two principle components based on genotypes. Ellipses represent Hotelling’s T2 95% confidence intervals for each population. d Cladogram of the soybean lines constructed using the neighbour-joining method. Different populations as determined by LML are indicated by identical symbols (triangle, circle and square) in all panels
Linkage disequilibrium decay rates varied across the soybean chromosomes, with the average 50% decay rate occurring between 150 and 200 kb. Exceptions were with chromosome 11, which hit its 50% decay mark at ~ 100 kb, and chromosomes 5, 15, 18 and 19, which had slower decay rates of approximately 325 kb (Fig. 4). The average starting linkage disequilibrium rate for SNPs within 100 bp was 0.48 and reached 50% of this value at approximately 180 kb.

Linkage disequilibrium decay plots across soybean chromosomes and the average decay across the genome. a Average LD between SNPs with a maximum distance of 5 Mb. b Zoom-in of average LD between SNPs with a maximum distance of 500 kb
Genome-wide association analysis of soybean traits
Genome-wide association analysis was performed for soybean agronomic traits of interest across two environments for up to 3 years. After removing SNPs in 100% LD and with MAF < 0.05, a total of 14,594 SNPs, 13,364 SNPs and 31,283 SNPs were selected for GWA analysis in the GBS, SoySNP50K microarray and merged data sets, respectively. To limit the effect of false positives in the analysis, population structure (covariate Q) or principle components (covariate P) were incorporated into the model as covariates. In the first approach, GWA analysis was performed using GLM analysis incorporating P or Q as covariates, while the second approach utilized a kinship matrix (covariate K) using CMLM analysis incorporating P or Q as covariates for a total of seven different models being performed on each trait to choose the optimal model for the population and given trait (Additional file 6). Models incorporating kinship and covariates P (CMLM) often resulted in model underestimation, while incorporation of only P or Q typically led to model overestimation. As such, the model incorporating only covariate Q (i.e. GLM with three populations) was used. The models for each trait were then adjusted with Bonferroni multiple corrections to reduce false positives, and further validated using pairwise comparisons between significant SNPs.
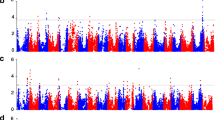
GWA analysis with the different data sets was performed for soybean days-to-flowering (DTF), days-to-maturity (DTM), days-to-pod-filling (DTPF), yield and 100 seed weight (SW). Loci detected as significant when analyzing the GBS or SoySNP50K microarray data sets alone were similar, but less comprehensive than those reported when analyzing the merged data (Table 2; Additional file 7). In general, loci detected as significantly affecting a trait in the GBS or SoySNP50K microarray analyses were detected in the merged GWA analyses; however, not all loci detected in the merged data set were present in the GBS or SoySNP50K microarray results (Table 2). Exceptions were for 100 seed weight and yield, where significant loci covered larger regions or were not detected in the merged data set (Additional file 8), with the latter typically occurring in loci that were just below the significance threshold in their respective data sets. In the merged data set, a total of 74 SNPs located on seven different chromosomes were found to be highly associated (Bonferroni-corrected P value < 0.01) with DTM (Fig. 5a). Of these, six loci are not reported in SoyBase or recent literature and are potentially novel maturity loci (Table 2). Association analysis with DTF revealed similar results with 41 SNPs located on five chromosomes (Fig. 5b), while DTPF resulted in six SNPs on four chromosomes (Fig. 5c), 100 seed weight contained 41 SNPs on four chromosomes (Fig. 5d), and three SNPs on two chromosomes for yield (Fig. 5e). Potentially novel loci were also detected for DTM, DTF and DTPF (Table 2); however, no novel loci were detected for 100 seed weight or yield. Of the SNPs that were significantly associated with a trait, eight were found to be pleiotropic for DTM and DTF and one for DTM and DTPF. No SNP detected as significantly associated with 100 seed weight was detected in the other traits. Comparison of traits with differing SNP marker haplotypes using Tukey’s HSD test confirmed the significance of all markers detected below the Bonferroni correction limit of P < 0.01 (data not shown).

Genome-wide association analysis Manhattan plots for (a) days to maturity (DTM), (b) days to flowering (DTF), (c) days to pod filling (DTPF), (d) 100 seed weight (SW), and (e) yield. Lines represent the significance threshold as determined by Bonferroni multiple comparisons corrections equivalent to P < 0.05 (blue lower line) or P < 0.01 (red upper line)
Among the SNPs detected as significantly associated with DTM and DTF, four were within 31.1 kb of E1, while one SNP was detected within 1 Mb of E2 for DTM. No other SNPs were found within or around other known soybean E genes for any trait. Interestingly, a total of 14 SNPs significantly associated with DTM were found between 29.0 and 31.3 Mb of Gm13, a region that has not been reported as associated with maturity.
Discussion
Genome-wide association studies are conducted to identify genetic loci associated with traits of interest [45]. This knowledge can then be utilized in breeding programs using marker-assisted breeding approaches. Although genotyping-by-sequencing is commonly used and has been shown to be reliable and efficient, this study shows that additional information obtained from other sources, such as microarrays, can help improve the depth of information obtained in GWA studies. Although intra-accession variation may exist within agricultural crop lines, the high concordance rate (98.8%) between overlapping SNPs between the GBS and SoySNP50K microarray data sets suggests that merging of two genotyping methods can be appropriate, particularly in self-pollinating plants such as soybean.
Population structure can greatly affect the statistical power during GWA analysis and as such several methods were tested to diminish false negative associations. In the present study, seven different statistical models were tested to determine the most appropriate model for the given data set (Additional file 6). Models utilizing population structure without kinship proved to result in the most appropriate distribution of P values compared to expected P values and was therefore chosen for GWA analyses. Population structure analysis showed that populations were highly similar to results obtained using principal component analysis and cladistics analysis (Fig. 3). The three reported populations could be tightly correlated with the country of origin and can be grouped into cultivars originating historically from Japan, China or a mix of unknown origins. Using the three main populations (GLM Q3) in the GWA analyses followed by adjustment of the significance threshold with Bonferroni multiple corrections resulted in a model providing SNPs that were significantly associated with and known to affect the various traits studied.
Inbreeding and selection can have large effects on linkage disequilibrium [18, 46]. As such, it has been estimated that the number of markers required to identify loci significantly affecting traits is in the tens of thousands for soybean [47]. In this study, a sufficient amount of markers (> 67,000) were obtained for detection of loci affecting various agronomic traits that have been highly selected for in breeding programs. Similar to other studies examining soybean LD [18, 20, 48], a diverse range of LD was observed across the various chromosomes (Fig. 4), with the highest LD occurring on chromosomes 19 and 5. As expected, chromosome 19 has been extensively utilized in soybean short-season breeding programs as it harbours the maturity locus E3. Chromosome 5, however, has been reported to affect maturity despite no known E genes being located on it [49,50,51]. The lowest levels of LD observed in this study were on chromosomes 8 and 11, with the former recently identified as harbouring the rare E10 allele [11]. Aside from E10 and possibly the as yet unidentified E8, no known maturity loci have been selected for on these chromosomes.
Complex traits such as maturity and flowering often make it difficult to assess the effectiveness of GWA analyses as they are often affected by multiple loci. Merging of GBS and microarray data not only resulted in a higher number of markers, but also proved to be complementary as not all regions detected as significant in one data set were detected in the other (Additional file 7). Additionally, merging of the data sets revealed loci not detected in either data set when analyzed alone, particularly for DTM (Additional file 7). A high number of significantly associated SNPs were observed for all traits, including days to maturity, a trait which has been highly selected for. Despite all of the lines being classified as 0 to 000, a high variation in the number of days to maturity (DTM), flowering (DTF) and pod filling (DTPF) was observed. Of the loci significantly associated with DTF, those on chromosomes 6 and 10 were also found to be significantly associated with DTM. This is not surprising as DTM and DTF were highly correlated (Table 1) and these two loci are known to have been highly selected for in early maturity breeding programs [9, 10]. Similarly, three of the four loci significantly associated with DTPF were also associated with DTM, two traits that were also highly correlated. Interestingly, no co-localization of loci was observed between 100 seed weight and the other traits despite a moderate correlation between DTF and seed weight. Therefore, selection for earlier maturing and high yielding varieties can be selected for independently of seed weight.
Of the known E genes affecting maturity, SNPs were only located in the region of E1 on chromosome 6 for DTM and DTF, and E2 for DTM (Table 2). This is in accordance with previous research demonstrating that E1 is a major determinant of early maturity and flowering under field conditions [52, 53]. Interestingly, another SNP located on Gm06 was found to be within 174 kb of GmWRKY76 (Glyma.06 g142000), a soybean transcription factor recently shown to affect flowering when introduced into Arabidopsis plants [54], suggesting that this gene merits further investigation for its role in soybean maturity and flowering. Despite known allelic variation for E3 [24] and E4 (Tardivel, unpublished data) in this collection of PIs, no SNP was significantly associated with these loci, most likely due to even distribution of the haplotypes within each population (data not shown). Interestingly, a maturity locus on chromosome 16 was located within 3.1 Mb of the recently reported E9 gene [55], suggesting that this chromosome could be further incorporated into soybean early maturity breeding programs. In addition to detecting loci associated with known alleles, several novel loci were detected for maturity traits (Table 2). These loci have most likely not been highly selected for and may show promise for utilization in early maturity breeding programs. As for 100 seed weight, no new loci were detected. Although novel loci were detected for DTM, DTF and DTPF, further work is required to fully validate the roles of these loci in controlling these traits.
Identification of genes involved in specific traits such as maturity is continuously advancing, as evidenced by recent publications identifying E9 [55], E10 [11], J [13] and FT5a [14], as well as the possibility of GmWRKY58 and GmWRKY76 [54], which demonstrated roles in flowering in transgenic Arabidopsis. Several SNPs associated with DTM were found in proximity to several Arabidopsis HAP5 (NUCLEAR FACTOR Y [NF-Y] family) orthologues, including HAP5A (Glyma.13 g207500), HAP5B (Glyma.13 g207700) and HAP5C (Glyma.13 g207600), and a SNF2/EDA16 orthologue (Glyma.13 g183900). NF-Y proteins have been shown to help induce flowering, particularly under long days [56, 57]. These SNPs were also detected in the DTF and DTPF analysis at the Bonferroni correction level P < 0.05 level. The SNPs identified as significant on Gm05 for DTM were within 45,000 bp of Glyma.05 g036300, a gene encoding SPERMIDINE/SPERMINE SYNTHASE. SPERMIDINE SYNTHASE genes have been found to be essential for Arabidopsis embryo development and survival [58], and the close proximity of significant markers to this gene in soybean may indicate its potential role in soybean maturity and seed development.
For DTF, significantly associated SNPs were found within or near genes with a variety of biological roles, results similar to those detected for DTPF and 100 seed weight. Of particular interest for SNPs associated with DTF were SNPs in close proximity to the soybean Glyma.15 g275100, an orthologue to the human breast cancer gene BRCA1 [59]. In Arabidopsis, this gene has been shown to be highly expressed in flower bud tissue [60], and is involved in homologous recombination and DNA repair [61]. For 100 seed weight, several SNPs were found in proximity to several AUXIN RESPONSIVE PROTEIN genes located on Gm04. Auxin has been shown to play a role in Arabidopsis embryo sac development as well as normal plant growth and development [62]. Several other SNPs associated with 100 seed weight SNPs on Gm19 were found within regions of a DOWNSTREAM NEIGHBOR OF SON (DONSON) or HUMPTY DUMPTY orthologue. DONSON proteins have been shown to be involved in DNA replication fork stability in humans [63], are required for ovary cell proliferation in Drosophila [64], and are linked with human microcephalic dwarfism [63]. The close proximity of SNPs associated with seed weight and the putative roles of DONSON in seed development and size suggest that this gene merits investigation for its role in plant seed development. Genes involved in carbohydrate metabolism, such as BETA-FRUCTOFURANOSIDASE and ALPHA-AMYLASE were also found to be in close proximity of SNPs associated with seed weight.
Conclusions
The following study demonstrates that combining GBS and microarray data prior to performing GWA analyses can not only improve the power of detection, but also help identify loci that are overlooked by either method. This study also supports the concept that small population sizes that are genetically diverse can successfully identify loci governing traits of interest as known loci (E1 and E2), as well as novel loci with Arabidopsis orthologues were identified. GWA analysis successfully identified loci with alleles previously known to affect the studied traits, but also novel loci containing genes orthologous to genes known to affect the traits in Arabidopsis. The GWA analyses have helped contribute to an enhanced understanding of known and novel genes affecting important soybean agronomical traits, which will be useful in future breeding programs utilizing marker-assisted selection. Further studies are currently ongoing to validate the roles of selected genes in soybean maturity.
Abbreviations
- CMLM:
-
Compressed mixed linear model
- DONSON :
-
DOWNSTREAM NEIGHBOR OF SON
- DTF:
-
Days to flowering
- DTM:
-
Days to maturity
- DTPF:
-
Days to pod filling
- FT :
-
FLOWERING LOCUS T
- GBS:
-
Genotyping-by-sequencing
- GI :
-
GIGANTEA
- GLM:
-
General linear model
- GWA:
-
Genome-wide association
- IBS:
-
Identity-by-state
- LD:
-
Linkage disequilibrium
- LML:
-
Log marginal likelihood
- MAF:
-
Minor allele frequency
- NF-Y :
-
NUCLEAR FACTOR Y
- PCA:
-
Principal component analysis
- PHYA :
-
PHYTOCHROME A
- PI:
-
Plant introduction
- SNP:
-
Single nucleotide polymorphism
- SW:
-
100 seed weight
References
Araki T. Transition from vegetative to reproductive phase. Curr Opin Plant Biol. 2001;4:63–8.
Watanabe S, Harada K, Abe J. Genetic and molecular bases of photoperiod responses of flowering in soybean. Breed Sci. 2012;61:531–43.
McBlain BA, Bernard RL. A new gene affecting the time of flowering and maturity in soybeans. J Hered. 1987;78:160–2.
Bonato ER, Vello NA. E6, a dominant gene conditioning early flowering and maturity in soybeans. Genet Mol Biol. 1999;22:229–32.
Cober ER, Voldeng HD. A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T. Crop Sci. 2001;41:698–701.
Liu C, Kanazawa A, Matsumura H, Takahashi R, Harada K, Abe J. Genetic redundancy in soybean photoresponses associated with duplication of the Phytochrome A gene. Genetics. 2008;180:995–1007.
Watanabe S, Hideshima R, Xia Z, Tsubokura Y, Sato S, Nakamoto Y, Yamanaka N, Takahashi R, Ishimoto M, Anai T, et al. Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics. 2009;182:1251–62.
Cober ER, Morrison MJ. Regulation of seed yield and agronomic characters by photoperiod sensitivity and growth habit genes in soybean. Theor Appl Genet. 2010;120:1005–12.
Watanabe S, Xia Z, Hideshima R, Tsubokura Y, Sato S, Yamanaka N, Takahashi R, Anai T, Tabata S, Kitamura K, et al. A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics. 2011;188:395–407.
Xia Z, Watanabe S, Yamada T, Tsubokura Y, Nakashima H, Zhai H, Anai T, Sato S, Yamazaki T, Lu S, et al. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci. 2012;109:12852–3.
Samanfar B, Molnar SJ, Charette M, Schoenrock A, Dehne F, Golshani A, Belzile F, Cober ER. Mapping and identification of a potential candidate gene for a novel maturity locus, E10, in soybean. Theor Appl Genet. 2016;130:377–90.
Zhao C, Takeshima R, Zhu J, Xu M, Sato M, Watanabe S, Kanazawa A, Liu B, Kong F, Yamada T, et al. A recessive allele for delayed flowering at the soybean maturity locus E9 is a leaky allele of FT2a, a FLOWERING LOCUS T ortholog. BMC Plant Biol. 2016;16:20.
Lu S, Zhao X, Hu Y, Liu S, Nan H, Li X, Fang C, Cao D, Shi X, Kong L, et al. Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat Genet. 2017;49:773–9.
Nan H, Cao D, Zhang D, Li Y, Lu S, Tang L, Yuan X, Liu B, Kong F. GmFT2a and GmFT5a redundantly and differentially regulate flowering through interaction with and upregulation of the bZIP transcription factor GmFDL19 in soybean. PLoS One. 2014;9:e97669.
Thakare D, Kumudini S, Dinkins RD. The alleles at the E1 locus impact the expression pattern of two soybean FT-like genes shown to induce flowering in Arabidopsis. Planta. 2011;234:933–43.
Cober ER, Tanner JW, Voldeng HD. Soybean photoperiod-sensitivity loci respond differentially to light quality. Crop Sci. 1996;36:606–10.
Morrison MJ, Voldeng HD, Cober ER. Agronomic changes from 58 years of genetic improvement of short-season soybean cultivars in Canada. Agron J. 2000;92:780–4.
Sonah H, O'Donoughue L, Cober E, Rajcan I, Belzile F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotech J. 2015;13:211–21.
Bartoli C, Roux F. Genome-wide association studies in plant pathosystems: toward an ecological genomics approach. Front Plant Sci. 2017;8:763.
Zhang J, Song Q, Cregan PB, Nelson RL, Wang X, Wu J, Jiang GL. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics. 2015;16:217.
Lin C-S, Poushinsky G. A modified augmented design for an early stage of plant selection involving a large number of test lines without replication. Biometrics. 1983;39:553–61.
Lin C-S, Poushinsky G. A modified augmented design (type 2) for rectangular plots. Can J Plant Sci. 1985;65:743–9.
R Core Team: R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/.
Tardivel A, Sonah H, Belzile F, O’Donoughue LS. Rapid identification of alleles at the soybean maturity gene E3 using genotyping by sequencing and a haplotype-based approach. Plant Genome. 2014; https://doi.org/10.3835/plantgenome2013.10.0034.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:e19379.
Torkamaneh D, Belzile F. Scanning and filling: ultra-dense SNP genotyping combining genotyping-by-sequencing, SNP array and whole-genome resequencing data. PLoS One. 2015;10:e0131533.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–4.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–23.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97.
Grant D, Nelson RT, Cannon SB, Shoemaker RC. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 2010;38:D843–D6.
Raj A, Stephens M, Pritchard JK. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics. 2014;197:573–89.
Sobota RS, Shriner D, Kodaman N, Goodloe R, Zheng W, Gao YT, Edwards TL, Amos CI, Williams SM. Addressing population-specific multiple testing burdens in genetic association studies. Ann Hum Genet. 2015;79:136–47.
Rosenburg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol. 2004;4:137–8.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Ruden DM, Lu X. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118 ; iso=2; iso-3. Fly. 2012;6:1–13.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Turner SD. qqman: an R package for visualizing GWAS results using QQ and manhattan plots. biorXiv. 2014; https://doi.org/10.1101/005165.
Brodie A, Azaria JR, Ofran Y. How far from the SNP may the causative genes be?. Nucleic Acids Res. 2016;44:6046–54.
Huang X, Han B. Natural variations and genome-wide association studies in crop plants. Annu Rev Plant Biol. 2014;65:531–51.
Li Y-H, Zhao S-C, Ma J-X, Li D, Yan L, Li J, Qi X-T, Guo X-S, Zhang L, He W-M. Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genomics. 2013;14:579.
Bastien M, Sonah H, Belzile F. Genome wide association mapping of Sclerotinia sclerotiorum resistance in soybean with a genotyping-by-sequencing approach. Plant Genome. 2014; https://doi.org/10.3835/plantgenome2013.10.0030.
Lam HM, Xu X, Liu X, Chen W, Yang G, Wong FL, Li MW, He W, Qin N, Wang B, et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet. 2010;42:1053–9.
Tasma I, Lorenzen L, Green D, Shoemaker R. Mapping genetic loci for flowering time, maturity, and photoperiod insensitivity in soybean. Mol Breed. 2001;8:25–35.
Kabelka E, Diers B, Fehr W, LeRoy A, Baianu I, You T, Neece D, Nelson R. Putative alleles for increased yield from soybean plant introductions. Crop Sci. 2004;44:784–91.
Panthee D, Pantalone V, Saxton A, West D, Sams C. Quantitative trait loci for agronomic traits in soybean. Plant Breed. 2007;126:51–7.
Abe J, Xu D, Miyano A, Komatsu K, Kanawaza A, Shimamoto Y. Photoperiod-insensitive Japanese soybean landraces differ at two maturity loci. Crop Sci. 2003;43:1300–4.
Xia Z, Zhai H, Liu B, Kong F, Yuan X, Wu H, Cober ER, Harada K. Molecular identification of genes controlling flowering time, maturity, and photoperiod response in soybean. Plant Syst Evol. 2012;298:1217–27.
Yang Y, Chi Y, Wang Z, Zhou Y, Fan B, Chen Z. Functional analysis of structurally related soybean GmWRKY58 and GmWRKY76 in plant growth and development. J Exp Bot. 2016;67:4727–42.
Kong F, Nan H, Cao D, Li Y, Wu F, Wang J, Lu S, Yuan X, Cober ER, Abe J, et al. A new dominant gene E9 conditions early flowering and maturity in soybean. Crop Sci. 2014;54:2529–35.
Cai X, Ballif J, Endo S, Davis E, Liang M, Chen D, DeWald D, Kreps J, Zhu T, Wu Y. A putative CCAAT-binding transcription factor is a regulator of flowering timing in Arabidopsis. Plant Physiol. 2007;145:98–105.
Kumimoto RW, Adam L, Hymus GJ, Repetti PP, Reuber TL, Marion CM, Hempel FD, Ratcliffe OJ. The Nuclear Factor Y subunits NF-YB2 and NF-YB3 play additive roles in the promotion of flowering by inductive long-day photoperiods in Arabidopsis. Planta. 2008;228:709–23.
Imai A, Matsuyama T, Hanzawa Y, Akiyama T, Tamaoki M, Saji H, Shirano Y, Kato T, Hayashi H, Shibata D. Spermidine synthase genes are essential for survival of Arabidopsis. Plant Physiol. 2004;135:1565–73.
Welcsh PL, King M-C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet. 2001;10:705–13.
Lafarge S, Montané MH. Characterization of Arabidopsis thaliana ortholog of the human breast cancer susceptibility gene 1: AtBRCA1, strongly induced by gamma rays. Nucleic Acids Res. 2003;31:1148–55.
Block-Schmidt AS, Dukowic-Schulze S, Wanieck K, Reidt W, Puchta H. BRCC36A is epistatic to BRCA1 in DNA crosslink repair and homologous recombination in Arabidopsis thaliana. Nucleic Acids Res. 2010;39:146–54.
Zhao Y. Auxin biosynthesis and its role in plant development. Annu Rev Plant Biol. 2010;61:49–64.
Reynolds JJ, Bicknell LS, Carroll P, Higgs MR, Shaheen R, Murray JE, Papadopoulos DK, Leitch A, Murina O, Tarnauskaitė Ž. Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism. Nat Genet. 2017;49:537–49.
Bandura JL, Beall EL, Bell M, Silver HR, Botchan MR, Calvi BR. Humpty dumpty is required for developmental DNA amplification and cell proliferation in Drosophila. Curr Biol. 2005;15:755–9.
Acknowledgements
The authors would like to thank Aurélie Tardivel, Martin Lacroix and Maryse Desrochers for their technical help in generating the phenotypic data. The authors would also like to thank the Germplasm Resources Information Network for providing the soybean lines used in this research.
Funding
This work was funded by The Canadian Field Crop Research Alliance and Agriculture and Agri-Food Canada under the Agri-Innovation Program, along with Genome Canada and Genome Québec funds awarded to the SoyaGen project. Funding bodies had no role in the design of the study, collection of data, data analysis and interpretation, or writing of the manuscript.
Availability of data and materials
Data pertaining to the study have been included in the article or as supplementary material.
Author information
Authors and Affiliations
Contributions
Conception and design of the study: LOD. Acquisition of data for the study: LOD. Analysis of data for the work: TC, MOD, LOD. Interpretation of data for the work: TC and LOD. Manuscript revision and approval: TC, MOD and LOD.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Plants used in this study were grown from seed provided from the Germplasm Resources Information Network. Sampling of plant material was performed in compliance with institutional guidelines. No further approvals, licences or permissions were required as no sampling was conducted from wild and/or native flora.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
List of 86 lines used in the GWA analyses, along with their corresponding maturity group and fastStructure group. (XLSX 24 kb)
Additional file 2:
Average trait values across the five site-years. (PPTX 87 kb)
Additional file 3:
Quantile-quantile (QQ) plots of phenotypic traits demonstrating normal distributions. (PPTX 131 kb)
Additional file 4:
Location of gaps and their distances in the genotyping-by-sequencing data set. (XLSX 11 kb)
Additional file 5:
Location of gaps and their distances in the merged data set. (XLSX 12 kb)
Additional file 6:
Quantile-quantile (QQ) plots of different genome-wide association study analytical approaches for various soybean agronomic traits. (PPTX 648 kb)
Additional file 7:
Genome-wide association analysis Manhattan plots for soybean agronomic traits of interest using various genotyping methods. (PPTX 941 kb)
Additional file 8:
Additional loci associated with important agronomic traits identified using genome-wide association analyses. (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Copley, T.R., Duceppe, MO. & O’Donoughue, L.S. Identification of novel loci associated with maturity and yield traits in early maturity soybean plant introduction lines. BMC Genomics 19, 167 (2018). https://doi.org/10.1186/s12864-018-4558-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4558-4