
Abstract
Background
Electrical status epilepticus during slow-wave sleep (ESESS) which is also known as continuous spike-wave of slow sleep (CSWSS) is type of electroencephalographic (EEG) pattern which is seen in ESESS/CSWSS/epilepsy aphasia spectrum. This EEG pattern can occur alone or with other syndromes. Its etiology is not clear, however, brain malformations, immune disorders, and genetic etiologies are suspected to contribute. We aimed to perform a systematic review of all genetic etiologies which have been reported to associate with ESESS/CSWSS/epilepsy-aphasia spectrum. We further aimed to identify the common underlying pathway which can explain it. To our knowledge, there is no available systematic review of genetic etiologies of ESESS/CSWSS/epilepsy-aphasia spectrum. MEDLINE, EMBASE, PubMed and Cochrane review database were searched, using terms specific to electrical status epilepticus during sleep or continuous spike–wave discharges during slow sleep or epilepsy-aphasia spectrum and of studies of genetic etiologies. These included monogenic mutations and copy number variations (CNVs). For each suspected dosage-sensitive gene, further studies were performed through OMIM and PubMed database.
Results
Twenty-six studies out of the 136 identified studies satisfied our inclusion criteria. I51 cases were identified among those 26 studies. 16 studies reported 11 monogenic mutations: SCN2A (N = 6), NHE6/SLC9A6 (N = 1), DRPLA/ ATN1 (N = 1), Neuroserpin/SRPX2 (N = 1), OPA3 (N = 1), KCNQ2 (N = 2), KCNA2 (N = 5), GRIN2A (N = 34), CNKSR2 (N = 2), SLC6A1 (N = 2) and KCNB1 (N = 5). 10 studies reported 89 CNVs including 9 recurrent ones: Xp22.12 deletion encompassing CNKSR2 (N = 6), 16p13 deletion encompassing GRIN2A (N = 4), 15q11.2–13.1 duplication (N = 15), 3q29 duplication (N = 11), 11p13 duplication (N = 2), 10q21.3 deletion (N = 2), 3q25 deletion (N = 2), 8p23.3 deletion (N = 2) and 9p24.2 (N = 2). 68 of the reported genetic etiologies including monogenic mutations and CNVs were detected in patients with ESESS/CSWSS/epilepsy aphasia spectrum solely. The most common underlying pathway was channelopathy (N = 56).
Conclusions
Our review suggests that genetic etiologies have a role to play in the occurrence of ESESS/CSWSS/epilepsy-aphasia spectrum. The common underlying pathway is channelopathy. Therefore we propose more genetic studies to be done for more discoveries which can pave a way for proper drug identification. We also suggest development of common cut-off value for spike-wave index to ensure common language among clinicians and researchers.
Similar content being viewed by others


Background
Electrical status epilepticus during slow-wave sleep (ESESS) which is also known as continuous spike-wave of slow sleep (CSWSS) is a type of an electroencephalogram (EEG) pattern which is characterized by presence of generalized bilateral and symmetric 1.5–3 Hz spike-waves [1]. The International League against Epilepsy (ILAE) defined it as the presence of significant activation of epileptiform discharges during sleep but no specific cut off-value for spike-wave index was indicated (Commission on Classification and Terminology of the International League Against Epilepsy 1989). Some authors have suggested the cut-off value of at least 85% [2, 3] but others have set the cut-off value at different percentage levels. This brings contradiction among clinicians, for instance Fernández IS et al. found in their survey that, 57.6% of the members of the Child Neurology Society and the American Epilepsy Society defined it by considering a cut-off value of 85% spike-wave index while 30.8% considered a cut-off value of 50% [4]. The ESESS/CSWSS pattern can be seen in different electroclinical syndromes with similar presentation including ESESS/CSWSS/epilepsy-aphasia spectrum. ESESS/CSWSS/epilepsy-aphasia spectrum is an acquired condition characterized by a triad of epilepsy, cognitive or behavioral impairment, and EEG abnormality of ESESS/CSWSS [1, 3, 5]. Epilepsy-aphasia spectrum is a spectrum of disorders ranging from severe epileptic encephalopathy with CSWSS and Landau- Kleffner syndrome (LKS) to the mild condition of childhood epilepsy with centrotemporal spikes [6,7,8]. ESESS/CSWSS/epilepsy-aphasia spectrum is age related and occurs commonly during the childhood usually in the first decade of life. It has a prevalence of about 0.5% of all childhood epilepsies [9]. It has long-term poor prognosis due to the persistence of neuropsychological impairment. Despite the fact that the ESESS/CSWSS pattern can be seen in ESESS/CSWSS/epilepsy-aphasia spectrum, it can also concur with other syndromes such as Rett syndrome, Costeff syndrome, Christianson syndrome, Tuberous sclerosis complex, Cohen syndrome and autism spectrum disorders [10,11,12,13,14,15].
The underlying etiology is unknown although brain malformations, immune disorders, and genetic factors have been reported. Brain malformations include porencephaly, polymicrogyria, pachygyria, cortical atrophy, and hydrocephalus [3, 9, 10, 16]. Immunity disorders with evidence of onconeuronal antibodies have been reported [17, 18]. Furthermore, few genetic causes have been reported including monogenic mutations and copy number variations (CNVs) [12, 19, 20]. Despite the availability of advanced technology in cytogenetic tests, few studies have focused on patients with ESESS/CSWSS/epilepsy-aphasia spectrum and the underlying mechanism for its occurrence remains unknown.
We aimed to perform a systematic review on all reported genetic etiologies of ESESS/CSWSS/epilepsy-aphasia spectrum including monogenic mutations and copy number variations. We further aimed to study the possible underlying mechanism for all reported genetic abnormalities especially for those associated with ESESS/CSWSS/epilepsy-aphasia spectrum solely. We believe this will help to identify the common genetic etiologies which can pave the way for the development of the appropriate therapy. This will help to reduce the burden of the complication of ESESS/CSWSS/epilepsy-aphasia spectrum owing to its long-term poor prognosis due to the persistence of neuropsychological impairment. Furthermore, our review will discover the existing gap and provide some suggestions. To our knowledge, there is no systematic review which has been done on the genetic etiologies of ESESS/CSWSS/epilepsy-aphasia spectrum.
Methods
Selection of studies
We developed search strategies for studies on genetic etiologies of electrical status epilepticus during sleep or continuous spike–wave discharges during slow sleep or epilepsy-aphasia spectrum in consultation with a librarian (Additional file 1). MEDLINE, EMBASE, PubMed and Cochrane review database were searched, using terms specific to electrical status epilepticus during sleep or continuous spike–wave discharges during slow sleep or epilepsy-aphasia spectrum and of studies of genetic etiologies. Studies reporting the monogenic mutations or copy number variations related to electrical status epilepticus during sleep or continuous spike–wave discharges during slow sleep were included. We included the studies which were done in human beings in all ages all over the world. We included case reports, case series, and cohort studies. Studies done in all years were included. Three independent reviewers screened the abstracts to determine if a full-text review should be performed. We included the studies published in English only and original peer-reviewed articles. We further performed hand searching of the references of articles that met eligibility criteria to ensure that additional relevant studies were not missed. We excluded the animal studies.
Data extraction
Data extraction for all studies was performed by three independent reviewers, and the accuracy of information extracted was confirmed by discussion. Collected data related to monogenic mutations included; gene information such as name/alternate name, gene location, the Online Mendelian Inheritance in Man (OMIM) number, type of mutation, the function of the gene, the possible underlying pathway, the number of reported cases, associated syndromes, authors and the years for publication. Collected information related to copy number variations included; the chromosomal location, coordinates when available, type of aberration, size, gene content, gene of interest for ESESS/ CSWSS, the possible underlying pathway, the number of reported cases, associated syndromes, authors and the years for publication. All the suspected pathogenic genes were further studied individually in OMIM and PubMed database to determine their functions and how do they relate to ESESS/CSWSS. Additionally, we collected information related to the diagnostic criteria (spike wave index) which was used to diagnose ESESS/CSWSS/epilepsy-aphasia spectrum.
Results
Results of the systematic literature review
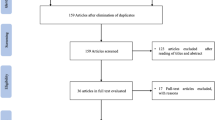
The combined searches yielded 136 abstracts, with 59 abstracts selected for full-text review. Of these 59, 33 studies were excluded because they reported ESESS/CSWSS/epilepsy-aphasia spectrum without underlying genetic etiologies or non-original data. 26 studies out of the 136 identified studies satisfied our inclusion criteria. This is summarized in PRISMA flowchart (Fig. 1). A total number of 151 cases were identified in those 26 studies. 124 cases were diagnosed with ESESS/CSWSS/epilepsy-aphasia spectrum solely.

PRISMA flowchart
Monogenic mutations which associate with ESESS/CSWSS/epilepsy-aphasia spectrum
We identified 11 monogenic mutations which have been reported to associate with ESESS/CSWSS/epilepsy-aphasia spectrum. These included: SCN2A (N = 6), NHE6/SLC9A6 (N = 1), DRPLA/ATN1 (N = 1), Neuroserpin/SRPX2 (N = 1), OPA3 (N = 1), KCNQ2 (N = 2), KCNA2 (N = 5), GRIN2A (N = 34), CNKSR2 (N = 2), SLC6A1 (N = 2) and KCNB1 (N = 5). Seven genes were noticed in ESESS/CSWSS/epilepsy-aphasia spectrum solely including SCN2A, KCNQ2, KCNA2, GRIN2A, CNKSR2, SLC6A1 and KCNB1. The underlying pathway for the SCN2A, KCNQ2, KCNB1, KCNA2 and GRIN2A was channelopathy (N = 52). Four genes were noticed in patients with certain syndromes (NHE6/SLC9A6, DRPLA/ ATN1, Neuroserpin/SRPX2, and OPA3). Those syndromes include Christianson syndrome, Dentatorubro-pallidoluysian atrophy, Familial encephalopathy with neuroserpin inclusion bodies, and Costeff syndrome. Table 1 summarizes this information.
Copy number variations which associate with ESESS/CSWSS/epilepsy aphasia spectrum
We identified 89 CNVs which have been reported to associate with ESESS/CSWSS/epilepsy-aphasia spectrum: 9 recurrent and 45 non recurrent. Recurrent CNVs included 15q11.2–13.1 duplication for 15 cases, 3q29 duplication for 11 cases, Xp22.12 deletion for 4 cases, 16p13 deletion for 4 cases, 11p13 duplication for 2 cases, 10q21.3 deletion for 2 cases, 3q25 deletion for 2 cases, 8p23.3 deletion for 2 cases and 9p24.2 duplication for 2 cases. 61 CNVs were noticed in patients with ESESS/CSWSS/epilepsy-aphasia spectrum solely. 4 of the 61 CNVs spanned gene involved in channel (GRIN2A), 8 CNVs spanned genes involved in synaptic connection (CNKSR2, SHANK3 and DLG2), and 14 CNVs spanned genes involved in cell adhesion (CDH13, CTNNA3, DIAPH3, CDH9, CDH6, KIF26B, CDH4, CNTNAP2, SGCZ, HIPK3, CSTF3 and CNTN6). Three CNVs were noticed in patients with certain syndromes including, 8p deletion syndrome and 9p duplication syndrome. Table 2 summarizes this information.
Discussion
Electrical status epilepticus during slow-wave sleep which is also known as continuous spike-wave of slow sleep is type of an EEG pattern which is seen in ESESS/CSWSS/epilepsy aphasia spectrum with an estimated prevalence of 0.5%. However, this prevalence might be inaccurate due to a few studies which have been done on it as well as the usage of different diagnostic criteria in making diagnosis. ESESS/CSWSS associates with long-term neuropsychological impairment. It can occur alone or with other syndromes. We aimed to review all reported genetic etiologies of ESESS/CSWSS/epilepsy-aphasia spectrum and to study their possible underlying pathway especially for ESESS/CSWSS/epilepsy aphasia spectrum which occurs alone. This review will provide an insight regarding the contribution of genetic etiologies in ESESS/CSWSS/epilepsy-aphasia spectrum and the possible common underlying pathway which can assist in identification of the appropriate therapy. Identification of the target therapy will help to reduce the long-term neuropsychological impairment.
We have observed that approximately 67.6% (N = 102/151) of the cases were diagnosed with ESESS/CSWSS/epilepsy-aphasia spectrum when they had spike-wave index > 50% clearly activated during sleep while 13.2% (N = 20/151) were diagnosed when they had spike-wave index > 85%. Our finding differs from the survey which was done by Fernández IS et al. in North America where they found 57.6% of the members of the Child Neurology Society and the American Epilepsy Society considered a cut-off value of 85% spike-wave index while 30.8% considered a cut-off value of 50% [4]. This difference could be due to the fact that our review involved multiple studies from different areas of the world. Currently, there is no specific criteria from ILAE for definition of ESESS/CSWSS pattern hence jeopardize communication among clinicians and research in this condition. We suggest development of common cut-off value.
A total number of 11 monogenic mutations and 89 CNVs were identified to associate with ESESS/CSWSS/epilepsy-aphasia spectrum. Monogenic mutations included SCN2A [21], NHE6/SLC9A6 [11], DRPLA/ATN1 [22], Neuroserpin/SRPX2 [23], KCNQ2 [24], OPA3 [14], KCNA2 [25,26,27], GRIN2A [28,29,30,31], CNKSR2 [32], SLC6A1 [33] and KCNB1 [34]. Seven genes were noticed in ESESS/CSWSS/epilepsy-aphasia spectrum solely including SCN2A, KCNQ2, KCNA2, GRIN2A, CNKSR2, SLC6A1 and KCNB1. Out of 89 CNVs, 9 were recurrent whilst 45 were non-recurrent. 4 CNVs spanned gene involved in channel (GRIN2A), 8 CNVs spanned genes involved in synaptic connection (CNKSR2, SHANK3 and DLG2), and 14 CNVs spanned genes involved in cell adhesion (CDH13, CTNNA3, DIAPH3, CDH9, CDH6, KIF26B, CDH4, CNTNAP2, SGCZ, HIPK3, CSTF3 and CNTN6). 68 of the reported genetic etiologies including monogenic mutations and CNVs were detected in patients with ESESS/CSWSS/epilepsy-aphasia spectrum solely. The most common underlying pathway was channelopathy (N = 56). The pathogenic genes included SCN2A, KCNQ2, KCNB1, KCNA2 and GRIN2A (Tables 1 and 2).
SCN2A gene encodes subunits of voltage-gated sodium channel which is responsible for generation and propagation of action potentials in neurons and muscles [35]. SCN2A mutations associate with two phenotypic spectrum related to epilepsy: the early onset (< 3 months) group which include benign familial neonatal or infantile seizures (BFNIS) and the late onset (> 3 months) group which include focal epilepsies with an ESESS/CSWSS-like picture [21]. Wolff M et al. studied the phenotypes of cases with SCN2A mutations in which they discovered three patients with A263V mutation who showed BFNIS phenotype, while three others with the same mutation had more severe phenotypes [21]. They concluded that, both the mutation itself and other genetic or environmental factors contribute to the individual phenotype. We identified only 6 patients with ESESS/CSWSS/epilepsy-aphasia spectrum who were reported to have SCN2A mutations. The few cases could be explained by other unknown genetic or environmental factors which could contribute to the phenotype. Additionally, it could be due to a few studies that have focused on identification of genetic etiologies in this particular condition.
KCNQ2 gene encodes for subunits of potassium channel which is highly expressed in brain neurons [36]. It produce M- current which prevents constant neuronal excitability and hence prevent seizures. KCNQ2 mutations associate with wide range of phenotypes: BFNIS, benign familial infantile seizures, neonatal onset epileptic encephalopathies, and ESESS/CSWSS. The wide range of phenotypes depend on the position and the features of the amino acid change which result to variable voltage sensitivity of the channel [37,38,39,40]. The change can promote increase and decrease of channel activity leading to different intensity levels hence different phenotypes. Other researchers have hypothesized that different phenotypes could be explained by an interplay of pathogenic mutations, modifier genes, and other environmental factors [41]. We identified only two reported cases with ESESS/CSWSS/epilepsy-aphasia spectrum who had KCNQ2 mutations [24]. The few cases could be explained by the kind of mutation, and other unknown modifier genes and environmental factors.
KCNA2 gene encodes potassium channel, voltage-gated, shaker-related subfamily and is highly expressed in brain and central nervous system [42]. It prevents abnormal action potential firing and regulates neuronal output. KCNA2 mutations associate with two types of phenotypes based on the severity of the encephalopathy and of the seizure disorder. The milder phenotypes correlates with loss-of-function mutations and more severe phenotypes with gain-of-function mutations [26]. Sachdev M et al. [25], Syrbe S et al [26] and Masnada S et al [27] reported a total number of five patients who were diagnosed with ESESS/CSWSS/epilepsy- aphasia spectrum and found to have KCNA2 mutations.
GRIN2A gene encodes N-methyl-D-aspartate (NMDA) glutamate receptor α2 subunit [43]. The NMDA receptor is a glutamate-activated ion channel permeable to sodium, potassium and calcium and is found at excitatory synapses throughout the brain. The current which is produced by NMDA receptor–mediated component of the excitation is crucial in the central nervous system as it determines the key features of the synaptic response and has important consequences for synaptic plasticity and cell physiology. Dysfunction of NMDA receptor–mediated signaling can trigger and/or influence numerous brain diseases, including epilepsy [44]. Four studies have reported 34 patients with GRIN2A mutations who were diagnosed with ESESS/CSWSS/epilepsy-aphasia spectrum [28,29,30,31]. Additionally, four patients with deletion at 16p13 spanning GRIN2A gene have been reported [12]. Miyamoto H et al. explained the relationship between NMDA receptor functioning and the modulation of ESESS/CSWSS [45]. Therefore, NMDA receptor can stand as a target for development of drug since GRIN2A mutations were reported in many patients with ESESS/CSWSS/epilepsy-aphasia spectrum (N = 38).
KCNB1 gene encodes a member of the potassium channel, voltage-gated, shab-related subfamily. It is highly expressed in brain neurons [46]. KCNB1 gene mutations associate with early infantile epileptic encephalopathies. de Kovel CGF et al. studied the phenotypes of 26 cases with KCNB1 gene mutations in which 5 of them were identified to have ESESS/CSWSS [34].
CNKSR2 gene encodes connector enhancer of KSR2 which is a synaptic protein involved in Ras signaling-mediated neuronal proliferation, migration and differentiation. Synaptic proteins are crucial for neuronal function in the brain, and their deficiency can lead to epilepsy and cognitive impairments. Damiano JA et al. reported a novel nonsense mutation (c.2314 C > T; p.Arg712*) in 2 siblings diagnosed with ESESS/CSWSS/epilepsy-aphasia spectrum [32]. Moreover, approximately 4 studies reported 6 patients with deletion at Xp22.12 spanning CNKSR2 gene [12, 47,48,49]. Hence CNKSR2 gene has a role to play in ESESS/CSWSS/epilepsy-aphasia spectrum.
SLC6A1 gene encodes voltage-dependent gamma-aminobutyric acid (GABA) transporter 1 (GAT-1), one of the main GABA transporters in central nervous system [50]. The dysfunction of this transporter leads to spontaneous spike-wave discharges and absence seizures [51]. Johannesen KM et al. reviewed the phenotypic spectrum of 34 cases with SLC6A1 mutations in which they identified two patients who presented with ESESS/CSWSS/epilepsy-aphasia spectrum [33]. This new finding suggests the role of GABA in pathogenesis of ESESS/CSWSS/epilepsy-aphasia spectrum.
Most of the reported CNVs span genes involved in cell adhesion (N = 14): CDH13, CTNNA3, DIAPH3, CDH9, CDH6, KIF26B, CDH4, CNTNAP2, SGCZ, HIPK3, CSTF3 and CNTN6. Hence we support Lesca G et al in suggesting that, these genes might explain the role of autoimmunity in the pathogenesis of ESESS/CSWSS/epilepsy-aphasia spectrum [12]. However, they have never been reported as monogenic mutation.
We have observed that SCN2A, KCNQ2, KCNB1, KCNA2 and GRIN2A contributed to the etiology of many patients with ESESS/CSWSS solely (Tables 1 and 2). The common underlying functions of these genes is to encode important channels in brain neurons. Their disturbances lead to ESESS/CSWSS/epilepsy aphasia spectrum. Therefore we think channelopathy plays a major role in pathogenesis of ESESS/CSWSS/epilepsy aphasia spectrum.
Several syndromes have been reported to associate with ESESS/CSWSS pattern including Christianson syndrome, Dentatorubro-pallidoluysian atrophy, Familial encephalopathy with neuroserpin inclusion bodies, Rolandic Epilepsy, Costeff syndrome, Landau-Kleffner syndrome, 8p deletion syndrome and 9p duplication syndrome. However, they have the separate possible underlying pathway (Tables 1 and 2). Our review was limited due to existing discrepancy in terms of diagnostic criteria (spike wave index) which are being used. Hence there is no common language.
Conclusion
Approximately 37% (56/151) of the reported cases diagnosed with ESESS/CSWSS/epilepsy-aphasia spectrum solely had pathogenic genes responsible for encoding channels in the brain neurons. Consequently, our review suggests channelopathy as a possible underlying cause which can be targeted for the development of appropriate therapy. However, this remains non-conclusive because less cytogenetic studies have been performed in this particular condition. We argue more research to be performed in patients who present with ESESS/CSWSS/epilepsy-aphasia spectrum solely so as discover more underlying causes which will facilitate in proper therapy identification. We also suggest development of diagnostic criteria (cut-off value for spike-wave index) which can be utilized worldwide to ensure common language among clinicians and researchers.
Abbreviations
- CNVs:
-
copy number variations
- CSWSS:
-
continuous spike-wave of slow sleep
- ESESS:
-
Electrical status epilepticus during slow-wave sleep
- OMIM:
-
Online Mendelian Inheritance in Man
References
Patry G, Lyagoubi S, Tassinari CA. Subclinical "electrical status epilepticus" induced by sleep in children. A clinical and electroencephalographic study of six cases. Arch Neurol. 1971;24(3):242–52.
Loddenkemper T, Fernández IS, Peters JM. Continuous spike and waves during sleep and electrical status epilepticus in sleep. J Clin Neurophysiol. 2011;28(2):154–64.
Tassinari CA, Rubboli G, Volpi L, Meletti S, d'Orsi G, Franca M, Sabetta AR, Riguzzi P, Gardella E, Zaniboni A, et al. Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin Neurophysiol. 2000;111(Suppl 2):S94–S102.
Fernández IS, Chapman KE, Peters JM, Kothare SV, Nordli DR Jr, Jensen FE, Berg AT, Loddenkemper T. The tower of babel: survey on concepts and terminology in electrical status epilepticus in sleep and continuous spikes and waves during sleep in North America. Epilepsia. 2013;54(4):741–50.
Tassinari CA, Cantalupo G, Rios-Pohl L, Giustina ED, Rubboli G. Encephalopathy with status epilepticus during slow sleep: "the Penelope syndrome". Epilepsia. 2009;50(Suppl 7):4–8.
Tsai MH, Vears DF, Turner SJ, Smith RL, Berkovic SF, Sadleir LG, Scheffer IE. Clinical genetic study of the epilepsy-aphasia spectrum. Epilepsia. 2013;54(2):280–7.
Deonna T, Roulet-Perez E. Early-onset acquired epileptic aphasia (landau-Kleffner syndrome, LKS) and regressive autistic disorders with epileptic EEG abnormalities: the continuing debate. Brain and Development. 2010;32(9):746–52.
Rudolf G, Valenti MP, Hirsch E, Szepetowski P. From rolandic epilepsy to continuous spike-and-waves during sleep and landau-Kleffner syndromes: insights into possible genetic factors. Epilepsia. 2009;50(Suppl 7):25–8.
Galanopoulou AS, Bojko A, Lado F, Moshé SL. The spectrum of neuropsychiatric abnormalities associated with electrical status epilepticus in sleep. Brain and Development. 2000;22(5):279–95.
Coppola G, Federico RR, Epifanio G, Tagliente F, Bravaccio C. Focal polymicrogyria, continuous spike-and-wave discharges during slow-wave sleep and Cohen syndrome: a case report. Brain and Development. 2003;25(6):446–9.
Zanni G, Barresi S, Cohen R, Specchio N, Basel-Vanagaite L, Valente EM, Shuper A, Vigevano F, Bertini E. A novel mutation in the endosomal Na+/H+ exchanger NHE6 (SLC9A6) causes Christianson syndrome with electrical status epilepticus during slow-wave sleep (ESES). Epilepsy Res. 2014;108(4):811–5.
Lesca G, Rudolf G, Labalme A, Hirsch E, Arzimanoglou A, Genton P, Motte J, de Saint MA, Valenti MP, Boulay C, et al. Epileptic encephalopathies of the landau-Kleffner and continuous spike and waves during slow-wave sleep types: genomic dissection makes the link with autism. Epilepsia. 2012;53(9):1526–38.
Nissenkorn A, Gak E, Vecsler M, Reznik H, Menascu S, Ben ZB. Epilepsy in Rett syndrome---the experience of a National Rett Center. Epilepsia. 2010;51(7):1252–8.
Carmi N, Lev D, Leshinsky-Silver E, Anikster Y, Blumkin L, Kivity S, Lerman-Sagie T, Zerem A. Atypical presentation of Costeff syndrome-severe psychomotor involvement and electrical status epilepticus during slow wave sleep. Eur J Paediatr Neurol. 2015;19(6):733–6.
Pacheva I, Panov G, Gillberg C, Neville B. A girl with tuberous sclerosis complex presenting with severe epilepsy and electrical status epilepticus during sleep, and with high-functioning autism and mutism. Cogn Behav Neurol. 2014;27(2):88–95.
Nickels K, Wirrell E. Electrical status epilepticus in sleep. Semin Pediatr Neurol. 2008;15(2):50–60.
Nabbout R. Autoimmune and inflammatory epilepsies. Epilepsia. 2012;53(Suppl 4):58–62.
Hu LY, Shi XY, Feng C, Wang JW, Yang G, Lammers SH, Yang XF, Ebrahimi-Fakhari D, Zou LP. An 8-year old boy with continuous spikes and waves during slow sleep presenting with positive onconeuronal antibodies. Eur J Paediatr Neurol. 2015;19(2):257–61.
Van Bogaert P. Epileptic encephalopathy with continuous spike-waves during slow-wave sleep including landau-Kleffner syndrome. Handb Clin Neurol. 2013;111:635–40.
Kevelam SH, Jansen FE, Binsbergen Ev, Braun KP, Verbeek NE, Lindhout D, Poot M, Brilstra EH. Copy number variations in patients with electrical status epilepticus in sleep. J Child Neurol 2012; 27(2):178–182.
Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, Lesca G, Ville D, Milh M, Villard L, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140(5):1316–36.
Kobayashi K, Hata H, Oka M, Ito M, Yoshinaga H, Kashihara K, Ohtsuka Y. Age-related electrical status epilepticus during sleep and epileptic negative myoclonus in DRPLA. Neurology. 2006;66(5):772–3.
Coutelier M, Andries S, Ghariani S, Dan B, Duyckaerts C, van Rijckevorsel K, Raftopoulos C, Deconinck N, Sonderegger P, Scaravilli F, et al. Neuroserpin mutation causes electrical status epilepticus of slow-wave sleep. Neurology. 2008;71(1):64–6.
Lee IC, Yang JJ, Li SY. A KCNQ2 E515D mutation associated with benign familial neonatal seizures and continuous spike and waves during slow-wave sleep syndrome in Taiwan. J Formos Med Assoc. 2017;116(9):711–9.
Sachdev M, Gaínza-Lein M, Tchapyjnikov D, Jiang YH, Loddenkemper T, Mikati MA. Novel clinical manifestations in patients with KCNA2 mutations. Seizure. 2017;51:74–6.
Syrbe S, UBS H, Riesch E, Djémié T, Müller S, Møller RS, Maher B, Hernandez-Hernandez L, Synofzik M, Caglayan HS, et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet. 2015;47(4):393–9.
Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, Klee EW, Lanpher BC, Gavrilova RH, Synofzik M, Bast T, et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain. 2017;140(9):2337–54.
Gao K, Tankovic A, Zhang Y, Kusumoto H, Zhang J, Chen W, XiangWei W, Shaulsky GH, Hu C, Traynelis SF, et al. A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS One. 2017;12(2):e0170818.
Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, Salmi M, Tsintsadze T, Addis L, Motte J, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. 2013;45(9):1061–6.
Carvill GL, Regan BM, Yendle SC, O'Roak BJ, Lozovaya N, Bruneau N, Burnashev N, Khan A, Cook J, Geraghty E, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. 2013;45(9):1073–6.
Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, Geider K, Laube B, Schwake M, Finsterwalder K, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet. 2013;45(9):1067–72.
Damiano JA, Burgess R, Kivity S, Lerman-Sagie T, Afawi Z, Scheffer IE, Berkovic SF, Hildebrand MS. Frequency of CNKSR2 mutation in the X-linked epilepsy-aphasia spectrum. Epilepsia. 2017;58(3):e40–3.
Johannesen KM, Gardella E, Linnankivi T, Courage C, de Saint Martin A, Lehesjoki AE, Mignot C, Afenjar A, Lesca G, Abi-Warde MT, et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. 2018;59(2):389–402.
de Kovel CGF, Syrbe S, Brilstra EH, Verbeek N, Kerr B, Dubbs H, Bayat A, Desai S, Naidu S, Srivastava S, et al. Neurodevelopmental disorders caused by De novo variants in KCNB1 genotypes and phenotypes. JAMA Neurol. 2017;74(10):1228–36.
Oliva M, Berkovic SF, Petrou S. Sodium channels and the neurobiology of epilepsy. Epilepsia. 2012;53(11):1849–59.
Robbins J, Passmore GM, Abogadie FC, Reilly JM, Brown DA. Effects of KCNQ2 gene truncation on M-type Kv7 potassium currents. PLoS One. 2013;8(8):e71809.
Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1(1):21–30.
Singh NA, Otto JF, Dahle EJ, Pappas C, Leslie JD, Vilaythong A, Noebels JL, White HS, Wilcox KS, Leppert MF. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J Physiol. 2008;586(14):3405–23.
Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, Taglialatela M. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci. 2015;35(9):3782–93.
Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Migliore M, Cilio MR, Taglialatela M. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K (v) 7.2 potassium channel subunits. Proc Natl Acad Sci U S A. 2013;110(11):4386–91.
Borgatti R, Zucca C, Cavallini A, Ferrario M, Panzeri C, Castaldo P, Soldovieri MV, Baschirotto C, Bresolin N, Dalla Bernardina B, et al. A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology. 2004;63(1):57–65.
Ovsepian SV, LeBerre M, Steuber V, O'Leary VB, Leibold C, Oliver Dolly J. Distinctive role of KV1.1 subunit in the biology and functions of low threshold K(+) channels with implications for neurological disease. Pharmacol Ther. 2016;159:93–101.
Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF. Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol. 2015;88(1):203–17.
Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7(8):742–55.
Miyamoto H, Katagiri H, Hensch T. Experience-dependent slow-wave sleep development. Nat Neurosci. 2003;6(6):553–4.
Bishop HI, Guan D, Bocksteins E, Parajuli LK, Murray KD, Cobb MM, Misonou H, Zito K, Foehring RC, Trimmer JS. Distinct cell- and layer-specific expression patterns and independent regulation of Kv2 channel subtypes in cortical pyramidal neurons. J Neurosci. 2015;35(44):14922–42.
Aypar U, Wirrell EC, Hoppman NL. CNKSR2 deletions: a novel cause of X-linked intellectual disability and seizures. Am J Med Genet A. 2015;167(7):1668–70.
Houge G, Rasmussen IH, Hovland R. Loss-of-Function CNKSR2 Mutation Is a Likely Cause of Non-Syndromic X-Linked Intellectual Disability. Mol Syndromol. 2012;2(2):60–3.
Vaags AK, Bowdin S, Smith ML, Gilbert-Dussardier B, Brocke-Holmefjord KS, Sinopoli K, Gilles C, Haaland TB, Vincent-Delorme C, Lagrue E, et al. Absent CNKSR2 causes seizures and intellectual, attention, and language deficits. Ann Neurol. 2014;76(5):758–64.
Madsen KK, Hansen GH, Danielsen EM, Schousboe A. The subcellular localization of GABA transporters and its implication for seizure management. Neurochem Res. 2015;40(2):410–9.
Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15(12):1392–8.
Verhoeven WM, Egger J, Feenstra I, de Leeuw N. A de novo 3.57 Mb microdeletion in 8q12.3q13.2 in a patient with mild intellectual disability and epilepsy. Eur J Med Genet. 2012;55(5):358–61.
Giorda R, Bonaglia MC, Beri S, Fichera M, Novara F, Magini P, Urquhart J, Sharkey FH, Zucca C, Grasso R, et al. Complex segmental duplications mediate a recurrent dup(X)(p11.22-p11.23) associated with mental retardation, speech delay, and EEG anomalies in males and females. Am J Hum Genet. 2009;85(3):394–400.
Reutlinger C, Helbig I, Gawelczyk B, Subero JI, Tönnies H, Muhle H, Finsterwalder K, Vermeer S, Pfundt R, Sperner J, et al. Deletions in 16p13 including GRIN2A in patients with intellectual disability, various dysmorphic features, and seizure disorders of the rolandic region. Epilepsia. 2010;51(9):1870–3.
Nakayama T, Nabatame S, Saito Y, Nakagawa E, Shimojima K, Yamamoto T, Kaneko Y, Okumura K, Fujie H, Uematsu M, et al. 8p deletion and 9p duplication in two children with electrical status epilepticus in sleep syndrome. Seizure. 2012;21(4):295–9.
Arkilo D, Devinsky O, Mudigoudar B, Boronat S, Jennesson M, Sassower K, Vaou OE, Lerner JT, Jeste SS, Luchsinger K, et al. Electroencephalographic patterns during sleep in children with chromosome 15q11.2-13.1 duplications (Dup15q). Epilepsy Behav. 2016;57(Pt A):133–6.
Acknowledgements
We are grateful for the support we received from the National Key Research and Development Program of China (NO. 2016YFC1306202, NO. 2016YFC0904400) and the National Natural Science Foundation of China (NO. 81370771).
Authors’ statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Funding
This work was supported by the National Key Research and Development Program of China (NO. 2016YFC1306202, NO. 2016YFC0904400) and the National Natural Science Foundation of China (NO. 81370771).
Availability of data and materials
All data are available in this manuscript.
Author information
Authors and Affiliations
Contributions
MK made substantial contributions to conception and acquisition of data. JP and LY participated in drafting the manuscript. JX and HD made substantial contributions to acquisition of data. NP and FY supervised and revised the manuscript critically. All the authors agreed to be accountable for all aspects of the work and gave final approval of the version to be published.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Search strategies which were used in MEDLINE, EMBASE, PubMed and Cochrane review database. (DOCX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kessi, M., Peng, J., Yang, L. et al. Genetic etiologies of the electrical status epilepticus during slow wave sleep: systematic review. BMC Genet 19, 40 (2018). https://doi.org/10.1186/s12863-018-0628-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-018-0628-5