
Abstract
Backgrounds
Bovine mastitis is a typical inflammatory disease causing seriously economic loss. Genome-wide association study (GWAS) can be a powerful method to promote marker assistant selection of this kind of complex disease. The present study aimed to analyze and identify single nucleotide polymorphisms (SNPs) and candidate genes that associated with mastitis susceptibility traits in Chinese Holstein.
Results
Forty eight SNPs were identified significantly associated with mastitis resistance traits in Chinese Holstein cows, which are mainly located on the BTA 14. A total of 41 significant SNPs were linked to 31 annotated bovine genes. Gene Ontology and pathway enrichment revealed 5 genes involved in 32 pathways, in which, TRAPPC9 and ARHGAP39 genes participate cell differentiation and developmental pathway together. The six common genome-wide significant SNPs are found located within TRAPPC9 and flanking ARHGAP39 genes.
Conclusions
Our data identified the six SNPs significantly associated with SCS EBVs, which suggest that their linked two genes (TRAPPC9 and ARHGAP39) are novel candidate genes of mastitis susceptibility in Holsteins.
Similar content being viewed by others

Background
Bovine mastitis is one of the most typical inflammatory diseases causing seriously economic loss in modern dairy farms and quality problems of dairy food worldwide [1]. Since the heritability of mastitis is low, genetic improvement on anti-mastitis by traditional selection is not very effective [2]. Moreover, it is not easy to measure mastitis in field scale. Somatic cell count (SCC) or log transformed SCC (somatic cell score, SCS) have relatively higher heritability compared to mastitis and are used as the first trait to improve mastitis resistance [3]. In addition, to avoid uncertain influences such as farms, seasons, sires and etc., estimated breeding values (EBVs) of somatic cell scores (SCSs) were normally used as pseudo-phenotypes of mastitis related traits in dairy cattle. Genome-wide association study (GWAS) is widely considered a potential method to promote marker assisted selection of mastitis related traits based on single nucleotide polymorphism (SNP) [4].
The previous GWAS for mastitis susceptibility showed multifarious results in different Holstein populations. Family-based association tests such as single locus regression analysis and transmission disequilibrium test have the robust advantage to population heterogeneity [5]. In 2011, Sodeland’s group detected QTLs for clinical mastitis on Bos taurus autosome (BTA) 2, 6, 14, and 20 in Norwegian red cattle [6]. In 2012, Meredith et al. reported that 9 SNPs located on BTA 6, 10, 15 and 20 were significantly associated with SCSs in Holstein sires and cows [7]. The same year, Wijga et al. [8] reported that SNPs relevant to log transformed lactation-average somatic cell scores or the standard deviation of test-day somatic cell score were mainly located on BTA 4, 6 and 18. In addition, strong associations of SNPs with clinical mastitis and SCS were reported on bovine BTA 6, 13, 14 and 20 in Nordic Holstein cattle by Sahana et al. [9]. Recently, GWAS performed in German Holstein cows identified significant SNPs on BTA 6, 13, 19 and X [10]. The studies in US Holstein dairy cows have shown that genetic variants on BTA 2, 14, 20 have impacts on clinical mastitis. The identified region on BTA 14 contains lymphocyte-antigen-6 complex (LY6) including LY6K, LY6D, LYNX1, LYPD2, SLURP1, PSCA genes in regulating the major histocompatibility complex [11]. The studies in Chinese population containing Chinese Holstein, Sanhe cattle and Chinese Simmental have analyzed that TLR4 gene (Toll-like receptor 4) and BRCA1 gene (Breast cancer 1) have the significant association with SCS [12, 13]. Even though many studies have identified significant SNPs, only one SNP (BTA-77077-no-rs, Position: 85527109) on BTA 6 was identical in the reports of Sahana et al. [9] and Abdel-Shafy et al [10]. These results implied that the significant SNPs associated with mastitis traits were not identified consistently and should be confirmed and validated in different Holstein populations.
In order to detect functional candidate genes for mastitis-related traits, GWAS was conducted with mixed model based single locus regression analysis (MMRA) in Chinese Holstein populations. Six common SNPs were identified by MMRA and two linked genes were disclosed with significant effects on mastitis-related traits in Chinese Holstein populations.
Results
Significant SNPs associated with SCSs EBVs
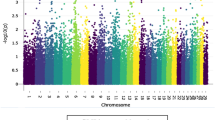
The –log10 P of all tested SNPs for SCS EBVs with MMRA is shown in Fig. 1. The significant SNPs associated with SCS EBVs were mainly located on BTA 14.

Manhattan plots of genome-wide association for SCS EBVs
The genomic association SNPs detected by MMRA were presented in Table 1. In total, 48 significant SNPs on chromosome level were detected including 13 SNPs on genome level. As shown in Table 1, 41 out of 48 SNPs were located within or near 31 known genes.
In the thirteen genome-wide significant SNPs, ARS-BFGL-NGS-100480 was located within TRAPPC9 gene (trafficking protein particle complex 9) on BTA 14 and showed lowest P-values of 1.24E-10. Two other significant SNPs, ARS-BFGL-NGS-56327 and UA-IFASA-5306 located within TRAPPC9 gene, were detected with P-values of 3.29E-08, and 3.64E-08, respectively. In addition, three other significant SNPs were identified linked with ARHGAP39 gene (Rho GTPase activating protein 39) (Table 2).
Linkage disequilibrium (LD) blocks of the significant SNPs on BTA 14
Linkage disequilibrium analysis for the total ten significant SNPs on BTA 14 showed two LD blocks (Fig. 2). Two significant SNPs (ARS-BFGL-NGS-57820 and ARS-BFGL-NGS-4939) in the block 1 were located on the upstream of ARHGAP39 gene, and three significant SNPs (BFGL-NGS-113575, ARS-BFGL-NGS-56327 and ARS-BFGL-NGS-100480) in the block 2 were located within TRAPPC9 gene.

Linkage disequilibrium (LD) pattern for 10 significant SNPs on BTA 14. Solid line triangles refer to linkage disequilibrium (LD). One square refers to LD level (r2) between two SNPs and the squares are colored by D’/LOD standard scheme (LOD is the logarithm of likelihood odds ratio and the reliable index to measure D’). D’/LOD standard scheme is that red refers to LOD > 2, D’ = 1; pink refers to LOD > 2, D’ < 1; blue refers to LOD < 2, D’ = 1; white refers to LOD < 2, D’ < 1
Two candidate genes for mastitis-related traits
TRAPPC9 and ARHGAP39 genes (each contains three significant SNPs on genome level) identified by MMRA can be considered potential candidate genes for mastitis-related traits. To decipher the effect of each genotype in each potential candidate gene on mastitis-related traits, the SCS EBVs of the cows with three genotypes were compared. As shown in the left panel of the Fig. 3, the cows with genotype AA in the two genes all owned significant higher SCS EBVs compared to the other genotypes (P < 0.001). These results appropriately confirmed the two genes (TRAPPC9 and ARHGAP39) as potential candidate genes for SCS EBVs. The right panel of the Fig. 3 showed the average original phenotypic SCC of the cows with three genotypes for each gene fluctuated with the days in milk (DIM). It was displayed that the cows with genotype AA had a tendency of higher SCC along DIM than the other two genotypes for the two genes especially for TRAPPC9 gene (Fig. 3).

The SCS EBVs and curves of SCC in different genotypes of TRAPPC9 and ARHGAP39 genes. **refers to P < 0.001
Gene ontology and pathway enrichment for the significant SNPs on genome level
Through the Gene Ontology (GO) analysis of GenCLiP 2.0 (http://ci.smu.edu.cn/GenCLiP2.0/analysis.php?random=new), we found that 5 genes perform mainly functions in 32 pathway terms presented in Table 3 and Fig. 4. Through enrichment of five genes, ARHGAP39 gene can totally participate 24 pathway terms including two pathway terms combined with TRAPPC9 gene (GO:0030154 and GO:0048869), which influence cell differentiation or cellular developmental process.

The cluster result of GO analysis
Discussion
The present study identified significant SNPs and novel candidate genes associated with mastitis-related traits in Chinese Holstein population with mixed model based single marker regression analysis (MMRA). Two genes (TRAPPC9 and ARHGAP39) identified by significant SNPs indicate that they are important candidate genes associated with mastitis-related traits. To our knowledge, this is the first study to decompose the genetic background of mastitis-related traits in Chinese dairy cattle using MMRA assay.
With regards to TRAPPC9 gene, it was reported that its product NIBP (NIK and IKKβ-binding protein) can enhance cytokine-induced NF-κB signaling pathway through interaction with NIK (NF-κB-inducing kinase) and IKKβ (IκB kinase-β) [14, 15]. In recent studies, TRAPPC9 gene was considered as candidate gene for autosomal recessive non-syndromic mental retardation [16, 17]. In the present study, the SCS EBVs (2.99) of the cows with AA genotype of SNP (ARS-BFGL-NGS-100480) in TRAPPC9 gene is significantly higher than the other two genotypes (P < 0.001). The similar tendency of the three genotypes was independently proved in a completely different Chinese Holstein population (n = 314, our unpublished data). As for ARHGAP39 gene, it was proved to be function to activate Rho GTPase which is known as new targets in cancer therapy [18]. Therefore, it is clear that the present study screened functional closely related genes to bovine mastitis resistance.
From the reported GWAS based on single locus regression analysis, it is not easy to identify the certain SNPs associated with SCS or mastitis-related traits. As shown in Table 1, 7 significant SNPs located on BTA 14 on whole genomic level (P < 1.14E-06) by MMRA in Chinese Holsteins were completely different from all the reported significant SNPs [7, 8], whereas significant SNPs on BTA 14 are consistent with other studies [6, 9–11, 19, 20]. In comparison, one significant SNP UA-IFASA-9288 (BTA 14, Position: 2201870) in Chinese Holstein was close to (147413 bp) the SNP ARS-BFGL-NGS-107379 (Position: 2054457) which was identified in Nordic Holstein [9]. However, Tiezz et al. [11] identified a region associated with clinical mastitis from 2,574,909 to 3,137,184 bp on BTA 14 which contains three genome-wide significant SNPs (ARS-BFGL-NGS-100480, ARS-BFGL-NGS-56327 and UA-IFASA-5306) covered by TRAPPC9 gene in this study. These GWAS studies suggest that mastitis-related traits as low heritable polygenetic traits are mainly controlled by multiple loci which distributed across the whole genome and each with relatively small genetic effect.
Although SCS is continuous trait which normally used as important indicator of mastitis, it is usually unstable and easily influenced by environment [21, 22]. Therefore, to disease indicator trait, current strategy has changed to performing association studies in cases and controls test [23], because of mastitis resistance or susceptibility can be considered as threshold traits [2]. In the current another study, we defined that the left and right parts of the population with half/one standard deviation of SCS EBVs were mastitis susceptibility group (case) and healthy group (control), respectively, and analyzed the two groups with ROADTRIPs (Robust Association-Detection Test for Related Individuals with Population Substructure) (version 1.2) (http://faculty.washington.edu/tathornt/software/ROADTRIPS2/) using bovine 54 k SNPs information. Although the decreased population size and increasing bias affect the testing power of the case-control association assay, we also have found two significant SNPs linked to two genes (TRAPPC9 and ARHGAP39) by ROADTRIPs of case-control test compared with MMRA results, which strongly suggest that these genes are novel candidate genes for mastitis traits.
The genes closed to or covered significant SNPs were further subjected to bioinformatics analysis. Results from Gene Ontology (GO) analysis (Table 3) indicated that TRAPPC9, ARHGAP39 and PTK2 genes play a role in regulation of cell differentiation (GO: 0030154, P = 0.033) or developmental process (GO: 0048869, P = 0.039). From the cluster result of GO analysis (Fig. 4), we found that ARHGAP39 and PTK2 genes are mostly close genes, which participate 24 pathway terms. However, TRAPPC9 gene has less result in GO analysis, thus the related pathways are needed to do further functional analysis.
Conclusions
Although lower detecting power exists in SCS EBVs and other mastitis resistance traits, results consistently support that the significant SNPs are mainly located on the BTA 14 in the Chinese Holstein cows. TRAPPC9 and ARHGAP39 genes reveal the two novel candidate genes associated with mastitis resistant traits in dairy cattle.
Methods
Ethics statement
All protocols for collection of the blood sample of experimental cows were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at China Agricultural University.
Animals and phenotype
A total of 2,093 cows from 14 sires were collected to construct the study population. The number of daughters of 14 sires range from 83 to 358 with an average of 150. Although the 14 sires were genotyped, they were not used in the association study in order to avoid double use of daughters’ information. These daughters were from 15 Holstein cattle farms in Beijing, China. No specific permissions were required for these locations/activities.
As closely following normal distribution, somatic cell scores (SCSs) are calculated from SCCs as (log2 (SCC/100,000) + 3). To avoid environment influence, EBVs of SCSs were provided as the phenotypes in the GWAS. These EBVs were obtained based on a multiple trait random regression test-day model [24] using the software RUNGE provided by Canadian Dairy Network (CDN) (http://www.cdn.ca).
DNA extraction and genotypes
Genomic DNA of the whole blood was extracted using the TIANamp Blood Genomic DNA Purification kit (Tiangen inc. Beijing, China). The criteria of DNA quality control were DNA concentration should be larger than 50 ng/μL, the ratio of OD260/OD280 in the range of 1.7–1.9 and the ratio of OD260/OD230 in the range of 1.5–2.1.
The cows were genotyped using Illumina Bovine SNP50 BeadChip [25]. The genotypes were edited according to the criteria: (1) call rate > = 90 %; (2) SNPs did not deviated extremely from Hardy-Weinberg equilibrium (P >10−6); (3) minor allele frequency > = 3 %). After quality control, a total of 43,885 SNPs were available for MMRA. Distribution of SNPs on each chromosome after quality control and the average distances between adjacent SNPs are shown in Additional file 1: Table S1.
Association analysis
Mixed model based single locus regression analysis (MMRA) applied to perform GWAS in our studies is as follows:
MMRA:
Where y is the vector of phenotypes (SCS EBVs), μ is the overall mean, b is the vector of coefficients of the regression on SNP genotypes, x is the vector of SNP genotypes, a ~ (0, Aσ 2a ) and e ~ (0, Wσ 2e ) are the vectors of the polygenic effects and residuals, where A is the additive genetic relationship matrix and W is a diagonal matrix with diagonal elements of 1/RELi to weight residuals variance for heterogeneity [26]. RELi is the reliability of EBV for the ith individual. σ 2a and σ 2e is the additive variance and residual error variance respectively. For each SNP, the estimated b and \( Var\left(\widehat{b}\right) \) are obtained via mixed model equations (MME). In addition, an approximate Wald chi-squared statistic \( {\widehat{b}}^2/Var\left(\widehat{b}\right) \) with df =1 is estimated for the SNPs significantly associated with phenotypes. This association analysis was conducted using a program written in FORTRAN language by our group [26].
Statistical inference
To decrease the false positive rate of multiple tests and screen more available SNPs as well as find more functional related genes, Bonferroni multiple testing (P < 0.05) was adopted to adjust for number of SNPs on genome and chromosome level. The results of Bonferroni threshold for genome and each chromosome divided by 0.05 were listed in Additional file 2: Table S2.
Linkage disequilibrium analysis for the significant SNPs on BTA 14 was performed using Haploview software (version 4.2) [27].
Student t-tests were conducted to compare the difference of cows SCS EBVs with different genotypes in each candidate gene.
Abbreviations
- GWAS:
-
Genome-wide association study
- SNP:
-
Single nucleotide polymorphism
- SCC:
-
Somatic cell count
- SCSs:
-
Somatic cell scores
- EBVs:
-
Estimated breeding values
- BTA:
-
Bos taurus autosome
- MMRA:
-
Mixed model based single locus regression analysis
- LY6 :
-
Lymphocyte-antigen-6 complex
- TLR4 :
-
Toll-like receptor 4
- BRCA1 :
-
Breast cancer 1
- TRAPPC9:
-
Trafficking protein particle complex 9
- ARHGAP39 :
-
Rho GTPase activating protein 39
- LD:
-
Linkage disequilibrium
- DIM:
-
Days in milk
- GO:
-
Gene Ontology
- NIBP:
-
NIK and IKKβ-binding protein
- NIK:
-
NF-κB-inducing kinase
- IKKβ:
-
IκB kinase-β
- ROADTRIPs:
-
Robust Association-Detection Test for Related Individuals with Population Substructure
- IACUC:
-
Institutional Animal Care and Use Committee
- CDN:
-
Canadian Dairy Network
- MME:
-
Mixed model equations
References
Hogeveen H, Huijps K, Lam TJ. Economic aspects of mastitis: new developments. N Z Vet J. 2011;59(1):16–23.
Heringstad B, Klemetsdal G, Ruane J. Selection for mastitis resistance in dairy cattle: a review with focus on the situation in the Nordic countries. Livest Prod Sci. 2000;64(2-3):95–103.
Shook GE, Schutz MM. Selection on somatic cell score to improve resistance to mastitis in the United States. J Dairy Sci. 1994;77(2):648–58.
Wiggans GR, Sonstegard TS, VanRaden PM, Matukumalli LK, Schnabel RD, Taylor JF, et al. Selection of single-nucleotide polymorphisms and quality of genotypes used in genomic evaluation of dairy cattle in the United States and Canada. J Dairy Sci. 2009;92(7):3431–6.
Thornton T, McPeek MS. Case-control association testing with related individuals: a more powerful quasi-likelihood score test. Am J Hum Genet. 2007;81(2):321–37.
Sodeland M, Kent MP, Olsen HG, Opsal MA, Svendsen M, Sehested E, et al. Quantitative trait loci for clinical mastitis on chromosomes 2, 6, 14 and 20 in Norwegian Red cattle. Anim Genet. 2011;42(5):457–65.
Meredith BK, Kearney FJ, Finlay EK, Bradley DG, Fahey AG, Berry DP, et al. Genome-wide associations for milk production and somatic cell score in Holstein-Friesian cattle in Ireland. BMC Genet. 2012;13:21.
Wijga S, Bastiaansen JW, Wall E, Strandberg E, de Haas Y, Giblin L, et al. Genomic associations with somatic cell score in first-lactation Holstein cows. J Dairy Sci. 2012;95(2):899–908.
Sahana G, Guldbrandtsen B, Thomsen B, Lund MS. Confirmation and fine-mapping of clinical mastitis and somatic cell score QTL in Nordic Holstein cattle. Anim Genet. 2013;44(6):620–6.
Abdel-Shafy H, Bortfeldt RH, Reissmann M, Brockmann GA. Short communication: validation of somatic cell score-associated loci identified in a genome-wide association study in German Holstein cattle. J Dairy Sci. 2014;97(4):2481–6.
Tiezzi F, Parker-Gaddis KL, Cole JB, Clay JS, Maltecca C. A genome-wide association study for clinical mastitis in first parity US Holstein cows using single-step approach and genomic matrix re-weighting procedure. PLoS One. 2015;10(2):e0114919.
Wang X, Xu S, Gao X, Ren H, Chen J. Genetic polymorphism of TLR4 gene and correlation with mastitis in cattle. J Genet Genomics. 2007;34(5):406–12.
Yuan Z, Chu G, Dan Y, Li J, Zhang L, Gao X, et al. BRCA1: a new candidate gene for bovine mastitis and its association analysis between single nucleotide polymorphisms and milk somatic cell score. Mol Biol Rep. 2012;39(6):6625–31.
Hu WH, Pendergast JS, Mo XM, Brambilla R, Bracchi-Ricard V, Li F, et al. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J Biol Chem. 2005;280(32):29233–41.
Kim JC, Kim SY, Roh SA, Cho DH, Kim DD, Kim JH, et al. Gene expression profiling: canonical molecular changes and clinicopathological features in sporadic colorectal cancers. World J Gastroenterol. 2008;14(43):6662–72.
Khattak NA, Mir A. Computational Analysis of TRAPPC9: Candidate Gene for Autosomal Recessive Non-Syndromic Mental Retardation. CNS Neurol Disord Drug Targets. 2013;13(4):699–711.
Mochida GH, Mahajnah M, Hill AD, Basel-Vanagaite L, Gleason D, Hill RS, et al. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am J Hum Genet. 2009;85(6):897–902.
Gomez del Pulgar T, Benitah SA, Valeron PF, Espina C, Lacal JC. Rho GTPase expression in tumourigenesis: evidence for a significant link. Bioessays. 2005;27(6):602–13.
Klungland H, Sabry A, Heringstad B, Olsen HG, Gomez-Raya L, Vage DI, et al. Quantitative trait loci affecting clinical mastitis and somatic cell count in dairy cattle. Mamm Genome. 2001;12(11):837–42.
Schulman NF, Viitala SM, de Koning DJ, Virta J, Maki-Tanila A, Vilkki JH. Quantitative trait Loci for health traits in Finnish Ayrshire cattle. J Dairy Sci. 2004;87(2):443–9.
de Haas Y, Ouweltjes W, ten Napel J, Windig JJ, de Jong G. Alternative somatic cell count traits as mastitis indicators for genetic selection. J Dairy Sci. 2008;91(6):2501–11.
Heringstad B, Rekaya R, Glanola D, Klemetsdal G, Welgel KA. Genetic change for clinical mastitis in Norwegian cattle: a threshold model analysis. J Dairy Sci. 2003;86(1):369–75.
Ferguson-Smith AC, Greally JM, Martienssen RA. Epigenomics. Dordrecht: Springer; 2009.
Schaeffer LR, Jamrozik J, Kistemaker GJ, Van Doormaal BJ. Experience with a test-day model. J Dairy Sci. 2000;83(5):1135–44.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009;4(4):e5350.
Jiang L, Liu J, Sun D, Ma P, Ding X, Yu Y, et al. Genome wide association studies for milk production traits in Chinese Holstein population. PLoS One. 2010;5(10):e13661.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (31272420), State High-Tech Development Plan of China (2008AA101002), Basic Research from the Ministry of Education of the People’s Republic of China (2011JS006), Modern Agro-industry Technology Research System (CARS-37), the Twelfth Five-Year plan of National Science and Technology Project in Rural Areas (2011BAD28B02) and the Program for Changjiang Scholar and Innovation Research Team in University (IRT1191). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
These authors declare that they have no competing interests.
Authors’ contributions
XW performed the genome-wide association analysis and prepared the manuscript. PM, JL, XD and LJ participated in the samples preparation and data analysis. QZ and YZ participated in the experiment design. YW, YZ, DS, SZ and GS participated in interpreting the result. YY conceived and designed the experiments and prepared the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1: Table S1.
Distribution of SNPs on each chromosome after quality control and the average distances between adjacent SNPs. These data were derived from Bos_taurus_UMD_3.1 assembly (http://www.ncbi.nlm.nih.gov/assembly/GCF_000003055.4/). SNPs which are not assigned to any chromosomes are noted as “0”. (DOCX 25.5 kb)
Additional file 2: Table S2.
Results of Bonferroni thresholds at genome-wide level and at chromosome-wide level for each chromosome. SNPs which are not assigned to any chromosomes are noted as “0”. (DOCX 24.8 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, X., Ma, P., Liu, J. et al. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet 16, 111 (2015). https://doi.org/10.1186/s12863-015-0263-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-015-0263-3