
Abstract
Background
The 100-pod fresh weight (PFW), 100-seed fresh weight (SFW), 100-seed dry weight (SDW) and moisture content of fresh seeds (MCFS) at the R6 stage are crucial factors for vegetable soybean yield. However, the genetic basis of yield at the R6 stage remains largely ambiguous in soybean.
Results
To better understand the molecular mechanism underlying yield, we investigated four yield-related traits of 133 soybean landraces in two consecutive years and conducted a genome-wide association study (GWAS) using 82,187 single nucleotide polymorphisms (SNPs). The GWAS results revealed a total of 14, 15, 63 and 48 SNPs for PFW, SFW, SDW and MCFS, respectively. Among these markers, 35 SNPs were repeatedly identified in all evaluated environments (2015, 2016, and the average across the two years), and most co-localized with yield-related QTLs identified in previous studies. AX-90496773 and AX-90460290 were large-effect markers for PFW and MCFS, respectively. The two markers were stably identified in all environments and tagged to linkage disequilibrium (LD) blocks. Six potential candidate genes were predicted in LD blocks; five of them showed significantly different expression levels between the extreme materials with large PFW or MCFS variation at the seed development stage. Therefore, the five genes Glyma.16g018200, Glyma.16g018300, Glyma.05g243400, Glyma.05g244100 and Glyma.05g245300 were regarded as candidate genes associated with PFW and MCFS.
Conclusion
These results provide useful information for the development of functional markers and exploration of candidate genes in vegetable soybean high-yield breeding programs.
Similar content being viewed by others


Background
Soybean (Glycine max (L.) Merr.) is a widely cultivated oil crop worldwide. Soybean seeds are used to supply edible oil and serve as a source of high-quality plant protein [1]. According to different harvest times and uses, soybean crops can be divided into grain or vegetable crops. Vegetable soybean is harvested during the R6 growth stage when the pods are still green and fully filled with seeds [2]. The characteristics of large pods and large grains are important visual qualities of vegetable soybeans [3, 4]. Therefore, yield has long been considered one of the most important traits in vegetable soybean breeding. The vegetable soybean yield is directly determined by yield components, including the number of pods per plant, seeds per pod, fresh seed weight and fresh pod weight. Furthermore, vegetable soybean seeds have a high moisture content of approximately 70.05%, which serves both as a yield component and as an influencing factor of sensory quality [5]. In maize, grain moisture has a higher h2 than does grain yield, and several quantitative trait loci (QTLs) are commonly associated with grain yield and grain moisture [6]. With economic development, the demand for vegetable soybeans has increased, but there are fewer available reports on the yield of vegetable soybean than grain soybean at present. Therefore, dissecting the genetic basis of soybean yield at the R6 stage is necessary and will help to improve the yield potential of vegetable soybean.
Yield-related traits are usually complex quantitative traits influenced by multiple QTLs. Previous studies were conducted to dissect the genetic basis of yield-related traits in biparental populations. Hundreds of QTLs were detected across the whole genome of soybean, many were simultaneously detected in multiple populations [7,8,9,10,11,12,13]. These studies demonstrated that the genetic mapping of quantitative traits using genetic linkage maps is an efficient approach for identifying QTLs. Compared with linkage mapping, a genome-wide association study (GWAS) is a more powerful method for dissecting the QTLs underlying agronomically important traits in natural populations with a high density of markers. Natural populations contain more genetic diversity than cross-derived segregating populations, which can be applied directly in GWAS analysis [14]. In addition, GWAS can effectively identify candidate genes that are closely linked to target traits, due to the low level of genomic linkage disequilibrium (LD) [15,16,17,18].
At present, association studies have been successfully performed in grain soybean for yield-related traits. For example, 19 SNPs and 5 haplotypes for yield and yield components were identified in a soybean landrace population [19]. For seed size and shape, a total of 59 large-effect QTLs and 31 QTL-by-environment interactions were identified in another study, which were closely related to seed yield and appearance quality [20]. Furthermore, multiple research groups have searched for QTLs related to flowering time and maturity dates that could influence soybean yield [21, 22]. Many of the above QTLs are located in or near QTLs reported in the previously linkage analysis. Based on the QTLs reported to date, several candidate genes have been identified. Gu et al. (2017) proposed SoyWRKY15a as a candidate gene locus for seed size, and differential expression of its orthologous genes GmWRKY15a and GsWRKY15a in soybean pods was correlated with the seed weight [23]. However, the molecular mechanism underlying yield-related traits in vegetable soybean remains unclear.
In this study, we genotyped a panel of 133 soybean landraces using 82,187 SNPs and surveyed four yield-related traits at the R6 stage in two consecutive years. The objectives of this study were to (1) reveal the genetic basis of yield-related traits in soybean at the R6 stage and (2) provide valuable markers and candidate genes for the molecular breeding of vegetable soybean.
Methods
Plant materials and field trials
A total of 133 soybean landraces came from the soybean mini core collection, and the soybean mini core collection were selected from 23,587 soybean germplasms in the Chinese National Soybean GeneBank. Thus the 133 soybean landraces had abundant genetic diversity and were suitable for association analysis [24]. The 133 soybean germplasms came from 24 provinces and were distributed in four ecoregions of China as follows: The Northeast region (NER), the North region (NR), the Huanghuai region (HHR) and the South region (SR) (Additional file 1: Table S1).
These germplasms included abundant genetic diversity due to geographic, climatic and cultivation factors present in China and could be used for GWAS analysis. They were planted at the Jiangpu Experimental Station of the Agricultural University of Nanjing, China (32.04°N 118.63°E) in late June 2015 and 2016, according to a completely randomized block design, with two years and three replications. Planting was performed with two rows per plot and 40 plants per row, with plant spacing of 10 cm and row spacing of 50 cm.
Phenotypic evaluation and statistical analysis
Four yield-related traits, the 100-pod fresh weight (PFW), 100-seed fresh weight (SFW), 100-seed dry weight (SDW) and moisture content of fresh seeds (MCFS), were investigated at the R6 growth stage during which the pods contain full-size green beans. At least fifty pods were harvested for each replication in each year. The pods were then weighed on the electronic scale, and PFW (g) was calculated. Next, the pod husks were stripped, and seed weight was measured to determine SFW (g). The seeds were then killed by heating at 110 °C for 30 min and dried at 65 °C to a constant weight to obtain the SDW (g). Finally, MCFS (%) was calculated using the following formula.
Statistical analyses for all traits were performed using SAS version 9.4 [25]. Analysis of variance (ANOVA) of the phenotypic data across multiple environments was performed using PROC GLM. The statistical model was as follows: yijk = μ + αi + βj + γkj + (αβ)ij + εijk, where μ is the overall mean, αi is the genetic effect of the ith genotype, βj is the effect of the jth environment, γkj is the random effect of the kth replicate in the jth environment, (αβ)ij is the interaction effect between the ith genotype and the jth environment, and εijk is the residual. As sources of variation, the environment, genotype, replication within environment, and genotype × environment were treated as random effects. The formula for calculating broad-sense heritability is:
\( {h}^2={\alpha}_g^2/\left({\alpha}_g^2+{\alpha}_{ge}^2/n+{\alpha}_{\varepsilon}^2/ rn\right) \), σ2g is the genotypic variance, σ2ge is the genotype by environment interaction variance, α2ε is the error variance, n is the number of environments, and r is the number of replications. All of the above variance values can be calculated using the REML method for the SAS VARCOMP procedure.
SNP genotyping
The association panel was genotyped using a 180 K AXIOM® SoyaSNP array [26], and a total of 169,028 high-quality single nucleotide polymorphisms (SNPs) were used for association mapping. In this study, SNPs with minor allelic frequencies (MAFs) of less than 5% and a missing rate of more than 10% were excluded from further analysis. As a result, 82,187 SNPs remained and were used in marker-trait association analysis. The density of the SNPs was estimated as one SNP per 11.76 kb for the 20 soybean chromosomes.
Population structure and linkage disequilibrium
We used PLINK V1.07 to perform SNP filtering by setting the MAF to 0.2 and the call rate to 0.1. The remaining data contained 8270 SNPs, which were used to construct a population structure in STRUCTURE 2.3.4. The number of subgroups (K) was set from 1 to 6, with 4 replications. The length of the burn-in period was set to 10,000, and the number of Monte Carlo Markov Chain (MCMC) replications was set to 100,000. The suitable K in this population was determined by the log probability of the data LnP(D) and delta K. In previous studies, the mini core collection was divided into two or three distinct subgroups depending on the markers used in the tests [24, 27, 28].
A total of 82,187 SNPs (MAF > 0.05) were employed to conduct principal component analysis (PCA) and construct a neighbor-joining (NJ) phylogenetic tree using PLINK V1.07 and PHYLIP. The kinship matrix was assessed using TASSEL V5.2.15 to determine the relatedness among individuals based on the SNP dataset [29]. Linkage disequilibrium parameters (r2) for estimating the degree of LD between pairwise SNPs (MAF > 0.2) were calculated using PLINK V1.07, and a figure showing average LD decay was drawn with R [30]. The LD decay rate of the population was measured as the chromosomal distance when the average r2 decreased to half its maximum value [31].
Association mapping
The population structure and relative kinship in natural populations always result in a high level of spurious positives in association mapping [32]. After assessment of the population structure (Q), PCA, and evaluation of the relative kinship (K) of 133 soybean landraces, the effects of these parameters on association analyses were evaluated with the following statistical models: (1) a general linear model (GLM) with Q; (2) GLM with PCA; (3) a mixed linear model (MLM) with PCA and K; (4) and MLM with Q and K. Genome-wide association analyses were performed by TASSEL V5.2.15. The significance threshold for SNP-trait associations was determined by 1/n where n is the number of markers in the association panel, and P ≤ 1/82,187, or –Log10(P) ≥ 4.91 [33].
Prediction of candidate genes
To identify candidate genes underlying the association signals, we selected significant SNPs associated with large-effect QTLs to search candidate genes in their candidate regions. The candidate regions were defined by the average LD decay distance or the LD block. The soybean reference genome was Wm82.a2.v1, and the functional annotations and tissue expression of genes located in the candidate regions were obtained from Phytozome (http://www.phytozome.net). Based on the soybean genomic annotations and expression data, potential candidate genes were predicted.
To determine the expression of potential candidate genes, we used quantitative real-time PCR (qRT-PCR) to analyze their expression in extreme materials with large phenotypic differences. Based on the phenotypic data in 2015 and 2016, the materials (ZDD21907 (PFW 198 g), ZDD20532 (PFW 39 g), ZDD01983 (MCFS 75.5%) and ZDD02315 (MCFS 61.7%)) showed stable and large phenotypic differences, therefore we chosen them as the extreme materials and cultivated in the field. Three replicate biological samples were collected in liquid nitrogen at three stages during soybean seed development (R5(3-mm-long seeds in a pod at one of the four uppermost nodes on the main stem, with a fully developed leaf), R6 (pods containing green seeds that fill the pod cavity, located at one of the four uppermost nodes on the main stem, with a fully developed leaf) and R7 (one normal pod on the main stem that has reached the mature pod color)), as defined by Fehr (1977) [34]. Total RNA was extracted from R5, R6, and R7 seeds using a RNA Simple Total RNA kit (TIANGEN, China). cDNA was synthesized using a Prime Script™ RT Reagent Kit (TaKaRa, Japan) with a standard protocol. The CDS sequences of the potential candidate genes were obtained from Phytozome (http://www.phytozome.net). The qRT-PCR primers were designed with Primer Premier 5.0 and were listed in Additional file 2: Table S2. GmEF1β (GenBank ID AK286947.1) was selected as the control gene, and the qRT-PCR assays were conducted three times using a Light Cycler 480 instrument. The relative expression level of the candidate genes was calculated using the comparative 2−△△CT method [35]. Statistical analyses were performed with Dunnett’s tests and Student’s t-tests.
Results
Phenotypic analysis of four yield-related traits
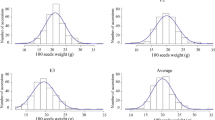
A total of 133 soybean landraces were planted in two consecutive years, and four yield-related traits were investigated. The average values of these traits across the two years showed a continuous distribution in the GWAS panel of 133 soybean landraces, with a wide range of variation (Table 1). PFW exhibited 9.25-fold variation, ranging from 35.9 g to 332.1 g, with an average of 118.2 ± 39.2 g. SFW and SDW showed approximately 8-fold differences, ranging from 8.7 g to 72.4 g and 2.7 g to 21.7 g, respectively. MCFS showed 1.38-fold variation, ranging from 57.0 to 79.0%, with an average of 66.0 ± 4.0%. The frequency distribution of the four yield-related traits displayed an approximately normal distribution except for a few materials that showed large deviation (Fig. 1). According to the method described by Wyman (1991) [36], the broad-sense heritability (h2) was calculated for the four traits. All traits presented an h2 above 82%, suggesting that genetic effects play a predominant role in the phenotype variation of these traits (Table 1). Phenotypic correlations were analyzed between the four traits, and most exhibited significant positive correlations with each other (p < 0.05; Table 2). Highly significant positive correlations were observed between PFW, SFW and SDW, with phenotypic correlation coefficients (rp) above 0.914. MCFS showed a significant positive correlation with PFW and SFW (rp = 0.205, rp = 0.245) but showed a nonsignificant negative correlation with SDW, suggesting that MCFS is an important factor influencing the yield of fresh pods.

Distribution of four yield-related traits, showing mean values across 2 years: a 100-pod fresh weight (g), b 100-seed fresh weight, c 100-seed dry weight, and d moisture content of fresh seeds
Distribution of markers and linkage disequilibrium
A total of 82,187 high-quality SNPs (MAF > 0.05, missing rate < 10%) were used for a GWAS of the four traits, with an average marker density of 11.76 kb/SNP at the genome-wide scale. The lowest marker density (16.28 kb/SNP) was found on Chr.14, and the highest marker density (9.57 kb/SNP) was found on Chr.16. Thus, the markers were unevenly distributed throughout the genome (Additional file 3: Table S3). The MAFs of the 82,187 SNPs are shown in Fig. 2. The average MAF was 0.24, and most of the SNPs (60.5%) exhibited an MAF higher than 0.2. The mean gene diversity (GD) was 0.37, and the values ranged from 0.34 to 0.40. The polymorphism information content (PIC) of all markers ranged from 0.29 to 0.33, with an average of 0.31 (Additional file 3: Table S3).

Minor allelic frequency distribution in 133 soybean landraces based on 82,187 SNPs
Genome-wide LD decay in the association panel was estimated. A rapid decline in LD was observed with increasing physical distance between pairwise SNPs. The mean length of LD decay decreased rapidly to 21 kb at a cut-off of r2 = 0.5. The overall LD decay for all chromosomes was estimated as 119.07 kb, where r2 = 0.375 (half of its maximum value) (Fig. 3).

Average linkage disequilibrium (LD) decay rate estimated among co-chromosome SNPs
Population structure analysis
Population structure analysis showed that the mean LnP (K) did not plateau at a single k value but instead continued to increase with relatively constant increments. Calculation of Delta K revealed a sharp peak at k = 2; therefore, the 133 soybean landraces were divided into two subgroups, designated subgroup1 and subgroup2 (Fig. 4a and c). The geographical origins of the 133 soybean landraces were the Northeast region (NER), the North region (NR), the Huanghuai region (HHR) and the South region (SR). Subgroup 1 contained 101 accessions; among these, 63 accessions belonged to SR, 21 accessions belonged to HHR, 5 accessions belonged to NR, and 12 accessions belonged to NER. Subgroup 2 was small and included only 32 accessions; among these, 2 accessions belonged to SR, 10 accessions belonged to HHR, 13 accessions belonged to NR, and 7 accessions belonged to NER (Additional file 4: Table S4). Notably, most accessions from SR (97%) were included in subgroup 1, whereas most accessions from NR (72%) were included in subgroup 2, suggesting that the population stratification of the 133 accessions essentially corresponded to their geographic origins. The NJ tree and PCA provided further support for the population structure results (Fig. 4b and d).

Population structure analysis of 133 soybean landraces. a The mean LnP(k) and Delta k values when k ranges from 1 to 6. b Two-dimensional scatter plot of PCA, the green dot represents subgroup 1 and the red dot represents subgroup 2. c Population structure of 133 soybean landraces, there are two colored segments and each segment represents the percentage of the individual in the population. d A neighbor-joining tree of the 133 soybean landraces that can be divided into two subgroups
Model comparison for the control of false associations
Association analyses for the four yield-related traits were performed to evaluate the effects of different models on the control of false associations. For PFW and SFW, the observed P values from the GLM(Q) model showed the greatest deviation from the expected P values assuming that no association exists, followed by the GLM (PCA) model. The P values from the MLM (Q + K) and MLM (PCA + K) models were similar and close to the expected P values, and the effects of the MLM (Q + K) and MLM (PCA + K) models on the controlling false associations were similar (Fig. 5). For SDW and MCFS, the observed P values from the MLM (PCA + K) and MLM (Q + K) models were lower than the expected P values, suggesting that the two models excessively corrected the observed P values; thus, no significant associations were identified. The observed P values from the GLM (PCA) and GLM (Q) models were higher than the expected P values, and the observed P values from GLM (PCA) were much closer to the expected P values than those from the GLM (Q) model, indicating that the GLM (PCA) model could effectively control false-positive associations and avoid false-negative associations. Thus, for PFW and SFW, the MLM (Q + K) model was chosen for subsequent association analyses, whereas for SDW and MCFS, the GLM (PCA) model was selected.

Quantile–quantile plots of estimated −log10 (P) from association analysis of four yield-related traits in two years (2015 and 2016): a 100-pod fresh weight, b 100-seed fresh weight, c 100-seed dry weight, and d moisture content of fresh seeds. Red line represents expected P values with no association. The black line represents the observed P values using the GLM (PCA) model. The green line represents the observed P values using the GLM (Q) model. The yellow line represents the observed P values using the MLM (PCA + K) model. The blue line represents the observed P values using the MLM (Q + K) model
Genome wide association analysis of four yield-related traits
Using GWAS, a total of 111 and 146 associations (−Log10(P) > 4.91) were evaluated for the four yield-related traits using the means across 2 years and within individual years, respectively (Additional file 5: Table S5). The resultant quantile–quantile plots and Manhattan plots are shown in Additional file 6: Figure S1, Additional file 7: Figure S2, Additional file 8: Figure S3 and Additional file 9: Figure S4. For PFW, fourteen SNPs were detected (Additional file 5: Table S5). Among these SNPs, nine were repeatedly detected in all environments and were distributed on 7 of 20 soybean chromosomes, and the contribution of a single marker to the observed phenotypic variation was 25.12–33.61% (Table 3). AX-90496773 presented the largest phenotypic difference of 16.33 g between alleles, with an effect on PFW (R2 = 29.99%). For SFW, fifteen significant SNPs were detected (Additional file 5: Table S5). Among these SNPs, only four were repeatedly detected in all environments, and each SNP could explain a large proportion (26.54–27.8%) of the phenotypic variance (Table 3). AX-90519309 had a large effect (R2 = 27.47%) on SFW, with variance of 1.74 g between alleles. Sixty-three SNPs were significantly associated with SDW (Additional file 5: Table S5). Among these, eight SNPs were repeatedly detected in all environments (Table 3). AX-90501040 had the largest effect (R2 = 24.87%) on SDW, associated with a difference of 5.81 g between alleles. For MCFS, a total of forty-eight SNPs were identified (Additional file 5: Table S5). Of these, twenty SNPs were repeatedly detected in all environments, and all were located in a range of 164 kb (41791118–41,955,229) on chromosome 5 (Table 3). AX-90435701 and AX-90460290 had the largest effect (R2 = 21.56%) on MCFS, associated with a difference of 3.51% between alleles. Altogether, thirty-five markers were repeatedly associated with one of the four yield-related traits in all environments. In addition, four markers (AX-90490395, AX-90481424, AX-90370125 and AX-90519309) were commonly associated with both PFW and SFW, and two markers (AX-90328574 and AX-90496773) were associated with both PFW and SDW in all environments (Table 3). However, no markers overlapped between MCFS and the other three traits.
Prediction of candidate genes
In this study, we were particularly interested in the markers with large effects, such as the PFW marker AX-90496773 (Gm16_1,617,227, MAF = 0.07) on chromosome 16, and the MCFS marker AX-90460290 (Gm05_41,927,984, MAF = 0.47) on chromosome 5. Compared with the alternative alleles, the PFW of the materials carrying the favorable allele (AA) at AX-90496773 was 16.33g higher than the materials carrying the unfavorable allele (GG), the MCFS of the materials carrying the favorable allele (GG) at AX-90460290 was 3.51% higher than the materials carrying the unfavorable allele (AA) (Fig. 6). LD analysis showed that AX-90496773 and AX-90460290 can be mapped to chromosomal regions of 34.5 kb on Gm16 and 189.1 kb on Gm05, respectively (Fig. 7). Within the regions of AX-90496773 and AX-90460290, there were five and twenty-seven putative genes, respectively. According to the functional annotations and the expression patterns of these putative genes from the Phytozome website (http://www.phytozome.net), we were able to initially predict potential candidate genes for PFW and MCFS. A total of six genes were considered potential candidate genes, and the functional annotations of these genes are listed in Table 4. To confirm the potential candidate genes whether participated in the accumulation of PFW or MCFS, we tested the expression patterns of the six genes via RT-qPCR in the seeds of extreme materials at three developmental growth stages (R5, R6 and R7). The genotype of extreme materials ZDD21907 (PFW 198 g) and ZDD20532 (PFW 39 g) at the AX-90496773 locus were AA (favourable allele) and GG (unfavourable allele), respectively. The genotype of extreme materials ZDD01983 (MCFS 75.5%) and ZDD02315 (MCFS 61.7%) at the AX-90460290 locus were GG (favourable allele) and AA (unfavourable allele), respectively. Among the three potential candidate genes associated with PFW, Glyma.16g018200 and Glyma.16g018300 showed significant differences in expression between ZDD21907 (PFW 198 g) and ZDD20532 (PFW 39 g) at the R5 and R6 stages (P ≤ 0.01) (Fig. 8). The potential candidate genes for MCFS were Glyma.05g243400, Glyma.05g244100 and Glyma.05g245300. These three genes showed significant differences in expression between ZDD01983 (MCFS 75.5%) and ZDD02315 (MCFS 61.7%) at the R5, R6 and R7 stages (P ≤ 0.05, P ≤ 0.01) (Fig. 8). The differential expression of these genes in extreme materials provided support for the identification of candidate genes. Therefore, we speculate that Glyma.16g018200 and Glyma.16g018300 may be the candidate genes for PFW and that Glyma.05g243400, Glyma.05g244100 and Glyma.05g245300 may be the candidate genes for MCFS. To analyze the genetic mechanism of yield in vegetable soybean, we still need to further study these five genes.

Phenotypic differences between accessions carrying different alleles. a The allele effects for the PFW marker AX-90496773 in soybean accessions. b The allele effects for MCFS marker AX-90460290 in soybean accessions. PFW means 100- pod fresh weight, MCFS means moisture content of fresh seeds

The candidate regions of the large-effect markers which associated with the 100-pod fresh weight (PFW) and moisture content of fresh seeds (MCFS) in soybean. a AX-90496773 which associated with PFW is located on Gm16. b AX-90460290 which associated with MCFS is located on Gm19. In the top panel, negative log10-transformed P values of SNPs from GWAS for PFW and MCFS are plotted against the physical positions of the given chromosomal regions. The bottom panel depicts the extent of LD in this region based on r2, and the color key displays r2 values. The horizontal dashed line (in blue) indicates the significant threshold of the genome wide association analysis (-log10(p) >4.91). The candidate region for the locus is indicated by two vertical dashed blue lines

Expression analysis of potential candidate genes in extreme materials at three growth developmental stages (R5, R6 and R7). The extreme materials include ZDD21907 (PFW 198 g), ZDD20532 (PFW 39 g), ZDD01983 (MCFS 75.5%) and ZDD02315 (MCFS 61.7%). The error bar indicates standard deviation. The results are representative of three biological replicates. ∗ Significant at P < 0.05; ∗∗ Significant at P < 0.01
Discussion
Vegetable soybean, or edamame, is a specialty soybean harvested at the R6-R7 stage when pods are green and seeds are immature [37]. The seeds of vegetable soybeans are larger, sweeter and tender than those of grain soybeans, and because of their rich protein (33–39%) and low fat (13–16%) contents, they are increasingly popular among young people who seek healthy diets, especially in developed countries [38]. In addition, vegetable soybean is a good source of soluble sugar, dietary fiber, vitamin C, vitamin E, calcium, and phytoestrogens [39, 40]. With the social and economic development, there is a growing global demand for vegetable soybeans. Since the 1990s, the demand for vegetable soybeans has grown in the US, reaching 10,000 tons in 2000 [41]. Japan is the largest importer of vegetable soybean, with a total demand of more than 176,000 tons annually [42] However, the demand for vegetable soybean cannot be met due to a lack of excellent varieties. China is the country of origin for soybeans and possesses the most soybean genetic resources worldwide. Based on the abundance of soybean resources, GWAS have been conducted to dissect the genetic architecture of vegetable soybean yield, providing functional markers, beneficial genes and specific materials for the molecular design and breeding of vegetable soybeans.
The acceptable distance between the markers and the candidate genes was determined based on LD, which varies with species and populations [43]. In this study, the overall LD decay distance for the 133 soybean landraces was 119.07 kb (r2 = 0.375) across the entire genome, which was within the reported range (90 kb ~ 574 kb), but slightly lower than the previously reported distance of 130 kb in cultivated soybean [44]. Greater diversity of geographic origins (NR, HR, SR, and NER) was included in our GWAS panel, and this difference in geographic origin may be responsible for the relatively low LD found in this study. A low LD decay rate was also identified in another recent GWAS of soybeans, involving widely distributed geographic origins (China, Korea, Japan) [45]. Moreover, the 975 Mb soybean genome includes 54,175 putative genes annotated in the cultivated soybean genome [44]. On average, every 18.42 kb contains a gene, and the average SNP spacing was approximately 11.76 kb in our study (Additional file 3: Table S3); thus, it was theoretically sufficient for efficient GWAS analysis.
In previous studies, a total of 294 QTLs for seed weight were reported across the 20 soybean chromosomes (http://www.soybase.org/). In addition, many QTLs have been identified for several traits that are highly related to yield, such as seed size, flowering time, maturity and plant height. These QTLs could be used to confirm the loci identified by GWAS. In this study, the genetic bases of four yield-related traits at the R6 stage were analyzed using association mapping, and a total of 116 significant SNPs were identified (Additional file 5: Table S5). Of these SNPs, 35 were repeatedly detected in all environments (Table 3). The data indicated that a large majority of the SNPs were environment specific, and phenotypic plasticity plays an important role in plant agronomic diversity [46]. Each SNP associated with the yield at the R6 stage could explain a large proportion (> 13.46%) of the observed phenotypic variance (Table 3). This finding differs from the reported low phenotypic variance (< 4%) of each locus associated with seed weight at maturity [47]. The results demonstrated that the soybean yield at the R6 stage is a typical quantitative trait that is genetically conditioned by many large-effect loci. Thirty-four of the repeatedly identified SNPs have been shown to colocalize with QTLs identified in previously studies (Table 3). Among these SNPs, AX-90496773 at the 1.62 Mb position on Gm16 (a region similar to a previously reported seed yield 23–6 and pod maturity 9–1 and 19–6 QTLs) was strongly associated with both PFW and SDW. Another SNP, AX-90435834 at the 1.4 Mb position on Gm04, has been reported to colocalize with QTLs related to seed weight and seed size (e.g., seed weight per plant 6–2, seed weight 47–3, seed length 1–13 and seed height 1–12). The SNP AX-90519309 on Gm17, associated with PFW and SFW, was mapped within an overlapping region of three seed weight QTLs, indicating that AX-90519309 might be located in the hottest region related to soybean yield. Twenty SNPs associated with MCFS were mapped to a small region on Gm05. Three QTLs were previously reported in a similar region with seed yield 15–3, seed thickness 1–3 and Ara/Gal 1–1. Ara/Gal represents the ratio of arabinose and galactose contents and is significantly and negatively correlated with the average concentration of pectin [48]. Pectin is multifunctional, including functions in cell wall deposition and assembly, cell expansion, cell wall swelling and softening during fruit development [49]. Therefore, the region containing twenty significant SNPs might have an effect on seed moisture content and seed thickness by affecting seed pectin. The seed moisture content and seed thickness may influence soybean yield at the R6 stage. Fine mapping of such co-localized chromosomal regions would help to determine the candidate genes responsible for the natural variation of these yield-related traits.
In this study, a total of five candidate genes associated with PFW and MCFS at the R6 stage were predicted within the LD blocks of two markers of large effect (Fig. 7 and Table 4). Among these 5 genes, Glyma.16g018200 and Glyma.16g018300 are proposed as the candidate genes for PFW. The large-effect marker AX-90496773 is located in the CDS region of Glyma.16g018200, whereas Glyma.16g018300 is located 1.9 kb downstream of AX-90496773. Glyma.16g018200 encodes a protein whose family membership is unknown, although the homologous gene of Arabidopsis thaliana is AT1g01080. The product encoded by this gene belongs to the RNA-binding (RRM motif) protein family, which may participate in the post-transcriptional regulation of genes, including pre-mRNA splicing and the cellular localization and stability maintenance of RNA [50]. Glyma.16g018300 is homologous to AT1g01090, and the proteins encoded by these genes share 80.3% amino acid sequence identity. Glyma.16g018300 encodes the pyruvate dehydrogenase E1 component alpha subunit and may be involved in two pathways, PWY-5173 (acetyl-CoA biosynthesis) and PWY-5464 (cytosolic glycolysis, pyruvate dehydrogenase and TCA cycle). In Arabidopsis thaliana, the WRINKLED1 (WRI1) transcription factor plays a role of utmost importance during oil accumulation in maturing seeds, and AT1g01090 is the putative target gene of WRI1 in the fatty acid synthesis pathway [51]. In addition, Glyma.05g243400, Glyma.05g244100 and Glyma.05g245300 are candidate genes for MCFS, and Glyma.05g243400 and Glyma.05g244100 are located 118 kb and 73 kb upstream of the large-effect marker AX-90460290, respectively. Glyma.05g243400 is homologous to AT1g1870, which encodes a putative EF-1-α-related GTP-binding protein. The vacuole is an essential organelle for plant life and plays important roles in storage (ions, metabolites, and proteins), digestion, pH and ion homeostasis, turgor pressure maintenance, biotic and abiotic defense responses, toxic compound sequestration, and pigmentation [52]. Analysis of the vegetative vacuole proteome of A. thaliana predicted that AT1g1870 may be related to vacuolar membrane fusion and remodeling [53]. Glyma.05g244100 shares 83.2% amino acid sequence identity with MOTHER OF FT AND TFL1 (MFT), which encodes a phosphatidylethanolamine-binding protein that regulates seed germination via the ABA and GA signaling pathways in Arabidopsis thaliana [54]. Glyma.05g245300 is homologous to the AT1g73660 gene, encoding a Raf-like MAPKKK. In Arabidopsis thaliana, the AT1g73660-encoded MAPKKK is a negative regulator of salt tolerance and may regulate targets involved in the salt stress response [55]. In the present study, the expression levels of the five abovementioned genes were significantly different between extreme materials during soybean seed development. Thus, we postulate that these five genes are candidate genes for PFW and MCFS. However, further evidence is needed to functionally validate this hypothesis.
Conclusion
In this study, we identified 14, 15, 63 and 48 markers associated with PFW, SFW, SDW and MCFS, respectively, via GWAS. Most markers co-localized with previously reported yield-related QTLs. We were particularly interested in the large-effect markers AX-90496773 and AX-90460290, which had an impact on yield-related traits at the R6 stage. According to genetic annotation and expression analyses, a total of five putative genes, including Glyma.16g018200, Glyma.16g018300 Glyma.05g243400, Glyma.05g244100 and Glyma.05g245300, are proposed as the candidate genes for PFW and MCFS, but further investigation is needed for verification of this hypothesis. These results provide insights into the yield improvement of vegetable soybean.
Abbreviations
- ANOVA:
-
Analysis of variance
- GD:
-
Gene diversity
- GLM:
-
A general linear model
- GWAS:
-
Genome-wide association study
- h 2 :
-
The broad-sense heritability
- HHR:
-
The Huanghuai region
- LD:
-
Linkage disequilibrium
- MAFs:
-
Minor allelic frequencies
- MCFS:
-
Moisture content of fresh seeds
- MCMC:
-
Monte Carlo Markov Chain
- MLM:
-
A mixed linear model
- NER:
-
The Northeast region
- NJ:
-
Neighbor-joining
- NR:
-
The North region
- PCA:
-
Principal component analysis
- PFW:
-
100-pod fresh weight
- PIC:
-
Polymorphism information content
- qRT-PCR:
-
Quantitative real-time PCR
- QTLs:
-
Quantitative trait loci
- r p :
-
Phenotypic correlation coefficients
- SDW:
-
100-seed dry weight
- SFW:
-
100-seed fresh weight
- SNPs:
-
Single nucleotide polymorphisms
- SR:
-
The South region
References
Graham PH, Vance CP. Legumes: importance and constraints to greater use. Plant Physiol. 2003;131(3):872–7.
Young G, Mebrahtu T, Johnson J. Acceptability of green soybeans as a vegetable entity. Plant Foods Hum Nutr. 2000;55(4):323–33.
Konovsky J, Lumpkin TA, Mcclary D. Edamame: the vegetable soybean. Understanding the Japanese food and agrimarket: a multifaceted opportunity 1994;173–181.
Delate K, Burcham R, Friedrich H, Wantate N, Wilson LA. "Edamame (vegetable soybeans) variety trial at the Neely-Kinyon farm, 2001". Iowa State Research Farm Progress Reports. 2002;1571. https://lib.dr.iastate.edu/farms_reports/1571.
Chen C. Evaluation, relationship, inheritance and variation study of summer-planted vegetable soybean's quality traits in middle and lower yangtze river valleys. MS diss., Nanjing Agricultural University. 2002.(in chinese).
Austin DF, Lee M, Veldboom LR, Hallauer AR. Genetic mapping in maize with hybrid progeny across testers and generations: grain yield and grain moisture. Crop Sci. 2000;40(1):30–9.
Orf JH, Chase K, Jarvik T, Mansur LM, Cregan PB, Adler FR, et al. Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Sci. 1999;39(6):1642–51.
Funatsuki H, Kawaguchi K, Matsuba S, Sato Y, Ishimoto M. Mapping of QTL associated with chilling tolerance during reproductive growth in soybean. Theor Appl Genet. 2005;111(5):851–61.
Palomeque L, Liu L, Li W, Hedges B, Cober ER, Rajcan I. QTL in mega-environments: II. Agronomic trait QTL co-localized with seed yield QTL detected in a population derived from a cross of high-yielding adapted × high-yielding exotic soybean lines. Theor Appl Genet. 2009;119(3):429–36.
Kim HK, Kim YC, Kim ST, Son BG, Choi YW, Kang JS, et al. Analysis of quantitative trait loci (QTLs) for seed size and fatty acid composition using recombinant inbred lines in soybean. J Life Sci. 2010;20:1186–92.
Liu W, Kim MY, Van K, Lee YH, Li H, Liu X, et al. QTL identification of yield-related traits and their association with flowering and maturity in soybean. J Crop Sci Biotechnol. 2011;14(1):65–70.
Han Y, Li D, Zhu D, Li H, Li X, Teng W, et al. QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theor Appl Genet. 2012;125(4):671–83.
Sun Y, Pan J, Shi X, Du X, Wu Q, Qi Z, et al. Multi-environment mapping and meta-analysis of 100-seed weight in soybean. Mol Biol Rep. 2012;39(10):9435–43.
Jannink JL, Lorenz AJ, Iwata H. Genomic selection in plant breeding: from theory to practice. Brief Funct Genomics. 2010;9(2):166–77.
Flint-Garcia SA, Thornsberry JM, Buckler ES IV. Structure of linkage disequilibrium in plants. Annu Rev Plant Biol. 2003;54(4):357–74.
Gupta PK, Rustgi S, Kulwal PL. Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol. 2005;57(4):461–85.
Mackay I, Powell W. Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 2007;12(2):57–63.
Li H, Ren X, Zhang X, Chen Y, Jiang H. Association analysis of agronomic traits and resistance to Aspergillus flavus in the ICRISAT peanut mini-core collection. Acta Agron Sin. 2012;38(6):935–46.
Hao D, Chen H, Yin Z, Cui S, Zhang D, Wang H, et al. Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean (Glycine max) landraces across multiple environments. Theor Appl Genet. 2012;124(3):447–58.
Niu Y, Xu Y, Liu X, Yang S, Wei S, Xie F, et al. Association mapping for seed size and shape traits in soybean cultivars. Mol Breed. 2013;31(4):785–94.
Tasma IM, Shoemaker RC. Mapping flowering time gene homologs in soybean and their association with maturity loci. Crop Sci. 2003;43(1):319–28.
Zhang J, Song Q, Cregan PB, Nelson RL, Wang X, Wu J, et al. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics. 2015;16(1):217.
Gu Y, Li W, Jiang H, Wang Y, Gao H, Liu M, et al. Differential expression of a WRKY gene between wild and cultivated soybeans correlates to seed size. J Exp Bot. 2017;68(11):2717–29.
Huang J, Guo N, Li Y, Sun J, Hu G, Zhang H, et al. Phenotypic evaluation and genetic dissection of resistance to phytophthora sojae in the chinese soybean mini core collection. BMC Genet. 2016;17(1):1–14.
SAS I. Base SAS 9.4 procedures guide: statistical procedures. Cary, NC, USA: SAS Institute Inc, 2013.
Lee YG, Jeong N, Kim JH, Lee K, Kim KH, Pirani A, et al. Development, validation and genetic analysis of a large soybean SNP genotyping array. Plant J. 2015;81(4):625–36.
Li Y, Wei L, Chen Z, Liang Y, Chang R, Gaut BS, et al. Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol. 2010;188(1):242–53.
Sun J, Guo N, Lei J, Li L, Hu G, Xing H. Association mapping for partial resistance to Phytophthora sojae in soybean (Glycine max (L.) merr.). J Genet. 2014;93(2):355–63.
Anderson AD, Weir BS. A maximum-likelihood method for the estimation of pairwise relatedness in structured populations. Genetics. 2007;176(1):421–40.
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, et al. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci U S A. 2001;98(20):11479–84.
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42(11):961–7.
Yu J, Pressoir G, Briggs WH, Vroh BI, Yamasaki M, Doebley JF, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38(2):203–8.
Yang N, Lu Y, Yang X, Huang J, Zhou Y, Ali F, et al. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet. 2014;10(9):e1004573.
Fehr WR, Caviness CE. Stages of soybean development. Spec Rep. 1977;87.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–8.
Nyquist WE, Baker RJ. Estimation of heritability and prediction of selection response in plant populations. Crit Rev Plant Sci. 1991;10(3):235–322.
Zhang Q, Gao Q, Herbert SJ, Li Y, Hashemi AM. Influence of sowing date on phenological stages, seed growth and marketable yield of four vegetable soybean cultivars in North-Eastern USA. Afr J Agric Res. 2010;5(18):2556–62.
Rao MS, Bhagsari AS, Mohamed AI. Fresh green seed yield and seed nutritional traits of vegetable soybean genotypes. Crop Sci. 2002;42(6):1950–8.
Li Y, Ming D, Zhang Q, Wang G, Hashemi M, Liu X. Greater differences exist in seed protein, oil, total soluble sugar and sucrose content of vegetable soybean genotypes [Glycine max (L.) Merrill] in Northeast China. Aust J Crop Sci. 2012;6(12):1681–6.
Miles CA, Lumpkin TA, Zenz L. Edamame Production. 2000.
Lin C. Frozen edamame: global market conditions. USA: Second International Vegetable Soybean conference; 2001. p. 93–7.
Nguyen VQ. Edamame (vegetable green soybean). Austrália: Rural Industries Research & Development. The new rural industries: a handbook for farmers and investors; 2001. p. 49–56.
Li Y, Reif JC, Hong H, Li H, Liu Z, Ma Y, et al. Genome-wide association mapping of QTL underlying seed oil and protein contents of a diverse panel of soybean accessions. Plant Sci. 2018;266:95–101.
Wang J, Chu S, Zhang H, Zhu Y, Cheng H, Yu D. Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Sci Rep. 2016;6(1):20728.
Hwang EY, Song Q, Jia G, Specht JE, Hyten DL, Jose C, et al. A genome-wide association study of seed protein and oil content in soybean. BMC Genomics. 2014;15(1):1–1.
Ungerer MC, Halldorsdottir SS, Purugganan MA, Mackay TFC. Genotype-environment interactions at quantitative trait loci affecting inflorescence development in Arabidopsis thaliana. Genetics. 2003;165(1):353–65.
Zhang J, Song Q, Cregan PB, Jiang G. Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean (Glycine max). Theor Appl Genet. 2016;129(1):117–30.
Stombaugh SK, Orf JH, Jung HG, Chase K, Lark KG, Somers DA. Quantitative trait loci associated with cell wall polysaccharides in soybean seed. Crop Sci. 2004;44(6):2101–6.
Willats WG, Mccartney L, Mackie W, Knox JP. Pectin: cell biology and prospects for functional analysis. Plant Mol Biol. 2001;47(1–2):9–27.
Du G, Yan Z, Yuan J, Qiang B. RRM RNA binding protein: structure and function. Prog Biochem Biophys. 1999;26(4):305–7.
Baud S, Mendoza MS, To A, Harscoët E, Lepiniec L, Dubreucq B. WRINKLED1 specifies the regulatory action of LEAFY COTYLEDON2 towards fatty acid metabolism during seed maturation in Arabidopsis. Plant J. 2010;50(5):825–38.
De DN. Plant Cell Vacuoles. Collingwood, Australia: CSIRO Publishing; 2000. p. 79–114.
Carter C, Pan S, Zouhar J, Avila EL, Girke T, Raikhel NV. The vegetative vacuole proteome of Arabidopsis thaliana reveals predicted and unexpected proteins. Plant Cell. 2004;16(12):3285–303.
Xi W, Liu C, Hou X, Yu H. MOTHER OF FT AND TFL1 regulates seed germination through a negative feedback loop modulating ABA signaling in Arabidopsis. Plant Cell. 2010;22(6):1733–48.
Lei G, Xiang C. The genetic locus At1g73660 encodes a putative MAPKKK and negatively regulates salt tolerance in Arabidopsis. Plant Mol Biol. 2008;67(1–2):125–34.
Acknowledgments
We thank Dr. Jianbo He for providing technical assistance in bioinformatics and for his critical review of the manuscript.
Funding
This work was supported by National Key R&D Program of China (2017YFD0101500, 2017YFD0102002), National Natural Science Foundation of China (31471519), Modern Agro-industry Technology Research System of China (CARS-04-PS10), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT_17R55), National Science and Technology Pillar Program of China (2014BAD11B01-X01) and Jiangsu Collaborative Innovation Center for Modern Crop Production (JCIC-MCP).
Availability of data and materials
The data sets supporting the results of this article are included within the article and its additional files.
Author information
Authors and Affiliations
Contributions
HX and JMZ conceived and designed the experiments. XNL, XLZ, XTW and NG performed the experiments. XNL, XFW, XZ and YZ analyzed the data. LJQ provided the genotype data. XNL wrote the paper. XLZ, LMZ, YPB, HX and JMZ revised the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. The ecological distribution of 133 soybean landraces. (XLSX 14 kb)
Additional file 2:
Table S2. qRT-PCR primers. (XLSX 9 kb)
Additional file 3:
Table S3. Summary of the polymorphic markers on the 20 chromosomes of Glycine max. (XLSX 10 kb)
Additional file 4:
Table S4. The ecological distribution of 133 soybean landraces from different subgroups. (XLSX 9 kb)
Additional file 5:
Table S5. SNPs significantly associated with the four yield-related traits. (XLSX 22 kb)
Additional file 6:
Figure S1. Manhattan and quantile–quantile (QQ) plots of the GWAS for 100-pod fresh weight (PFW) in soybean at the R6 stage. The horizontal blue line indicates the genome-wide significance threshold (−log10(P) > 4.91); a, b and c represent 2015, 2016 and the means across the two years, respectively. (TIF 10418 kb)
Additional file 7:
Figure S2. Manhattan and quantile–quantile (QQ) plots of the GWAS for 100-seed fresh weight (SFW) in soybean at the R6 stage. The horizontal blue line indicates the genome-wide significance threshold (−log10(P) > 4.91). a, b and c represent 2015, 2016 and the means across the two years, respectively. (TIF 5303 kb)
Additional file 8:
Figure S3. Manhattan and quantile–quantile (QQ) plots of GWAS for 100-seed dry weight (SDW) in soybean at the R6 stage. The horizontal blue line indicates the genome-wide significance threshold (−log10(P) > 4.91). a, b and c represent 2015, 2016 and the means across the two years, respectively. (TIF 5874 kb)
Additional file 9:
Figure S4. Manhattan and quantile–quantile (QQ) plots of GWAS for moisture content of fresh seeds (MCFS) in soybean at the R6 stage. The horizontal blue line indicates the genome-wide significance threshold (−log10(P) > 4.91). a, b and c represent 2015, 2016 and the means across the two years, respectively. (TIF 10454 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, X., Zhang, X., Zhu, L. et al. Genome-wide association study of four yield-related traits at the R6 stage in soybean. BMC Genet 20, 39 (2019). https://doi.org/10.1186/s12863-019-0737-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-019-0737-9