
Abstract
Background
Dissecting the role copy number variants (CNVs) play in disease pathogenesis is directly reliant on accurate methods for quantification. The Shar-Pei dog breed is predisposed to a complex autoinflammatory disease with numerous clinical manifestations. One such sign, recurrent fever, was previously shown to be significantly associated with a novel, but unstable CNV (CNV_16.1). Droplet digital PCR (ddPCR) offers a new mechanism for CNV detection via absolute quantification with the promise of added precision and reliability. The aim of this study was to evaluate ddPCR in relation to quantitative PCR (qPCR) and to assess the suitability of the favoured method as a genetic test for Shar-Pei Autoinflammatory Disease (SPAID).
Results
One hundred and ninety-six individuals were assayed using both PCR methods at two CNV positions (CNV_14.3 and CNV_16.1). The digital method revealed a striking result. The CNVs did not follow a continuum of alleles as previously reported, rather the alleles were stable and pedigree analysis showed they adhered to Mendelian segregation. Subsequent analysis of ddPCR case/control data confirmed that both CNVs remained significantly associated with the subphenotype of fever, but also to the encompassing SPAID complex (p < 0.001). In addition, harbouring CNV_16.1 allele five (CNV_16.1|5) resulted in a four-fold increase in the odds for SPAID (p < 0.001). The inclusion of a genetic marker for CNV_16.1 in a genome-wide association test revealed that this variant explained 9.7 % of genetic variance and 25.8 % of the additive genetic heritability of this autoinflammatory disease.
Conclusions
This data shows the utility of the ddPCR method to resolve cryptic copy number inheritance patterns and so open avenues of genetic testing. In its current form, the ddPCR test presented here could be used in canine breeding to reduce the number of homozygote CNV_16.1|5 individuals and thereby to reduce the prevalence of disease in this breed.
Similar content being viewed by others


Background
The role copy number variants (CNVs) play in genomic processes spanning from evolution [1–4], through population genetic diversity [5–8] and disease susceptibility [9, 10] is under constant investigation. Whilst CNVs are less common than single nucleotide polymorphisms (SNPs), these genomic gains and losses are more likely to have an impact on phenotypic diversity [9]. One of the key mechanisms of action for CNVs is through the disruption of gene or regulatory element dosage, leading to perturbed gene expression. In order to dissect the impact these structural variants play, it is essential that they be accurately detected and quantified. Methods such as fluorescence in situ hybridisation (FISH), multiplex ligation-dependent probe amplification (MLPA) and array comparative genomic hybridisation (aCGH) can all be used to detect copy number variants, but the establishment of mode of transmission or genotype may fall to alternate PCR based methods such as quantitative PCR (qPCR), or more recently, droplet digital PCR (ddPCR).
The detection of canine CNVs has advanced dramatically from the first array CGH which revealed 155 variants [11], through to a catalogue of more than 1,600 polymorphisms which have been assayed in both domestic dogs and a variety of members from the Canidae family [5–8]. Through these studies and others (summarised in [12]), we have gained insight into what sets domestic dogs apart from their wild ancestors (e.g. a CNV gain in a region encompassing AMY2B drives increased amylase activity and the ability for domestic animals to digest a starch rich diet [1]) and an understanding of dog phenotypic diversity and disease susceptibility (e.g. a copy number gain encompassing FGF3, FGF4 and FGF19 which results in the breed defining dorsal ridge of the Rhodesian and Thai Ridgeback, but also predisposes these breeds to dermoid sinus [13]). The link between genotype and phenotype relies not only on the ability to identify CNVs, but also on the accuracy of variant quantification. If the CNV is to be used diagnostically, the importance of the latter cannot be overestimated.
This study focuses on two breed specific CNVs found in the Chinese Shar-Pei; the “traditional” (14.3 kb) variant and the “meatmouth” (16.1 kb) variant, located in an overlapping region of chromosome 13 [14]. It has been reported that a high copy number of the 16.1 kb variant is associated with the increased expression of Hyaluronan Synthase 2 (HAS2), the driver of long-chain hyaluronan (HA) synthesis [14, 15]. The elevated expression of HAS2 results in cutaneous hyaluronosis, creating the Shar-Pei’s distinctive thickened and folded skin [16–18]. The increased copy number of the 16.1 kb variant was also shown to be significantly correlated to the occurrence of a breed specific fever syndrome [14]. It was hypothesised that the recurring fever followed the cyclical over production and subsequent degradation of HA and that low molecular weight HA was acting as a danger associated molecular pattern (DAMP) triggering the release of inflammatory interleukins [14, 19].
More recently it was reported that this fever syndrome was actually one of a spectrum of clinical signs including arthritis, ear (otitis) and skin (vesicular hyaluronosis) inflammation and amyloidosis that were encompassed by the larger Shar-Pei Autoinflammatory Disease (SPAID) [20]. In a genetic study which utilised 250 carefully phenotyped individuals, genome wide association analyses showed that the five clinical signs of SPAID overlapped one genetic locus; a region which also encompassed HAS2 and both the 14.3 kb and the 16.1 kb copy number variants [20].
When the 14.3 kb and the 16.1 kb CNVs were first measured, the methodology employed was quantitative PCR (qPCR) [14]. This was a primer-limited multiplex qPCR where the result of an unknown individual was calibrated to the result of an individual with known diploid copy number (i.e. CNV = 2). The error in this measurement was therefore a composite of the errors from four PCR reactions (two individuals for both a target CNV and reference PCR) and quadruplet replications. When applied to the 16.1 kb variant this resulted in a continuum of copy number estimates, from two to fifteen or more [14]. An alternative to qPCR is droplet digital PCR (ddPCR). In the latter case, restriction digested DNA and a multiplex PCR mix are evenly partitioned across many thousands of oil droplets. After the completion of thermocycling, the initial concentration of template DNA is determined from the Poisson distribution of positive (those containing amplified target, be it CNV or reference) and negative (those containing no amplified target) reaction droplets [21]. Results comparing the two PCR methodologies have generally shown gains in precision and reproducibility when using ddPCR versus qPCR [22–24] although, as noted by others, this can come at an increased cost per reaction [25].
With the recent advancement in defining Shar-Pei Autoinflammatory Disease and the availability of alternate methods of quantifying copy number variants, the aims of the current study were three-fold, i) to identify a reliable CNV measurement method (quantitative PCR versus droplet digital PCR), ii) to apply that method to investigate the relationship between CNV load, the occurrence of SPAID and SPAID sub-phenotypes and iii) to assess the utility of CNV variants as a genetic test for SPAID.
Results
Droplet digital PCR has reduced variability compared to quantitative PCR
Copy numbers were measured for 196 Shar-Pei (Additional file 1: Table S1) with three assays (Assay-CNV-East, Assay-CNV-759 and Assay-CNV-E, Fig. 1a) and two methodologies, droplet digital PCR (ddPCR) and quantitative PCR (qPCR). The relationship between assays and methodologies is illustrated in Fig. 1. The linear correlation between methodologies was found to be the highest for CNV-East (r 2 = 0.72, Fig. 1b), followed by CNV-759 (r 2 = 0.64) and was the lowest for CNV-E (r 2 = 0.44). The value for CNV_14.3 as measured by Assay-CNV-East was on average 0.14 CNVs smaller when measured by ddPCR than qPCR. For CNV_16.1 the pattern was discordant with CNV measures on average 3.44 and 0.77 larger (Assay-CNV-759 and Assay-CNV-E respectively), when measured with ddPCR compared to qPCR. Whilst qPCR CNV measures for one individual were not always in agreement between Assay-CNV-759 and Assay-CNV-E (Average CNV difference = 2.66 ± 2.60), they were in extremely close agreement when measured with ddPCR (Average CNV difference = 0.01 ± 0.52). Examples of these inconsistencies in qPCR compared to ddPCR at an individual level are illustrated in Additional file 2: Figure S1. From this point forward, the results of Assay-CNV-759 and Assay-CNV-E will be presented as the result for CNV_16.1, as the former were interchangeable.

a The genomic location of the two CNVs and b the comparison of results obtained using quantitative PCR (qPCR) and droplet digital PCR (ddPCR) for assays in each (CNV_14.3: Assay-CNV-East; CNV_16.1: Assay-CNV-759 and Assay-CNV-E). Regression analysis showed Assay-CNV-East reported reasonably similar results (r 2 = 0.72), whilst concordance was lower for Assay-CNV-759 (r 2 = 0.64) and Assay-CNV-E (r 2 = 0.44). In each assay it can be seen ddPCR reports tighter clustering at higher qPCR values
Breed specific CNVs are stable and inherited in a bi-allelic fashion
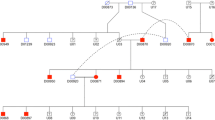
ddPCR showed clearly that the results for both CNV_14.3 and CNV_16.1 formed three clusters (CNV_14.3: 2, 4, 6; CNV_16.1: 2, 6, 10) suggesting that these copy number variants may be acting as stably inherited alleles. We tested this hypothesis in extended pedigrees (n = 92, Fig. 2; Additional file 1: Table S1; Additional file 2: Figure S2) and found that this was in fact true. CNV_14.3 was demonstrated to have two alleles comprising either 1 or 3 copies and for CNV_16.1, alleles of 1 or 5 copies were shown. For the full set of individuals included in the study (n = 327), no alternate combinations were observed.

An example pedigree illustrates the segregation of CNV alleles across generations. The second generation shows a distribution that is in keeping with Mendelian segregation. CNV results are coded to reflect the number of alleles per chromosome. Using the male in generation one as an example, the result for CNV_14.3 (copy number: 4; alleles: 1|3) is written above that for CNV_16.1 (copy number: 6; alleles: 5|1)
A pattern of CNV genotype segregation was observed whereby CNV_14.3 = 2/CNV_16.1 = 10 accounted for 64.5 % of the CNV pairs, followed by CNV_14.3 = 4/CNV_16.1 = 6 (30.3 %) and CNV_14.3 = 6/CNV_16.1 = 2 (4.3 %). However we also recorded the following pairs, CNV_14.3 = 4/CNV_16.1 = 2 (0.6 %) and CNV_14.3 = 2/CNV_16.1 = 6 (0.03 %).
CNV_16.1 is associated with increased HAS2 expression and SPAID risk
Olsson et al., [14] showed that the expression of genes HAS2 and HASas increased with increasing CNV_16.1 copy number. This experiment was replicated using their gene expression measures and new ddPCR calculations of copy number for the assayed fibroblast DNA. The same trend of increased gene expression with CNV_16.1 copy number was observed (Additional file 2: Figure S3).
The ddPCR method provided the resolution required to demonstrate that both duplications were stably transmitted. In order to assess disease correlation, phenotype positive and negative genotype and allele counts were made (Table 1), and 2x2 allele risk- and odds- ratio calculations performed (Table 2). As reported above, 64.5 % of duplication pairs form the pattern of CNV_16.1 = 10/CNV_14.3 = 2. This was patterned carried forward to Table 1, where a high proportion both CNV_16.1 = 10 and CNV_14.3 = 2 were recorded in affected individuals in each disease set. It was shown previously that CNV_14.3 and CNV_16.1 are breed specific variants, and that a copy number of two is observed at both genomic locations in all other breeds [14]. We therefore assessed the link between CNV_16.1 and disease, and not CNV_14.3.
As reported in Table 1, the CNV_16.1 allele 5 (CNV_16.1|5) is the variant highly significantly associated to SPAID (p < 0.0001), and confered a greater than four fold increase in odds-ratio (Table 2). Similar odds ratios were noted for four of the five phenotypes of SPAID, but not for Amyloidosis.
CNV_16.1 is predictive of disease risk
The utility of CNV_16.1 and CNV_14.3 in a genetic test for SPAID was evaluated using random forest (RF) [26] and J48 decision trees [27]. Given the low membership of C1 (n = 34), a comparison with C2 (n = 80) was also calculated. The best model was generated using the data set containing the allelic information from both CNVs and the maximum number of participants (C2 RF AUC = 0.613, Table 3), although the performance of all models compared using the C2 control group were similar (maximum AUC difference 0.014).
Local genomic architecture may explain lower than expected AUC
In the genome wide association analysis (GWAS) of SPAID, Olsson et al. [20] noted high linkage disequilibrium (LD, r 2 > 0.8) across the genomic regions associated with all five SPAID sub-phenotypes. A GWAS was repeated with the addition of CNV_16.1 as a biallelic marker to the genome wide set. With our reduced cohort (SPAID, n = 155; C1, n = 34) and the same methodology as reported previously [20], we found that whilst not reaching genome-wide significance (λ = 1.04; p CNV_16.1 = 2.78 × 10-5; p Bonferroni = 4.55 × 10-7), the CNV marker explained approximately 10 % of the genetic variance (CNV_16.1 = 9.7 %) and close to 25 % of the genetic heritability (CNV_16.1 = 25.8 %).
Discussion
We assessed the ability of two PCR methods, quantitative PCR (qPCR) and droplet digital PCR (ddPCR), to accurately quantify two breed specific CNVs in relation to the susceptibility of Shar-Pei Autoinflammatory Disease (SPAID). We found that whilst disease association remained highly significant whether the CNV assays were measured with either qPCR or ddPCR (All assays, either control group; Mann Whitney p-value < 0.001), only ddPCR allowed for the true bi-allelic pattern of inheritance to be followed. In fact, ddPCR also revealed that whilst CNV pairs were typically inherited in a predictable manner, e.g. CNV_14.3 = 2 copies with CNV_16.1 = 10 copies), recombination does occur at this part of the genome and other combinations of results are observable, albeit at a much lower frequency. The continuum of CNV values we observed when using the qPCR method has also been noted by others using similar means [28]. It is likely that their ability to resolve copy number alleles would also improve with ddPCR. This point is extremely important in the context of genetic testing and future breeding programs.
The reasons as to why ddPCR was able to more clearly resolve copy number in this setting may simply be due to the mechanics of that method versus qPCR. For example, in our hands and with a subset of 50 samples, we generated and read approximately 13,000 droplets per individual tested. Of those accepted droplets, 6,000 on average contained the VIC labelled reference PCR product which equates to 6,000 separate C7orf28b 90 bp PCR products (Additional file 2: Table S3). This is in comparison to the four PCR replicates that were measured for qPCR. The number of FAM droplets is dependent on the number of 16.1 kb or 14.3 kb segments and so averaging this value was not appropriate. There are also differences in the way the two methods estimate errors and also CNV results, e.g. the normalisation to a reference individual for the delta delta CT method of qPCR versus absolute concentration calculation of ddPCR [21].
However, the differences in copy number results (Fig. 1b) may also be a reflection of the architecture in surveyed genome region. It is known that PCR can be more challenging in GC rich regions, but this does not seem to be a contributing factor in this case. Both Shar-Pei copy number elements, and the 100 kb region encompassing them, are estimated to slightly less than the average GC content for dog and a set of primates (36 % versus 41 % in dog, 43 % in human and 42 % in chimpanzee and macaque [29]). It seems more likely that there are complex secondary structure issues that are resolved when the target genomic DNA is digested into small five kb blocks as part of the ddPCR protocol. This may facilitate easier primer binding and product elongation.
Shar-Pei Autoinflammatory Disease (SPAID) is both a phenotypically and genetically complex condition [20]. The 16.1 kb CNV is the marker most associated with disease, explaining 25 % of the genetic heritability in the studied population, however it is the interplay between this genomic region and as yet other undiscovered genes, plus the effect of the dog’s environment, that ultimately determines that individual’s clinical disease status.
In summary, carrying the CNV_16.1|5 allele will increase a dog’s odds of developing disease by four-fold (Table 2), but this predictive measure does not mean that the same carrier will present with clinical disease, be it fever, arthritis, otitis, vesicular hyaluronosis or amyloidosis. For that reason, we would suggest that the results of the CNV_16.1 measurement be used to inform mating strategies, preferentially breeding a homozygous CNV_16.1|5 individual (i.e. CNV_16.1 = 10 copies) with either a CNV_16.1 heterozygote (i.e. CNV_16.1 = 6 copies) or CNV_16.1|1 homozygote (i.e. CNV_16.1 = 2 copies). However, there appear to be very few CNV_16.1|1 homozygotes (CNV_16.1 = 2 copies) in the general population; we found only 16 within our tested set of 327 Shar-Pei. Whilst it may seem prudent to use these individuals widely in order to quickly reduce the number of homozygous CNV_16.1|5 dogs, this may have dire results for the breed. The overuse of CNV_16.1|1 homozygotes could serve to reduce the overall genetic diversity of the breed and perhaps even enrich for as yet unknown diseases.
Conclusions
These results clearly illustrate the potential for ddPCR to quantify the true count of alleles at CNVs. This gain of precision revealed a previously unknown pattern of allele segregation for two Shar-Pei Specific CNVs and in doing so allowed for the evaluation of a genetic test. This test could now be used in carefully managed breeding programs to methodically reduce the number of individuals carrying the disease associate allele (CNV_16.1|5) without dramatically reducing the breed’s overall genetic variation.
Method
SPAID phenotype characterisation
Purebred pet Shar-Pei were sampled from France, the Netherlands, Sweden and the United States following owner consent and ethical approvals (See Declaration). Owners submitted a standardised questionnaire regarding the overall health of their animal. Where possible they also provided detailed medical records and pedigree information. This information was compiled, and in conjunction with veterinarians, used to determine an individual’s case or control status. As per Olsson et al. [20], cases were defined based on the clinical signs of SPAID. These were, SPAID (S, n = 155): Any one or more of the five inflammatory signs of SPAID; Fever (F, n =114): Recurrent bouts of fever lasting 6–72 h with no underlying infection; Arthritis (Ar, n = 79): Recurrent or prolonged bouts of joint (hock) inflammation with no known underlying infection; Vesicular Hyaluronosis (V, n = 44): Dermatological vesicular changes to the skin leading to recurrent or persistent secondary inflammation; Otitis (O, n = 34): Recurrent or chronic inflammation of the ears; Amyloidosis (Am, n = 31): Congo Red stained amyloid deposits observed in a post mortem kidney biopsy. These categories were not discrete (Additional file 2: Table S2).
Three control groups were defined. The first, Control 1 (C1, n = 34), encompassed individuals older than 60 months with no signs of SPAID, whilst Control 2 (C2, n = 80) was more relaxed and contained all dogs free from SPAID, irrespective of age. Control 3 (C3, n = 17) was specific for the sub-phenotype of Amyloidosis and included only those healthy individuals that were negative for Congo Red stained amyloid deposits in post mortem kidney tissue and were also free from a clinical history of unexplained inflammation. Additional pedigree material (n = 92, Additional file 1: Table S1) was used to test the stability and transmission of both CNVs assayed.
Genotyping and genetic analysis
Two breed-specific copy number variants were identified in Olsson et al., [14]. These were termed Traditional (14.3 kb; CanFam3.1 chr13:20,706,841-20,721,149) and Meatmouth (16.1 kb; CanFam3.1 chr13: 20,709,024-20,725,124). To aid clarity, in this manuscript they are named CNV_14.3 and CNV_16.1 to reflect their length as opposed to Shar-Pei breed subtypes.
Olsson et al., [14] designed two assay sets (primer pair and fluorescently labelled probe) that are also utilised in the current analysis. The first was unique to CNV_16.1, Assay-CNV-E, and the second acts as a housekeeper for normalisation, Assay-C7orf28b. Two new assay pairs were designed for the current work. One set was designed to a unique region of CNV_14.3, Assay-CNV-East, and the other is an additional set for CNV_16.1, Assay-CNV-759. All primer and probe assay sets are listed in Additional file 2: Table S3 and illustrated in Fig. 1.
We utilised two methodologies, quantitative real-time PCR (qPCR) and droplet digital PCR (ddPCR) to estimate the number of CNV_14.3 and CNV_16.1 copies present in each DNA sample tested. For qPCR, the (ΔΔCT) relative quantification method and a reference individual with known copy number status (German Shepherd 95, GSP95, Additional file 1: Table S1) was used. The multiplex reaction contained a primer limited target copy number assay for one of the following target assays, Assay-CNV-East or Assay-CNV-759 or Assay-CNV-E, plus the housekeeper assay, Assay-C7orf28b. A target assay comprised 300nM of forward and reverse primer plus 250nM of FAM labelled probe (LifeTechnologies) whilst the housekeeper contained 900nM of forward and reverse primer plus 250nM of VIC labelled probe (LifeTechnologies). These multiplex reactions were performed in quadruplet using 10 ng of gDNA, Genotyping Master Mix (LifeTechnologies) and a 7900HT Real-Time PCR machine (LifeTechnologies) following manufacturers specifications.
For ddPCR, absolute quantification was performed using 15 ng of DraI (NEB) pre-restriction digested DNA in a 20ul reaction mix containing 1x ddPCR Supermix for Probes (BioRad) with 900nM target and reference primers and 250nM of target and reference probes. The primers and probes were the same used for qPCR (Additional file 2: Table S3). The ddPCR reaction mix was portioned into oil droplets following the manufacturer’s (BioRad) specifications [30], amplified at 58 °C in a C1000 Touch thermocycler (BioRad) and quantified using a QX100 instrument (BioRad). QuantaSoft v1.3.1.0 was used for the visualisation of digital droplet results and for the calculation of template per droplet based on a Poisson distribution.
Statistical analysis
In order to assess the utility of either Assay-CNV-E or Assay-CNV-759 as a diagnostic test for SPAID, we used the allelic values of each assay as variables and constructed predictive models using WEKA software [27]. We built the models using two different statistical-learning algorithms: J48 implementation [27] of Quinlan’s [31] C4.5 decision trees and Breiman’s [26] Random Forest (RF). The two models were selected to overcome any biases in classifier choice. RF-based classifiers have proven robust and reliable on most types of data and as such have become the primary algorithm of choice. RFs, being an ensemble method, provide only a limited insight into the actual classification process in their basic version. This limits our ability to obtain additional insight into the nature of the modelled phenomenon. For this reason we also constructed an extra set of classifiers using another popular and robust statistical-learning algorithm, decision trees. Both types of classifiers were evaluated in a standard 10-fold cross-validation.
Genome wide association study
Bialellic results from the ddPCR CNV_16.1 assay were combined with existing Illumina CanineHD array data [20] for the available 155 SPAID cases and 34 control individuals. GenABEL v1.7-2 [32] in R v2.15.0 was used to perform the analysis. Quality control involved tests for missing genotype calls for single SNP and individuals (<5 %), minor allele frequency (<0.05) and strong deviations from Hardy-Weinberg Equilibrium (HWE, p > 1 x10-8). A FDR rate of 0.2 was applied to the controls. From the starting set of 173,663 markers genotyped, 109,966 remained for analysis. To correct for population stratification, a polygenic mixed model was fitted which encompassed the Identity-By-State matrix.
Ethics statement
Samples were collected following owner consent and with the following ethical approvals: DEC Utrecht University Ethical Committee, permit #10813; Ethical Board for Experimental Animals in Uppsala, permit #C103/10; Massachusetts Institute of Technology Committee for Animal Care, permit #0910-074-13.
Availability of data and material
The dataset supporting the conclusions of this article is included within the article (and its additional files).
Abbreviations
- aCGH:
-
array comparative genomic hybridisation
- AMY2B :
-
amylase, alpha 2B
- AUC:
-
area under the curve
- CNV:
-
copy number variant
- DAMP:
-
danger associated molecular pattern
- ddPCR:
-
droplet digital polymerase chain reaction
- FDR:
-
False discovery rate
- FGF3, FGF4 and FGF19 :
-
Fibroblast Growth Factor 3, 4 and 19
- FISH:
-
fluorescence in situ hybridisation
- GWAS:
-
genome wide association analysis
- HA:
-
hyaluronan
- HAS2 :
-
Hyaluronan Synthase 2
- HWE:
-
Hardy-Weinberg Equilibrium
- kb:
-
kilobase
- LD:
-
linkage disequilibrium
- MLPA:
-
multiplex ligation-dependent probe amplification
- nM:
-
nanomolar
- PCR:
-
polymerase chain reaction
- qPCR:
-
quantitative polymerase chain reaction
- RF:
-
random forest
- SNP:
-
single nucleotide polymorphisms
- SPAID:
-
Shar-Pei Autoinflammatory Disease
References
Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, Liberg O, Arnemo JM, Hedhammar A, Lindblad-Toh K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495(7441):360–4.
Lou H, Lu Y, Lu D, Fu R, Wang X, Feng Q, Wu S, Yang Y, Li S, Kang L, Guan Y, Hoh BP, Chung YJ, Jin L, Su B, Xu S. A 3.4-kb Copy-Number Deletion near EPAS1 Is Significantly Enriched in High-Altitude Tibetans but Absent from the Denisovan Sequence. Am J Hum Genet. 2015;97(1):54–66.
Radke DW, Lee C. Adaptive potential of genomic structural variation in human and mammalian evolution. Brief Funct Genomics. 2015;14(5):358–68.
Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, Jorde LB, Posukh OL, Sahakyan H, Watkins WS, Yepiskoposyan L, Abdullah MS, Bravi CM, Capelli C, Hervig T, Wee JT, Tyler-Smith C, van Driem G, Romero IG, Jha AR, Karachanak-Yankova S, Toncheva D, Comas D, Henn B, Kivisild T, Ruiz-Linares A, Sajantila A, Metspalu E, Parik J, Villems R, Starikovskaya EB, Ayodo G, Beall CM, Di Rienzo A, Hammer MF, Khusainova R, Khusnutdinova E, Klitz W, Winkler C, Labuda D, Metspalu M, Tishkoff SA, Dryomov S, Sukernik R, Patterson N, Reich D, Eichler EE. Global diversity, population stratification, and selection of human copy-number variation. Science. 2015;349(6253):aab3761.
Molin AM, Berglund J, Webster MT, Lindblad-Toh K. Genome-wide copy number variant discovery in dogs using the CanineHD genotyping array. BMC Genomics. 2014;15:210.
Nicholas TJ, Cheng Z, Ventura M, Mealey K, Eichler EE, Akey JM. The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Res. 2009;19(3):491–9.
Nicholas TJ, Baker C, Eichler EE, Akey JM. A high-resolution integrated map of copy number polymorphisms within and between breeds of the modern domesticated dog. BMC Genomics. 2011;12:414.
Ramirez O, Olalde I, Berglund J, Lorente-Galdos B, Hernandez-Rodriguez J, Quilez J, Webster MT, Wayne RK, Lalueza-Fox C, Vilà C, Marques-Bonet T. Analysis of structural diversity in wolf-like canids reveals post-domestication variants. BMC Genomics. 2014;15:465.
Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–55.
Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–83.
Chen WK, Swartz JD, Rush LJ, Alvarez CE. Mapping DNA structural variation in dogs. Genome Res. 2009;19(3):500–9.
Alvarez CE, Akey JM. Copy number variation in the domestic dog. Mamm Genome. 2012;23:144–63.
Salmon Hillbertz NH, Isaksson M, Karlsson EK, Hellmén E, Pielberg GR, Savolainen P, Wade CM, von Euler H, Gustafson U, Hedhammar A, Nilsson M, Lindblad-Toh K, Andersson L, Andersson G. Duplication of FGF3, FGF4, FGF19 and ORAOV1 causes hair ridge and predisposition to dermoid sinus in Ridgeback dogs. Nat Genet. 2007;39(11):1318–20.
Olsson M, Meadows JR, Truvé K, Rosengren Pielberg G, Puppo F, Mauceli E, Quilez J, Tonomura N, Zanna G, Docampo MJ, Bassols A, Avery AC, Karlsson EK, Thomas A, Kastner DL, Bongcam-Rudloff E, Webster MT, Sanchez A, Hedhammar A, Remmers EF, Andersson L, Ferrer L, Tintle L, Lindblad-Toh K. A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet. 2011;7(3):e1001332.
Itano N, Kimata K. Mammalian hyaluronan synthases. IUBMB Life. 2002;54:195–9.
Docampo MJ, Zanna G, Fondevila D, Cabrera J, López-Iglesias C, Carvalho A, Cerrato S, Ferrer L, Bassols A. Increased HAS2-driven hyaluronic acid synthesis in shar-pei dogs with hereditary cutaneous hyaluronosis (mucinosis). Vet Dermatol. 2011;22:535–45.
Zanna G, Fondevila D, Bardagí M, Docampo MJ, Bassols A, Ferrer L. Cutaneous mucinosis in shar-pei dogs is due to hyaluronic acid deposition and is associated with high levels of hyaluronic acid in serum. Vet Dermatol. 2008;19(5):314–8.
Zanna G, Docampo MJ, Fondevila D, Bardagí M, Bassols A, Ferrer L. Hereditary cutaneous mucinosis in shar pei dogs is associated with increased hyaluronan synthase-2 mRNA transcription by cultured dermal fibroblasts. Vet Dermatol. 2009;20:377–82.
Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, Grant EP, Coyle AJ, Misaghi A, Hoffman HM, Gallo RL. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem. 2009;284:12762–71.
Olsson M, Tintle L, Kierczak M, Perloski M, Tonomura N, Lundquist A, Murén E, Fels M, Tengvall K, Pielberg G, Dufaure de Citres C, Dorso L, Abadie J, Hanson J, Thomas A, Leegwater P, Hedhammar Å, Lindblad-Toh K, Meadows JR. Thorough investigation of a canine autoinflammatory disease (AID) confirms one main risk locus and suggests a modifier locus for amyloidosis. PLoS One. 2013;8(10):e75242.
Pinheiro LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S, Emslie KR. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem. 2012;84:1003–11.
Gutiérrez-Aguirre I, Rački N, Dreo T, Ravnikar M. Droplet digital PCR for absolute quantification of pathogens. Methods Mol Biol. 2015;1302:331–47.
Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang S, Stumbo DP, Hodges SP, Romine S, Milanovich FP, White HE, Regan JF, Karlin-Neumann GA, Hindson CM, Saxonov S, Colston BW. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–10.
Te SH, Chen EY, Gin KY. Multiplex assay for two bloom-forming cyanobacteria, Cylindrospermopsis and Microcystis - A comparison between qPCR and ddPCR. Appl Environ Microbiol. 2015;81(15):5203–11.
Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, Vessella RL, Tewari M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10:1003–5.
Breiman L. Random Forests. Mach Learn. 2001;45(1):5–32.
Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH. The WEKA Data Mining Software: An Update. SIGKDD Explorations. 2009;11(1):10–18.
Metzger J, Distl O. A study of Shar-Pei dogs refutes association of the ‘meatmouth’ duplication near HAS2 with Familial Shar-Pei Fever. Anim Genet. 2014;45(5):763–4.
Karro JE, Peifer M, Hardison RC, Kollmann M, von Grünberg HH. Exponential decay of GC content detected by strand-symmetric substitution rates influences the evolution of isochore structure. Mol Biol Evol. 2008;25(2):362–74.
Mazaika E, Digital HJ, Droplet PCR. CNV Analysis and Other Applications. Curr Protoc Hum Genet. 2014;82:7.24.1–7.24.13.
Quinlan, JR. C4.5: Programs for Machine Learning. San Francisco, USA: Morgan Kaufmann Publishers; 1993.
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23(10):1294–6.
Acknowledgements
We thank the global community of Shar-Pei owners, breeders, breed clubs and veterinarians who supported this study and contributed samples. Max Fels, Tekla Kolbow and Sabine Bolte are acknowledged for their technical assistance. Canine sample collection was facilitated by the Cani-DNA biobank (France) and the Canine Biobank at Uppsala University and the Swedish University of Agricultural Sciences (Sweden).
Funding
JRSM was supported by the Swedish Research Council, FORMAS (221-2012-1531). French samples were collected by the Cani-DNA biobank which is part of the CRB-Anim infrastructure, ANR-11-INBS-0003, funded by the French National Research Agency in the frame of the ‘Investing for the Future’ program. These funding sources did not play a role in the design, analysis or interpretation of results.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors disclose the following: Caroline Dufaure de Citres and Anne Thomas are employed by ANTAGENE and Mia Olsson, Linda Tintle, Kerstin Lindblad-Toh and Jennifer Meadows have filed a patent (Y/Ref. BI-2012/088; O/Ref. BINS.P0002US SN 13/161,213) related to the current material. The authors state that these interests do not alter their adherence to policies on sharing data and materials.
Authors’ contributions
Conceived and designed the experiments: JRSM, MKi. Performed the experiments: MKo, AT, CDdC, MKi, ÅK, JJ, JA, JRSM, MO. Analysed the data: MKi, MO, JRSM. Contributed reagents/materials/analysis tools: JRSM, AT, CDdC, PL, ÅH, LT, KLT. Wrote the paper: JRSM. All authors read and approved the final manuscript.
Additional files
Additional file 1: Table S1.
Summary information for each individual included in the current study. (XLSX 69 kb)
Additional file 2: Table S2.
Phenotypic overlap observed within the genetic test cohort. Table S3. Primers and Probes used in analysis. Figure S1. Illustrative plots for two individuals showing the results obtained for each assay and methodology. Figure S2. Five pedigrees illustrating the segregation of copy number variant (CNV) alleles. Figure S3. The relationship of increased HAS2 and HAS2as gene expression with increased CNV_16.1 copy number holds with droplet digital PCR (ddPCR) CNV measures. (DOCX 1628 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Olsson, M., Kierczak, M., Karlsson, Å. et al. Absolute quantification reveals the stable transmission of a high copy number variant linked to autoinflammatory disease. BMC Genomics 17, 299 (2016). https://doi.org/10.1186/s12864-016-2619-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-2619-0