
Abstract
Biogeographic barriers to gene flow are central to studies of plant phylogeography. There are many physical and geographic barriers in China, but few studies have used molecular ecological evidence to investigate the natural geographic isolation barrier of the Qinling Mountains-Huaihe River Line (QHL). Allium macrostemon is a precious Chinese perennial herb belonging to the Amaryllidaceae family. It is used as a food and medicine, with a variety of health and healing properties. Five SSR markers, three chloroplast DNA (cpDNA) markers (psbA-trnH, rps16 and trnL-F), one nuclear ribosomal DNA (nrDNA) marker (ITS), and simplified genome GBS sequencing were used to analyse the genetic diversity and structure of A. macrostemon. Combining SSR, cpDNA, nrDNA ITS data and GBS analysis results, we divided A. macrostemon populations into northern and southern groups, with the southern group further divided into southwestern and central-southeastern groups. Niche simulation results reveal that the distribution area of A. macrostemon will reach its maximum in the future. These data indicate that the regional separation of A. macrostemon has been maintained by the combined influence of a geographical barrier and Quaternary climate, and that the back-and-forth fluctuations of QHL and Quaternary climate have played an important role in this process. QHL acts as a north-south dividing line in phylogeography and population genetic structure, promoting physical geographic isolation. This study provides a theoretical basis for the conservation, development, and utilization of A. macrostemon resources. It further provides a reference for understanding the systematic geographical pattern of the large-scale spatial distribution of plants in China and enriches our understanding of Quaternary plant evolution in areas with complex terrain.
Similar content being viewed by others


Introduction
Geological history and climatic oscillations are important drivers of evolution and influence the genetic structure of plant species [1, 2]. Since the beginning of the Quaternary glacial period, global temperatures have generally decreased, but fluctuations between cold and warm conditions, repeated alternation between glacial and interglacial periods, and geological events and monsoon fluctuations have strongly influenced the geographic distribution, population dynamics, and genetic diversity of species [3,4,5,6]. The uplift of the Tibetan Plateau was an important geological event that changed the global climate and stimulated the East Asian Monsoon, increasing the meridional zonation of Chinese flora [7,8,9]. Thus, alternations between dry and wet climatic conditions have strongly influenced the evolution and distribution of Chinese plants. Stable climate, mature plant communities across a vast landscape, high spatial heterogeneity, and a long evolutionary history established the foundation for the formation of a rich and diverse Chinese flora. As such, China is an important region of species diversity in the Northern Hemisphere [10] and important for species conservation, speciation, and evolution [11].
The impact of environmental changes since the beginning of the Quaternary on speciation, genetic diversity, population genetic structure, and demographic history can be studied using phylogeography and population genetics techniques [11, 12]. Characterizing biogeographical barriers to gene flow are central to plant phylogeography. Geographic isolation resulting from the uplift of mountain ranges and climate fluctuations associated with glacial oscillations can drive dramatic changes in the morphologies and geographic distributions of many species [13]. Biological populations have increasing been found to coincide with known geographic boundaries, such as mountains, river systems, or straits, which act as physical barriers to dispersal, facilitating systematic intraspecific geographic subdivision and divergence. China has multiple geographic distribution zones separated by biogeographic boundaries. Molecular ecological data suggest that China has at least seven genealogical discontinuity locations across China, including the Mekong-Salween Divide in the Himalaya-Hengduan Mountain region [14]; the Tanaka-Kaiyong Line in the southwest [15]; the Sichuan Basin [16]; the region near 105° E [17]; the second and third step dividing line [18]; the North China region [19]; and the East China Sea/Tsushima–Korean straits [20,21,22]. However, few studies have investigated the impact of the Qinling Mountain-Huaihe River Line (QHL), a natural geographic isolation boundary, on the phylogeographical patterns and genetic structure of plant species.
The Qinling-Huaihe River Line (QHL), which extends from the Qinling Mountains to the Huaihe River, is located at about 33° N and forms the natural geographic division between North and South China [23]. It includes the Qinling and Dabie Orogenic Belt (QDB), the Yangtze River Plain, and the southern mountains of Anhui [24,25,26]. Climatic conditions and geographic features vary markedly across this boundary. For example, the southern region (i.e., south of QHL) has subtropical and tropical climates with greater precipitation and higher temperatures than the northern region (i.e., north of QHL), which has a temperate climate with obvious seasonal variation across four distinct seasons [27]. Previous research has shown that areas on both sides of the QHL have retained glacial refugia for plants, with these sanctuaries acting as sites of extended range in the late interglacial or post-glacial periods [28,29,30]. However, little is known regarding the impact of the QHL on the phylogeographic patterns and genetic structure of plant species.
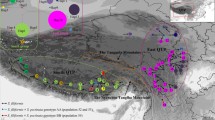
Allium macrostemon is a perennial herb belonging to the family Amaryllidaceae. It is a precious Chinese herb used for food and medicinal purposes, with a variety of health benefits and healing properties [31]. Current research on A. macrostemon tends to focus on its chemical composition, pharmacological action, and its potential for use as a source of crude materials for drug manufacturing [32,33,34]. It is distributed widely throughout the mountains and fields straddling the QHL in eastern and western China (Fig. 1). A. macrostemon is a polyploid species with a polyploid series of 2x, 3x, 4x, 5x, 6x, and 7x. Populations with 5n ploidy have been found in Japan, and populations with 3x, 4x, 5x, 6x and 7x ploidy have been found in China, with the remaining ploidies yet to be discovered in nature [35,36,37]. Analysis of the systematic taxonomic status of A. macrostemon using nrDNA (ITS) and cpDNA (trnL-F) sequencing data indicate that the species may represent a new group (Huang et al., 2015). Previous work from our group distinguishes A. macrostemon from other related Allium plants and supports A. macrostemon as an independent monophyletic group [38].

Growth environment and morphology of A. macrostemon. A, E, F. Plant morphology of A. macrostemon; B. Flowering morphology of A. macrostemon; C. Seed morphology of A. macrostemon; D. Root morphology of A. macrostemon.
However, few reports are available on the systematic geography of A. macrostemon. As an herbaceous plant, A. macrostemon has a wide distribution, spanning different climate zones and multiple tropical biodiversity regions in China. In recent years, various molecular markers have been developed, including microsatellite markers, which are co-dominant markers that can be used to distinguish homozygosity from heterozygosity and to detect multiple alleles. They have the advantages of rich polymorphism, simple operation, reliable results, and good repeatability [39]. With the rise of high-throughput sequencing technology, single nucleotide polymorphism (SNP) marker analysis with simplified genotyping by sequencing (GBS) technology is increasingly common. GBS combines genotyping data and a large number of SNP sites to reveal speciation and evolutionary relationships, germplasm conservation, and population genetic structure [40]. The evolution rate of cpDNA is very slow, with low recombination and mutation rates, and the unique geographic distribution and evolutionary history of natural populations is usually evident in cpDNA lineages. As such, they are used widely in systematic geographical studies [41]. Organelle markers can be used as powerful tools for studying the phylogeography and migratory footprints of species; parental genetic markers can be combined with single-parent organelle markers for population genetics studies; and cpDNA and ITS sequence variations are effective methods to identify glacial refuges of plants [42]. Because the lifecycles of herbaceous plants are much faster than those of long-lived woody plants, they are generally expected to respond more quickly to changing environmental conditions [43, 44]. Thus, herbaceous plants may provide a better opportunity to evaluate the drivers of diversification and speciation [45, 46]. However, to our knowledge, only a few herbs from this region, such as Dysosma versipellis, Primula ovalifolia, and Oryza stavia, have been studied to date [26, 45, 47, 48]. Moreover, phylogeographic research is still lacking for plants with wide distributions spanning different climate zones and multiple tropical biodiversity regions, especially in China.
Here, we collected samples from both sides of the QHL and spanning east to west across China. Simple sequence repeats (SSRs), GBS sequencing, cpDNA genes (psbA-trnH, rps16, and trnL-F), and nrDNA (ITS) fragments were used to analyze the phylogeographic patterns and genetic structure of A. macrostemon. We also combined geographic distribution and climate data with niche simulation to predict the distribution of A. macrostemon in different time periods. In this study, we aimed to address the following questions: (1) What role did Quaternary climate history play throughout the phylogeographic history of A. macrostemon? (2) Is the influence of the QHL evident in the phylogeographic structure of A. macrostemon? (3) Does genetic diversity vary across populations of A. macrostemon? These analyses contribute to the theoretical basis for the conservation, development, and use of A. macrostemon resources as well as to the theoretical basis for further discussion of plant evolution and species diversity in China. They also advance understanding of the molecular systematic evolution and biogeography of East Asian herbaceous plants.
Materials and methods
Sampling, experimental design, and DNA extraction
A sampling plan was formulated after consulting sample records. Between 2015 and 2020, individuals ≥ 5 m were collected from a total of 50 populations across the Allium macrostemon Bunge distribution area following population ecology sampling methods. Sampled populations were separated by at least 15 km to avoid sampling bias caused by including closely related individuals. Plants were identified by Associate Professor Zhao Cai and specimens were preserved in the Department of Botany, Guizhou University (deposition numbers: ZhaocX1-ZhaocX50). Following collection, fresh leaves were dried and preserved with silica gel. A global positioning system device was used to record the longitude, latitude, and altitude of each population, a total of 50 natural A. macrostemon populationwere collected (Table S1). To improve experimental efficiency and conserve resources, 24 representation populations of A. macrostemon with regional representation were selected for SSR molecular marker analysis to investigate their genetic diversity and structure (Table S2). 13 representative populations were selected for GBS analysis (Table S3).
Allium Chinense G.Don from Xifeng County, Guiyang City, Guizhou Province were selected as the outgroup for phylogenetic reconstructions (population JTW). Chloroplast DNA (cpDNA) and nuclear ribosomal DNA (nrDNA) segments were sequenced from 50 natural populations to analyze population structure and genetic diversity. Total genomic DNA was isolated from dried leaves using the modified CTAB method [49].
SSR marker amplification and cpDNA and nrDNA sequencing
Primers were synthesized according to the primer sequences of Allium L. Five geographically distant A. macrostemon populations were selected for PCR amplification. Primers that produced clear and repeatable DNA fragments were first identified using agarose gel electrophoresis, followed by further selection by polyacrylamide gel electrophoresis. Five pairs of primers consistent with A. macrostemon SSR markers were obtained (Table S4) [50,51,52]. FAM fluorophore was added to the 5’ end of the final primer sequence selected for PCR amplification. Amplification consisted of the following steps: pre-denaturation at 94℃ for 2 min; a total of 30 cycles of denaturation at 94℃ for 1 min and extension at 72℃ for 45 s; and final extension at 72℃ for 4 min. The annealing temperature of each primer is presented in Table S5. Amplification products were stored at 4℃ prior to capillary electrophoresis using an ABI 3730XL gene sequencer at Qing Ke Biotechnology Co., Ltd. Three chloroplast gene fragments were amplified using the following primers: rps16, psbA-trnH, and trnL-F. One nuclear gene fragment was amplified using ITS as the primer. Primer design and PCR amplification were performed using the methods described by Hamilton [53], Oxelman et al., [54] Taberlet [55], and Wendel et al. [56] (Table S4). The PCR amplification reaction mixture consisted of 12.5 µL 2×Taq PCR Master Mix (Beijing Tiangen), 2 µL DNA template, 1 µL upstream primer, and 1 µL downstream primer; ddH2O was added to a final volume of 25 µL. Amplification followed the protocol outlined by the primers’ source literature (Table S4). Sequencing reactions were performed on an ABI 3730 automated sequencer at Sangon Biotech (Shanghai, China).
GBS library preparation and high-throughput sequencing
We performed GBS analysis to obtain genome-wide SNP data [57]. A. macrostemon DNA was treated with restriction endonuclease HincII, after which the endonuclease was repaired and the A-tail was added. To complement the notch cut in the DNA by the enzyme, splices were added at both ends of the small fragments. PCR was used to amplify the tag sequence containing the splice. DNA fragments were merged, and required fragments were recovered using agarose gel electrophoresis. Recovered fragments were further purified using a DNA molecular purification kit. DNA concentrations were assessed using Qubit fluorescence quantification to ensure the quality of GBS sequencing. An Illumina NovaSeq 6000 sequencing platform was used, with the sequencing mode set to PE150 for high-throughput sequencing of qualified libraries.
Ecological niche modelling
The geographic distribution of A. macrostemon was obtained from field surveys and the Virtual Herbarium of China (CVH, http://www.cvh.ac.cn). To prevent model overfitting caused by data duplication and spatial autocorrelation, we eliminated spatial coincidence data points within 10 km of each other [58]. 196 distribution points were obtained covering the distribution of A. macrostemon in China.
Nineteen bioclimatic factors from the last glacial maximum (LGM), mid-Holocene, and current and future periods were downloaded from the WorldClim database (http://www.worldclim.org) with a spatial resolution of 10 arc-minutes. Variables corresponding to species distribution points were extracted and then imported into MaxEnt v.3.4.1 along with species data for suitability prediction. Climate variables whose ecological contribution rate was greater than 1% in modeled predictions were retained [59]. To eliminate multicollinearity effects, variables with Pearson’s |R|≥ 0.7 were excluded [60]. Eight variables were ultimately selected as climate predictors to model potentially suitable areas for past, present, and future climates.
Data analysis
GenAlex 6.5 software [61] was used to calculate genetic parameters, such as expected heterozygosity (He), observed heterozygosity (Ho), effective number of alleles (Ne), observed number of alleles (Na), Shannon Information index (I), and percentage of polymorphic loci (PPL), were calculated for each population based on SSR data. The polymorphism information content (PIC) of each SSR locus was calculated using Cervus 3.0 software [62]. Analysis of molecular variance (AMOVA) was conducted using Arlequin version 3.5 [63] to further calculate assess variation and genetic differentiation between and within populations. GenAlex was also used to complete Nei’s genetic distance detection, and MEGA 7.0 was used to perform cluster analysis to obtain the adjacent tree (NJ). GenAlex was also used to conduct an IBD (Isolation-by-Distance) analysis between microsatellite and geographic distance data using a Mantel test and to complete principal coordinate analysis (PCoA) of all A. macrostemon groups and individuals to further evaluate the genetic structure of A. macrostemon. STRUCTURE v2.3.3 based on Bayes cluster analysis [64] was used to predict the geographic distribution of genetic variation among A. macrostemon populations. K values were set as 1–10, with each K value iterated 10 times, with model settings “Admixture Model” and “Allele Frequencies Correlated” selected. Burnin period length was set to 200, 000number of MCMC Reps after Burnin was set to 1000, 000. After the model was run, the rate of change for L (K) between continuous K values was obtained, and the most possible K value was determined by Delta K between continuous K values. Finally, the STRUCTURE cluster diagram was obtained [65]. Barrier v2.2 [66] was used to compute Monmonier’s maximum-difference algorithm for identifying biogeographic boundaries or areas exhibiting the largest genetic discontinuities between population pairs. In Barrier analysis, a total of 48 populations were selected for analysis, and there were three sampling points in Zhejiang, so only one population was selected for analysis.
Chromas software was used to view the base peak map of cpDNA and nrDNA sequence data, and DNAstar software [67] was used to perform para-alignment and positive direction splicing. CLUSTAL W in MEGA7.0 software [68] was used to manually correct sequences and to remove primers at both ends. Haplotype number, haplotype diversity (Hd), and nucleotide diversity (π) of cpDNA joint sequences and nrDNA sequences were analyzed using DnaSP v.5.0 [69] software. Total genetic diversity (HT) and intra-population mean genetic diversity (HS) were calculated using PERMUT 2.0 [70]. Haplotype NETWORK diagrams were constructed using NETWORK 5.0 software [71]. A haplotype phylogenetic tree was constructed using MEGA 7.0. The haplotype distribution map was drawn using Arc Map in ArcGIS 10.2. The phylogenetic tree was constructed using the Maximum Parsimony (MP) method based on cpDNA haplotype data using MEGA 7.0, and statistical support analysis was performed with the Bootstrap value set to 1000. The constructed MP tree was imported into GeneMiner 2.1, with replacement mode set to HKY85. Genealogies and model parameters were sampled every 20,000 iterations [72], and the correction point was 1.87-7.54Ma [73]. Finally, FigTree 1.4.2 was used to view and configure the constructed phylogenetic tree. Arlequin 3.5 was used to complete AMOVA [63] to calculate the degree of genetic variation within and between populations and the coefficient of genetic differentiation (Fst) between populations. DnaSP v.5.0 was used to conduct mismatch analysis and the neutral test for cpDNA association fragments and nrDNA sequences, respectively, to verify whether this species has undergone rapid expansion in the past.
After the GBS sequencing data was disassembled, quality control was performed using FastP 0.20.0 [74], after which all sample reads were clustered. Finally, BWA 0.7.13 [75] was used to compare the data with the consistency sequence for each individual. GATK [76] and Samtools [77] were used in tandem to detect SNPs, and quality filtering was conducted where mutations were detected. SNP data was used to perform evolutionary analysis, and MEGA 7.0 [68] was used to analyze genetic evolution and phylogeny of the samples.
Results
Genetic diversity and structure based on SSR
SSR amplified at five loci in 288 A. macrostemon individuals from 24 different populations were used to estimate genetic diversity. He, Ho, Ne, Na, I, and PPL were 0.498, 0.808, 2.357, 3.008, 0.871, and 80.8, respectively (Table 1). The BXS, SYS, THS, JAS, and QDS populations had high levels of genetic diversity, while the HDS, NXS, and JGX populations had low levels of genetic diversity (Table 1). The five SSRs all exhibited high polymorphism at the species level, with the highest genetic diversity occurring in SSR ACE039 and the lowest occurring in SSR ACM096 (Table S6). AMOVA based on SSR markers revealed that genetic variation occurred mainly within populations, accounting for 76% of the total variation (Table 2).
The Mantel test revealed that there was no significant correlation between geographic and genetic distance among A. macrostemon groups (r = 0.0714) (Fig. S1), indicating that geographic distance was not the main factor driving genetic differentiation in A. macrostemon. PCoA demonstrated that individuals from the same population clustered together, while only a few individuals from ZTS, SHS, QDS, and SYS had crossover with individuals from other populations (Fig. S2). The optimal K value determined by genetic structure analysis using STRUCTURE divided the 24 populations into three groups: Group A (northern group), Group B (central-southeastern group), and Group C (southwestern group). Some individuals in the populations were mixed to different degrees, indicating some gene exchange between populations (Fig. 2A and B). Nei’s genetic distance and geographic distribution were used to divide the 24 populations into three groups: north, southwest, and central-southeast (Fig. 2C and D). These groupings are consistent with those determined by STRUCTURE analysis. Calculations of gene flow and genetic distance revealed that average gene flow was smaller in the northern and southern groups (Group A, Group B, and Group C) than in the southern populations (Group B and Group C). The genetic distance of Group A, Group B, and Group C was much greater than average genetic distance of the either the northern populations (Group A) or southern populations (Group B and Group C), indicative of some isolation between northern and southern groups (Table S7). Barrier analysis were suggestive of a trend of genetic boundaries between northern and southern populations (Fig. S3A).

Genetic structure of A. macrostemon population using STRUCTURE. A. The 288 A. macrostemon individuals were divided into 24 populations. From top to bottom, K = 2, K = 3, K = 4, K = 5. Each A. macrostemon population is represented by a vertical bar; B. Delta K values for K from 1 to 10; C. Population genetic structure of A. macrostemon;D. NJ tree of A. macrostemon populations based on Nei’s genetic distance
Genetic diversity and structure based on cpDNA and nrDNA sequences
By concatenating alignments from three cpDNA sequences (psbA-trnH, 539 bp; rps16, 739 bp; trnL-F, 652 bp), we obtained 1930 bp of total cpDNA sequences from 574 individuals, containing 66 variant sites with G + C content of 32.99%. A total of 42 chloroplast haplotypes (H1-H42) were identified (Fig. 3B; Table S8). Haplotype H1 was the oldest haplotype and the most common, appearing in 144 individuals. It was also the most widely distributed, appearing in 14 populations (Table S8, Fig. 3C). In addition, multiple chloroplast haplotypes were found in 12 populations. The remaining 38 populations were monomorphic. The species showed high haplotype and nucleotide diversity (Hd = 0.904, π = 2.08 × 10− 3) at the species level. HT of chloroplast segments was 0.860, and HS within population was 0.121. At the population level, some populations in eastern and northeastern regions had higher genetic diversity. DPS, JHS, HCS, BXS, SHS, SMX and XYS all had higher haplotype and nucleotide diversity. Hd of SHS was the highest (0.758), and the nucleotide diversity (π) of JHS was the highest (1.700 × 10− 3). Using cpDNA haplotype data (Fig. S4), we estimated A. macrostemon separated from the outgroup in the late Pliocene (about 3.16 Ma), indicating that A. macrostemon originated much earlier than the beginning of the Quaternary. As evident in the figure, intraspecific divergence time was 0.100 Ma to 3.16 Ma, spanning from the late Pliocene epoch of the Tertiary to the late Pleistocene of the Quaternary, during which the Quaternary climate oscillation and the LGM occurred.

Geographic distribution of haplotypes and haplotype network based on nrDNA, ITS, and cpDNA sequences. A. The geographic distribution of haplotypes according to phylogenetic grouping based on nrDNA sequencing data. B. The geographic distribution of all nrDNA haplotypes. Population numbers 1–50 match those listed in Table S1. C. Haplotype network based on nrDNA. Circle size is proportional to the number of individuals bearing the haplotype. Colors indicate region: red, southwestern region (Group A); blue, central-southeastern region (Group B); and green, northern region (Group C). D. Phylogenetic tree based on ITS using MP and ML methods. E. Geographic distribution of haplotypes based on cpDNA sequences according to phylogenetic grouping. F. Geographic distribution of all cpDNA haplotypes. G. Haplotype network based on cpDNA. Circle size is proportional to the number of individuals bearing the haplotypes. Colors indicate region: green, northern region (Group I); yellow, southern region (Group II). H. Phylogenetic tree based on cpDNA using MP and ML methods
The 633 bp nrDNA ITS sequence of A. macrostemon was obtained from 581 individuals containing 391 variant sites, with a G + C content of 50.43%. These polymorphic sites revealed a total of 65 haplotypes (H1-H65) (Fig. 3F). Of these, the H7 haplotype had the widest distribution and highest frequency, occurring in 96 individuals. The core haplotype of the ITS network center was H7, which was presumed to be the oldest haplotype (Table S8, Fig. 3G). Seven populations (TSS, XAS, YCS, SMX, NXS, HCS, and SYS) contained more than three haplotypes, and 34 populations had only one haplotype. CpDNA analysis revealed that there was no haplotype sharing between different groups (northern, southwest, and center-southeast), but only within the same population (Table S8, Fig. 3D). Our nrDNA analysis suggested that haplotype sharing occurred between different groups. For example, it revealed that the northern and center-southeast groups of the BXS and HCS populations were mixed (Fig. 3F and H), suggesting that there was gene flow between different groups. These findings suggest that different populations in the same area often experienced genetic exchange at cpDNA and ITS loci. Compared with chloroplast gene sequences, ribosome gene sequences showed higher haplotype diversity and nucleotide diversity at the species level (Hd = 0.957, π = 9.162 × 10− 2) (Table S8). Populations in southwestern China exhibited high genetic diversity, in contrast to results from our cpDNA analysis.
AMOVA based on cpDNA and ITS sequence data further revealed the genetic structure of A. macrostemon. For cpDNA sequences, inter-population genetic variation (93.45%) was significantly higher than intra-population variation (6.55%), and FST was 0.93445 (P < 0.05). The results of AMOVA based on ITS were similar (FST = 0.94058), with most variation occurring between populations (Table 2). Nst genetic differentiation coefficients were not significantly larger than Gst (cpDNA: Nst = 0.930, Gst = 0.859, p > 0.05; nrDNA: Nst = 0.937, Gst = 0.808, p > 0.05), indicating that A. macrostemon had no significant systematic geographic structure. However, barrier analysis suggested there are multiple genetic boundaries within northern and southern populations, respectively (Fig. S3).
Phylogenetic tree analysis based on GBS
GBS analysis was performed using data from 13 individuals from 13 populations representing the entire geographic distribution of A. macrostemon, with A. chinense used as the outgroup. This data was used to construct phylogenetic trees, with A. macrostemon individuals clustering into northern, southwestern, and central-southeastern groups, similar to SSR and cpDNA and ITS analysis, further supporting the results of our fragment sequencing analysis (Fig. 4A and B).

Neighbor-joining tree and population genetic structure based on GBS. A. Neighbor-joining phylogenetic tree based on SNPs; B. Population genetic structure of A. macrostemon
Inference of demographic history
Based on mismatch distribution analysis of cpDNA an nrDNA sequences, the Tajima’s D values for the overall population were negative and nonsignificant. Tajima’s D was − 1.42056 (p > 0.10) for the chloroplast sequences and − 0.71303 (p > 0.10) for the nuclear sequences. The Fu’s Fs value was − 6.394 for the chloroplast sequences and 37.290 for the nuclear sequences. Mismatch distribution analysis produced multimodal curves, and observed values did not align with expected values (Fig. S5). This violated the population expansion model, indicating that A. macrostemon did not experience significant population expansion but was in dynamic equilibrium.
Analysis of suitable establishment areas for A. macrostemon
MaxEnt software was used to forecast the potential distribution of A. macrostemon in China. We estimated the mean value of test AUC as 0.983, indicating high-accuracy discrimination. The results suggest that global cooling drove marked contraction and southwestern migration toward areas of high suitability during the Quaternary glacial period. During the warm and humid middle Holocene, which was similar to the modern climate, the distribution of A. macrostemon expanded, and the predicted distribution range is similar to the present-day distribution. Our analysis suggests that the distribution of A. macrostemon will expand slightly in the future to attain its widest distribution (Fig. 5). Warm season precipitation had the highest contribution (41.2%), with mean temperature in the coldest season (16.1%) and variation coefficient of precipitation (13.7%) also making notable contributions, indicating that temperature, precipitation, and season strongly influence the distribution of A. macrostemon (Table S9).
Inhabitable area first decreased before increasing during different periods. In the future, the total suitable area of A. macrostemon will reach its maximum, and the center of distribution may move northward.

Simulation of the distribution of A. macrostemon under different climatic conditions in China. A. Last Glacial Maximum (LGM); B. Mid-Holocene; C. Present-day; D. Future (2070)
Discussion
Population genetic diversity
Our analysis of cpDNA and ITS data indicated that genetic variation in the chloroplast and nuclear ribosome genes of A. macrostemon was 0.67 and 0.137 higher than the average genetic diversity of angiosperm chloroplast and nuclear ribosome DNA, respectively [78]. Genetic diversity estimated by chloroplast genes was higher than that of other herbs, such as Allium mongolica (HT = 0.693) [79] and Fritillaria pallidifora (HT = 0.670) [80]. According to SSR data, A. macrostemon possesses greater genetic diversity than other herbaceous plants (He = 0.498; I = 0.871), such as Monochasma savatieri (He = 0.342, I = 0.535) [39]; and Dactylorhiza hatagirea (He = 0.158, I = 0.239) [81]. A population’s genetic diversity is influenced by a number of factors related to a species’ geographical distribution, biological characteristics, population size, and reproductive system [78]. Many studies have employed various molecular markers to explore the genetic diversity of herbaceous plants, such as Typha domingensis and Typha latifolia [82]. The low genetic diversity estimated for these species may be related to their high self-breeding rates and sizable vegetative reproductive capacity [83]. The genetic and nucleotide diversity of A. macrostemon genes were higher than those of chloroplast genes, which may be related to the relative conservation of chloroplast genes and their maternal inheritance pattern [84].
In general, species with a wide geographic range have high genetic diversity, large population sizes, high adaptability, and a rich gene pool. The high genetic diversity of A. macrostemon may be related to its breeding system and geographic distribution [85, 86]. First, A. macrostemon can reproduce sexually or via bulb tillering, in which the underground mother plant grows out a peanut stem and produces an air bulb for propagation [87]. The reproductive mode of A. macrostemon is the main factor affecting its genetic diversity. Sexual reproduction can provide genetic information and increase genetic variation driving high genetic diversity in this species. Secondly, A. macrostemon is widely distributed throughout China, occurring in all provinces except Xinjiang and Qinghai. This wide distribution may be one of the causes of its high genetic diversity. In summary, A. macrostemon has maintained relatively rich genetic diversity [88].
There may be several reasons for the contrary results of SSR molecular variance in this study. First, the fruits of A. macrostemon are not favored by most animals, its ability to spread seeds is very limited, and most seeds occur near the mother plant. Second, the formation time of A. macrostemon is very long. In this study, it is estimated that the intraspecific divergence time of A. macrostemon is about 3.16 Ma, which is very close to the formation time of some woody plants. The phenomenon that genetic variation within populations is greater than that between populations occurs mostly in woody plants [13, 89]. Therefore, A. macrostemon may have a similar genetic variation pattern to that of woody plants. Thirdly, gene exchange among A. macrostemon populations is more limited due to habitat fragmentation and human activities. However, the results of CPDNA and ITS markers in this study showed that the genetic variation among A. macrostemon populations was greater than within the population. Therefore, we considered that the genetic variation among A. macrostemon populations was greater than within population.
Contributions of geographic separation (QHL) to the phylogeographic structure of A. macrostemon
Geographic structure is common in plants with continuous distributions, usually emerging because of distance or environmental isolation [90, 91]. Here, SSR data indicated that genetic variation mainly occurred within populations. However, low levels of gene flow may lead to a population adapting to local environmental conditions and thus accelerating genetic differentiation among populations. Genetic drift can also be a major factor affecting population genetic structure [92,93,94]. In addition, FST analysis of both cpDNA and nrDNA sequencing data indicated that the contribution of inter-population genetic differentiation to total genetic diversity was about 0.93445 and 0.94058, respectively (Table 2). According to Write [94], inter-population genetic differentiation is extremely high in A. macrostemon (FST > 0.25). The Mantel test showed that geographic and genetic distance were not significantly correlated between A. macrostemon groups (r = 0.0714) (Fig. S1), indicating that geographic distance was not the main factor leading to genetic differentiation of A. macrostemo. Isolation between populations is caused by physical separation, such as the complex terrain and mountains in China. In addition, the mismatched distributions generated from cpDNA and ITS data exhibited a multimodal curve. The observed and expected values also did not match, violating the population expansion model and indicating that the population of A. macrostemon had not undergone a significant expansion. Barrier analysis results indicated that multiple genetic boundaries do exist between the northern and southern groups of the QHL line, which suggests that the QHL plays a role in promoting the genetic differentiation of A. macrostemon.
The QHL is a dividing line forming the boundary between China’s subtropical and temperate monsoon climate zones, separating the warm, wet southeast from the cold, dry northwest. The relatively mild Pleistocene climate provided a relatively stable microclimate environment for a range of habitats at lower elevations [95]. Previous geological studies have shown that the Qinling orogenic belt in east Asia has experienced a long evolutionary history and has a complex composition. The Qinling orogenic belt formed in what is now Shaanxi Province during the Mesozoic and Cenozoic Eras and extends more than 1,600 km from east to west, exerting a complex influence on the genetic patterns of east Asian species [96, 97]. Our study demonstrated that intraspecific divergence in A. macrostemon began in 3.16 Ma based on previous studies on its emergence at 4.55 Ma [73]. These estimates suggest that the genetic divergence time within A. macrostemon much earlier than the rapid uplift of the Qinling Mountains (about 1.84 Ma) [98], and indicated that the genetic model and distribution of A. macrostemon have been maintained and strengthened by the formation and rapid uplift of the QHL.
We constructed phylogenetic trees based on SSR, cpDNA, nrDNA, and GBS data, and divided A. macrostemon into north and south groups. The south group was further divided into the southwest and central-southeast group. Northern and southern populations were divided by the QHL. A. macrostemon populations of similar geographic origin were relatively clustered and belonged to the same group. Conversely, populations that had different geographic origin were somewhat geographically isolated, and clustering based on SNPs was related to geographic origin. This suggests that QHL formed a barrier that has affected the geographic distribution of A. macrostemon and prevents genetic exchange between southern and northern populations, although there is significant gene flow across the QHL from north to south. A. macrostemon is widely distributed and has three reproductive modes (bud, bulb, and seed), affording it high ecological adaptability and reproductive ability. Therefore, we believe that the geographic structure of A. macrostemon is mainly influenced by the geographic barrier formed by the QHL, which hinders gene exchange between different populations. As such, we speculate that the formation of the current geographic structure of A. macrostemon may have resulted from allogeneic differentiation caused by its long-term adaptation to different geological histories, climatic conditions, and elevational differences between the Qinling-Huaihe and Wushan-Xuefeng regions. Consistent with this idea, previous work has shown that the QHL is key in shaping plant dispersal and forms an important boundary separating ecologically distinct habitats in China [99, 100]. Taken together, our results support the role of the QHL as a geographic barrier in influencing patterns of intraspecies genetic differentiation between A. macrostemon populations located in the south and north. These findings underscore the critical role of QHL in shaping the genetic landscape of A. macrostemon by isolating populations from one another and facilitating different evolutionary tracks.
The influence of Quaternary climate change history on phylogeographical patterns of A. macrostemon
Fluctuations between cold and warm climates in the Quaternary have strongly impacted the geographic distribution of plants in China [101, 102]. Several phylogeographical studies suggest that Quaternary climatic oscillations drove expansion and intraspecific divergence in most plant species in China [103], such as Allium przewalskianum [104] and Pteroceltis tatarinowii [105]. A leading hypothesis is that plants were confined to low latitudes and high elevations during periods of expansive glaciation, with interglacial periods characterized by plant distributions similar to those that exist in the present day [12, 106]. High cpDNA haplotype and nucleotide diversity are characteristic of ice age sanctuaries [107], and these stable and diverse environments may have helped maintain species richness. Both haplotype and nucleotide diversity of A. macrostemon were high in select populations north (TSS, SMX, HCS, and SHS) and south (DPS and SNX) of the QHL. Thus, these northern and southern areas were potential habitats for the species despite their great north-south geographical distance. Consistent with this idea, some research demonstrates that areas on either side have served as glacial refugia for other species [29, 30]. Niche simulations indicate that the distribution of A. macrostemon during the LGM was significantly more geographically confined compared to the current distribution, which may be related to lower global temperatures. During the LGM, areas highly suited to A. macrostemon tended to shrink to the south and north. Therefore, we speculate that populations of A. macrostemon may have been confined to inhabitable zones in high altitude or low altitude areas. This is consistent with the results of SSR, cpDNA, nrDNA, and GBS analyses, which suggest that the species may have split into separate populations inhabiting at least two glacial refugia straddling the QHL, one to the south and one to the north. In MaxEnt simulations, A. macrostemon expanded in the middle of the Holocene, migrating south or to lower altitudes during warmer climates and thus making it better suited to growth in relatively hot and humid environments. The distribution of this species is predicted to continue to shrink in the future, with the center of its distribution moving northward. A. macrostemon is widely distributed, and past and future changes in its distribution may signal future changes in populations of other species with similar ecological habits. Other studies have shown that climate change affects species distribution patterns [108, 109].
Our results show that the A. macrostemon group is partially mixed between different groups. For example, STRUCTURE SSR analysis shows that the THS population of the first group located north of the QHL is divided into the second group south of the QHL, while the XWX population of the third group located south of the QHL is divided into the first group north of QHL. Similar results were also found using cpDNA and nrDNA sequencing data, and intergroup mixing was particularly evident in the phylogenetic tree, where the same haplotype was shared between different groups and confusion in geographical distribution of haplotypes and phylogenetic trees. For example, cpDNA haplotypes H1, H5 and H6 shared in groupsIand II, the geographic distribution of haplotypes H41, H42, H3, H4, H9, H24, H18, H19, H20 and H28 does not correspond to the phylogenetic tree(Fig. 3D); nrDNA haplotypes H8 were shared in groups B and C, the geographic distribution of haplotypes H1, H2, H34, H35, H36, H37, H60, H61, H62, etc. does not correspond to the phylogenetic tree(Fig. 3H). There may be four causes for this structure. First, repeated expansion and contraction of the area occupied by A. macrostemon populations caused by glacial cycles caused overlap between the distributions of different groups, and repeated contact between refuge and expanding populations caused lineage fusion. This pattern has been confirmed in other plants, such as Paeonia decomposita, P. rotundiloba, P. rockii, Ostryopsis davidiana [6, 29]. This study shows that A. macrostemon intraspecific divergence in the Late Pliocene (3.16 Ma), earlier than the last glacial maximum, which suggests that its genetic pattern was influenced by Quaternary climate oscillation. Therefore, the complex climatic oscillations of the Quaternary and the history of repeated contractions and expansions of the distribution area of A. macrostemon likely contribute to this species’ existing geographical pattern. Second, cultivation of this plant for its medicinal and nutritional value may have influenced the genetic pattern of A. macrostemon after a long period of long-term cultivation. Examples of this type of anthropogenic effect have been documented in other species, such as Allium sativum [41], Mango cultivars [110] and Fagus hayatae [111]. Geological events, geography, and climate change may also contribute to this genetic structure. Ecological factors such as temperature, rainfall, and other climatic conditions influence interspecific and intraspecific variation [112, 113]. Third, infiltration and hybridization may offer another explanation for inconsistencies in the geographic distribution of haplotype and phylogenetic trees of A. macrostemon. The diverse reproduction modes of this plant, which include bulb, bud, and seed propagation, contributes to its wide distribution and its adaptability to different habitats and hybridization and infiltration, resulting in its geographic distribution of haplotype and phylogenetic trees [114]. Finally, incomplete lineage sorting (ILS) is a potential explanation for the geographic inconsistencies between phylogenetic trees and haplotypes. ILS is easily observable when effective population size is large [115, 116] and affects species lineage patterns. Previous studies have reached similar conclusions [114]. Therefore, the existing geographical pattern can be explained by this plant’s diverse reproduction modes, ILS, and the complex climatic oscillations of the Quaternary period that caused repeated contraction and expansion of distributions of A. macrostemon populations. Together, these explain gene flow between southern and northern populations of A. macrostemon across the QHL.
Conservation strategies for A. macrostemon
A. macrostemon has been valued since antiquity for its medicinal and nutritional uses. In recent years, however, its genetic diversity has declined because of overharvesting and habitat fragmentation, which interferes with the regeneration of wild populations and can reduce population size. Small populations that do not exchange genes with the outside world tend to experience greater genetic drift. In the long run, A. macrostemon may face the danger of extinction and we should therefore move to protect A. macrostemon. Southwest China is an important center for biodiversity and as such is the site of many conservation efforts. This study also showed that A. macrostemon had high genetic and haplotype diversity (populations JLQ, SNX, and SYS) in southwest China. We also found that some boreal populations have high genetic and haplotype diversity (populations BXS, HCS, and THS), so northern populations should also be protected. We believe that areas with high genetic and haplotype diversity can be protected in situ as conservation units, and areas with high genetic diversity can be classified as nature reserves to reduce the impacts of human disturbance. Secondly, ex situ protection can facilitate the reproduction of A. macrostemon through introduction and protection. Such conservation measures require transplantation and naturalization of protected plants to cover different geographical units that preserve the various characteristics of the plants. Plants in different regions experience different environmental conditions, and other factors affect their adaptation and variation. Therefore, in addition to customized plant protection measures, specific plant and habitat characteristics should also be considered [117]. In addition, tissue culture technology can solve the problems related to plant regeneration, breeding, and cultivation; promote rapid growth; and increase population sizes. Some germplasm resources of A. macrostemon, such as large rhizomes, high growth rate, and strong insect resistance, can be screened out with good quality. These plants can then be cultivated and propagated rapidly using tissue culture approaches. Finally, raising awareness of resource conservation among residents is also key to protecting this important species.
Conclusions
In this study, the genetic structure and geographic distribution of A. macrostemon were analyzed by combining cpDNA, ITS, SSR, and GBS data generated from 50 sampled populations. The QHL acts as a north-south dividing line, mediating phylogeography and population genetic structure and promoting physical geographic isolation, with gene exchange occurring on both sides of the QHL. In addition, Quaternary climate fluctuation also contributed to the formation of the genetic patterns observed in A. macrostemon, indicating that the formation of the geographic pattern and genetic structure of A. macrostemon is influenced by climate fluctuation and environmental heterogeneity. This work shows that the genetic structure and spatial distribution of A. macrostemon reflect the combined influence of geographic history, climate change, and biological characteristics, and emphasizes the importance of using multi-line evidence to infer the Quaternary evolutionary history of plant species widely distributed across complex terrains. It further provide a reference for the systematic geographic patterns of the large-scale spatial distribution of plants in China and enriches our understanding of the evolutionary history of east Asian plants.
Data availability
Sequeence data that support the findings of this study have been despostied in the GenBank with the accession code OM102763-OM102953, PQ144907-PQ144971.
Abbreviations
- cpDNA:
-
Chloroplast DNA
- nrDNA:
-
Ribosomal DNA
- SSR:
-
Simple Sequence Repeats
- GBS:
-
Genotyping by sequencing
- QHL:
-
Qinling Mountains-Huaihe River Line
- F st :
-
Fixation index
- He :
-
Expected heterozygosity
- Ho :
-
observed heterozygosity
- Ne :
-
effective number of alleles
- Na :
-
observed number of alleles
- I :
-
Shannon information index
- PPL:
-
percentage of polymorphic loci
- AMOVA:
-
Analysis of molecular variance
- H d :
-
Haplotype diversity
- π:
-
Nucleotide diversity
- H T :
-
Total genetic diversity
- H S :
-
Averages within-population diversity
- N st, G st :
-
Parameters of differentiation among populations
- LGM:
-
The last glacial maximum
References
Fukushima C, Mendoza JI, West RC, et al. Species conservation profiles of tarantula spiders (Araneae, Theraphosidae) listed on CITES. Biodivers Data J. 2019;7:e39342. https://doi.org/10.3897/BDJ.7.e39342. Published 2019 Nov 8.
Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci. 2004;359(1442):183–95. https://doi.org/10.1098/rstb.2003.1388.
Bennett KD, Provan J. What do we mean by ‘refugia’? Q Sci Rev. 2008;2449–55. https://doi.org/10.1016/j.quascirev.2008.08.019. 27.27-28.
Stewart JR, Lister AM, Barnes I, Dalén L. Refugia revisited: individualistic responses of species in space and time. Proc Biol Sci. 2010;277(1682):661–71. https://doi.org/10.1098/rspb.2009.1272.
Wang YH, Jiang WM, Comes HP, Hu FS, Qiu YX, Fu CX. Molecular phylogeography and ecological niche modelling of a widespread herbaceous climber, Tetrastigma hemsleyanum (Vitaceae): insights into Plio-Pleistocene range dynamics of evergreen forest in subtropical China. New Phytol. 2015;206(2):852–67. https://doi.org/10.1111/nph.13261.
Liu G, Xue G, Zhao T et al. Population structure and phylogeography of three closely related tree peonies [published correction appears in Ecol Evol. 2023;13(6):e10237]. Ecol Evol. 2023; 13(6):e10073. https://doi.org/10.1002/ece3.10073
Zhisheng A, Kutzbach JE, Prell WL, Porter SC. Evolution of Asian monsoons and phased uplift of the Himalaya-Tibetan plateau since late miocene times. Nature. 2001;411(6833):62–6. https://doi.org/10.1038/35075035.
Raymo ME, Ruddiman WF. Tectonic forcing of late cenozoic climate. Nature. 1992;359(6391):117–22. https://doi.org/10.1038/359117a0.
Spicer RA, Harris NB, Widdowson M, et al. Constant elevation of southern Tibet over the past 15 million years. Nature. 2003;421(6923):622–4. https://doi.org/10.1038/nature01356.
Ying JS. Species diversity and distribution pattern of seed plants in China. J Biol Divers. 2001;9(4):6. 10.3321 / j.issn: 1005-0094.2001.04.011.
Axelrod DI. Poleward Migration of Early Angiosperm Flora: Angiosperms only displaced the relict jurassic-type flora at high latitudes in late cretaceous time. Science. 1959;130(3369):203–7. https://doi.org/10.1126/science.130.3369.203.
Qiu YX, Fu CX, Comes HP. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogenet Evol. 2011;59(1):225–44. https://doi.org/10.1016/j.ympev.2011.01.012.
Liu J, Möller M, Provan J, Gao LM, Poudel RC, Li DZ. Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phytol. 2013;199(4):1093–108. https://doi.org/10.1111/nph.12336.
Li Y, Zhai SN, Qiu YX, Guo YP, Ge XJ, Comes HP. Glacial survival east and west of the ‘Mekong-Salween divide’ in the Himalaya-Hengduan Mountains region as revealed by AFLPs and cpDNA sequence variation in Sinopodophyllum hexandrum (Berberidaceae). Mol Phylogenet Evol. 2011;59(2):412–24. https://doi.org/10.1016/j.ympev.2011.01.009.
Tian B, Zhou Z, Du FK, et al. The Tanaka Line shaped the phylogeographic pattern of the cotton tree (Bombax ceiba) in southwest China. Biochem Syst Ecol. 2015. https://doi.org/10.1016/j.bse.2015.04.014.
Luo SJYH et al. Genetic diversity and genetic structure of different populations of the endangered species Davidia involucrata in China detected by inter-simple sequence repeat analysis. TREES-STRUCT FUNCT, 2011, 2011, 25(6) (-):1063–1071.https://doi.org/10.1007/s00468-011-0581-7
Ye JW, Bai WN, Bao L, et al. Sharp genetic discontinuity in the aridity-sensitive Lindera obtusiloba (Lauraceae): solid evidence supporting the Tertiary floral subdivision in East Asia. J Biogeogr. 2017. https://doi.org/10.1111/jbi.13020.
Kou Y, Cheng S, Tian S, et al. The antiquity of Cyclocarya paliurus (Juglandaceae) provides new insights into the evolution of relict plants in subtropical China since the late early miocene. J Biogeogr. 2016;43(2):351–60. https://doi.org/10.1111/jbi.12635.
Bai WN, Wang WT, Zhang DY. Phylogeographic breaks within Asian butternuts indicate the existence of a phytogeographic divide in East Asia. New Phytol. 2016;209(4):1757–72. https://doi.org/10.1111/nph.13711.
Li EX, Yi S, Qiu YX, Guo JT, Comes HP, Fu CX. Phylogeography of two east Asian species in Croomia (Stemonaceae) inferred from chloroplast DNA and ISSR fingerprinting variation. Mol Phylogenet Evol. 2008;49(3):702–14. https://doi.org/10.1016/j.ympev.2008.09.012.
Qiu YX, Sun Y, Zhang XP, Lee J, Fu CX, Comes HP. Molecular phylogeography of east Asian kirengeshoma (Hydrangeaceae) in relation to quaternary climate change and landbridge configurations. New Phytol. 2009a;183(2):480–95. https://doi.org/10.1111/j.1469-8137.2009.02876.x.
Qiu YX, Qi XS, Jin XF, Tao XY, Fu CX, Naiki A, et al. Population genetic structure, phylogeography, and demographic history of platycrater arguta (Hydrangeaceae) endemic to East China and South Japan, inferred from chloroplast DNA sequence variation. Taxon. 2009;1226–41. https://doi.org/10.1002/tax.584014.
Wu QL, Shen XJ, He LM, et al. Windborne migration routes of newly-emerged fall armyworm from Qinling Mountains-Huaihe River region, China. J Integr Agric. 2021;20(3):694–706. https://doi.org/10.1016/S2095-3119(20)63207-5.
Chen L. The precise biogeographic division of palaearctic and oriental realm in the East of China: based on data of amphibians. Zoological Res. 2002;25(5):369–77. https://doi.org/10.3321/j.issn:0254-5853.2004.05.001.
Tan Z. The calculation and simulation of Chinese South-North demarcation based on GIS (Master thesis). Lanzhou University. 2011.
Zhu X, Cai Y, Wang J. Spatial dimensions of mountains, faults and river basins of continental China. J Mt Sci. 2003;21(3):311–7. https://doi.org/10.3969/j.issn.1008-2786.2003.03.009.
Liu J, Yang Q, Liu J, et al. Study on the spatial differentiation of the populations on both sides of the Qinling-Huaihe line in China. Sustainability. 2020;12. https://doi.org/10.3390/su12114545.
Qiu YX, Guan BC, Fu CX, Comes HP. Did glacials and/or interglacials promote allopatric incipient speciation in east Asian temperate plants? Phylogeographic and coalescent analyses on refugial isolation and divergence in Dysosma Versipellis. Mol Phylogenet Evol. 2009;51(2):281–93. https://doi.org/10.1016/j.ympev.2009.01.016.
Tian B, Liu R, Wang L, et al. Phylogeographic analyses suggest that a deciduous species (Ostryopsis Davidiana Decne. Betulaceae) survived in northern China during the last glacial Maximum. J Biogeogr. 2010;36(11):2148–55. https://doi.org/10.1111/j.1365-2699.2009.02157.x.
Zhao C, Wang CB, Ma XG, Liang QL, He XJ. Phylogeographic analysis of a temperate-deciduous forest restricted plant (Bupleurum Longiradiatum Turcz.) Reveals two refuge areas in China with subsequent refugial isolation promoting speciation. Mol Phylogenet Evol. 2013;68(3):628–43. https://doi.org/10.1016/j.ympev.2013.04.007.
He,Quan. Huang,et al.Comparative study on the composition of free amino acids and derivatives in the two botanical origins of an edible Chinese herb Xiebai, i.e. Allium chinense G. Don and Allium Macrostemon Bunge. species.Food Research International; 2018.
Ishimaru K, Usui A, Ohshima K, Fukuda S. Phenolics concentration of Allium macrostemon bunge collected in various area of Japan. Japanese J Food Chem Saf. 2018.
Feng H, Wang Z, Wang C, et al. Effect of Furostanol Saponins from Allium Macrostemon Bunge Bulbs on platelet aggregation rate and PI3K/Akt pathway in the rat model of Coronary Heart Disease. Evid Based Complement Alternat Med. 2019;2019:9107847. https://doi.org/10.1155/2019/9107847. Published 2019 Jun 23.
Wu Z, Zhao Z, Chen T, et al. Study on the prevention and treatment of ulcerative colitis and antioxidant activity of allium macrostemon powder. Fourteenth Natl Conf Laser Technol Optoelectron. 2019. https://doi.org/10.1117/12.2533718.
Wang A, Wang Y, Ren X, Zhang L. Discovery of triploid Allium macrostemon Bunge in Laizhou City of Shandong Province. J Anhui Agricultural Sci. 2009. https://doi.org/10.3969/j.issn.0517-6611.2009.09.054.
Zhu SM, Xu JM. Karyotypic differentiation in Allium Macrostemon Bunge. J Syst Evol. 1999;37(3):269–78.
Zhao LL, Zhang ZZ, Chen LL, Liang L, Ao CQ. Discovery of heptaploid Allium macrostemon in Daluo mountain. Zhejiang J Northwest Plants. 2012. https://doi.org/10.3969/j.issn.1000-4025.2012.11.010.
Yang P, Shu J, Cai S, Zhao C. nrDNA ITS sequence analysis and genetic relationship of Allium macrostemon from different geographical regions in Guizhou. Plant Sci J. 2017;35(2):171–6. https://doi.org/10.11913/PSJ.2095-0837.2017.20171.
Yang W, Bai Z, Wang F, et al. Analysis of the genetic diversity and population structure of Monochasma Savatieri Franch. Ex Maxim using novel EST-SSR markers. BMC Genomics. 2022;23(1):597. https://doi.org/10.1186/s12864-022-08832-x. Published 2022 Aug 16.
Oyundelger K, Harpke D, Herklotz V, et al. Phylogeography of Artemisia frigida (Anthemideae, Asteraceae) based on genotyping-by-sequencing and plastid DNA data: Migration through Beringia. J Evol Biol. 2022;35(1):64–80. https://doi.org/10.1111/jeb.13960.
Li WW, Liu LQ, Zhang QP, Zhou WQ, Fan GQ, Liao K. Phylogeography of Prunus armeniaca L. revealed by chloroplast DNA and nuclear ribosomal sequences. Sci Rep. 2021;11(1):13623. https://doi.org/10.1038/s41598-021-93050-w.
Avise JC. Phylogeography: retrospect and prospect. J Biogeogr. 2009;36:3–15. https://doi.org/10.1111/j.1365-2699.2008.02032.x.
You J, Qin X, Ranjitkar S, et al. Response to climate change of montane herbaceous plants in the genus Rhodiola predicted by ecological niche modelling. Sci Rep. 2018;8(1):5879. https://doi.org/10.1038/s41598-018-24360-9. Published 2018 Apr 12.
Compagnoni A, Levin S, Childs DZ, et al. Herbaceous perennial plants with short generation time have stronger responses to climate anomalies than those with longer generation time. Nat Commun. 2021;12(1):1824. https://doi.org/10.1038/s41467-021-21977-9.
Xie XF, Yan HF, Wang FY, et al. Chloroplast DNA phylogeography of Primula Ovalifolia in central and adjacent southwestern China: past gradual expansion and geographical isolation. J Syst Evol. 2012;50:284–94. https://doi.org/10.1111/j.1759-6831.2012.00204.x.
Comes HP, Kadereit JW. The effect of quaternary climatic changes on plant distribution and evolution. Trends Plant Sci. 1988;3(11):432–8. https://doi.org/10.1016/S1360-1385(98)01327-2.
Guan BC, Fu CX, Qiu YX, Zhou SL, Comes HP. Genetic structure and breeding system of a rare understory herb, Dysosma Versipellis (Berberidaceae), from temperate deciduous forests in China. Am J Bot. 2010;97(1):111–22. https://doi.org/10.3732/ajb.0900160.
Liu ZW, Zhang SQ, Zhai D, et al. Molecular phylogeny of the Chinese weedy rice based on nrDNA ITS and two cpDNA sequences. Indian J Genet Plant Breed. 2016;76(01):57–63. https://doi.org/10.5958/0975-6906.2016.00008.0.
Doyle J. DNA protocols for plants-CTAB total DNA isolation [J]. Mol Techniques Taxonomy, 1991.
Z Li H. Study on SRAP and SSR markers and their application in cultivated varieties of Allium fistulosum L. Tai ‘an: Shandong Agricultural Univ. 2007. https://doi.org/10.7666/d.y1093935.
Tian BH. Genetic diversity of Allium germplasm resources [D]. Taiyuan: Shanxi University; 2011.
Li MT, Zhang SL, Deng P, et al. SSR analysis and polymorphism of onion transcriptome. J Gardening. 2015;42(6):1103–11. doi: 10.16420 / j.i SSN. 0513 – 353 - x. 2015-0046.
Hamilton MB. Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Mol Ecol. 1999;8:521–2. https://doi.org/10.1046/j.1365-294X.1999.00510.x.
Oxelman B, Lidén M, Berglund D. Chloroplast rps 16 intron phylogeny of the tribe Sileneae (Caryophyllaceae). Plant Syst Evol. 1997;206:393–410. https://doi.org/10.1007/BF00987959.
Taberlet P, Gielly L, Pautou G, Bouvet J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol. 1991;17:1105–9. https://doi.org/10.1007/BF00037152.
Wendel JF, Schnabel A, Seelanan T. Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc Natl Acad Sci. 1995;92(1):280–4.
Elshire RJ, Glaubitz JC, Sun Q, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6(5):e19379. https://doi.org/10.1371/journal.pone.0019379.
Dai X, Wu W, Ji L, et al. MaxEnt model-based prediction of potential distributions of Parnassiawightiana (Celastraceae) in China. Biodivers Data J. 2022;10:e81073. https://doi.org/10.3897/BDJ.10.e81073. Published 2022 Mar 16.
Shi YH, Ren ZX, Wang WJ, et al. Predicting the spatialdistribution of three Astragalus species and their pollinating bumblebees in the sino-himalayas. Biodivers Sci. 2021;29(6):759–69. https://doi.org/10.17520/biods.2020268.
Yusup S, Sulayman M. Prediction of potential distribution of Didymodon (bryophyta, pottiaceae) in xinjiang based on the maxent model. Plant Science Journal; 2018.
Smouse RPP, Peakall R. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update.BIOINFORMATICS -OXFORD-, 2012.https://doi.org/10.1093/bioinformatics/bts460
Kalinowski ST, Taper ML, Marshall TC. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol. 2007;16(5):1099–106. https://doi.org/10.1111/j.1365-294X.2007.03089.x.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–7. https://doi.org/10.1111/j.1755-0998.2010.02847.x.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–87. https://doi.org/10.3410/f.1015548.197423.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20. https://doi.org/10.1111/j.1365-294X.2005.02553.x.
Manni F, et al. Geographic patterns of (genetic, Morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol. 2004;76:173–90. https://doi.org/10.1353/hub.2004.0034.
Burland TG. DNASTAR’s lasergene sequence analysis software. Bioinf Methods Protocols, 1999, 71–91.
Kumar S, Stecher G, Tamura K. Mol Biol Evol. 2016;33(7):1870–4. https://doi.org/10.1093/molbev/msw054. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets.
Librado P, Rozas J. Bioinformatics. 2009;25(11):1451–2. https://doi.org/10.1093/bioinformatics/btp187. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data.
Pons O, Petit R. Measwring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996;144(3):1237–45. https://doi.org/10.1016/S1050-3862(96)00162-3.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91. https://doi.org/10.1093/molbev/msm088.
Yang J, Kim SH, Gil HY, Choi HJ, Kim SC. New insights into the phylogenetic relationships among wild onions (Allium, Amaryllidaceae), with special emphasis on the subgenera Anguinum and Rhizirideum, as revealed by plastomes. Front Plant Sci. 2023;14:1124277. https://doi.org/10.3389/fpls.2023.1124277.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinf (Oxford England). 2018;34(17):i884–90. https://doi.org/10.1093/bioinformatics/bty560.
Heng, Li. Richard, Durbin. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009.
McKenna A, Hanna M, Banks E, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.1101/gr.107524.110.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinf (Oxford England). 2011;27(21):2987–93. https://doi.org/10.1093/bioinformatics/btr509.
Petit RJ, Duminil J, Fineschi S, Hampe A, Salvini D, Vendramin GG. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol. 2005;14(3):689–701. https://doi.org/10.1111/j.1365-294X.2004.02410.x.
Zhang Y, Yu Q, Zhang Q, et al. Regional-scale differentiation and phylogeography of a desert plant Allium mongolicum (Liliaceae) inferred from chloroplast DNA sequence variation. Plant Syst Evol. 2017;303(4):451–66. https://doi.org/10.1007/s00606-016-1383-6.
Su Z, Pan B, Sanderson SC, et al. Conservation genetics and geographic patterns of genetic variation of the endangered officinal herb Fritillaria Pallidiflora Nordic. J Bot. 2015;33(4):506–12. https://doi.org/10.1111/njb.00677.
Sharma S, Chhabra M, Singh SK, Parmar R, Kapila RK. Genetic diversity and population structure of critically endangered Dactylorhiza hatagirea (D. Don) Soo from North-Western Himalayas and implications for conservation. Sci Rep. 2022;12(1):11699. Published 2022 Jul 9. https://doi.org/10.1038/s41598-022-15742-1
Zhang XH, Tapia M, Webb JB, Huang YH, Miao S. Molecular signatures of two cattail species, Typha domingensis and Typha latifolia (Typhaceae), in South Florida. Mol Phylogenet Evol. 2008;49(1):368–76. https://doi.org/10.1016/j.ympev.2008.03.032.
Krattinger K. Genetic mobility in Typha. Aquat Bot. 1975;1:57–70.
Tate JA, Simpson BB. Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Syst Bot. 2003;28(4):723–37. https://doi.org/10.1043/02-64.1.
Frankham R, Ballou JD, Briscoe DA. Introduction to Conservation Genetics. 2nd ed. Cambridge: Cambridge University Press; 2010.
Luan S, Chiang TY, Gong X. High genetic diversity vs. low genetic differentiation in Nouelia insignis (Asteraceae), a narrowly distributed and endemic species in China, revealed by ISSR fingerprinting. Ann Botany. 2006;98(3):583–9. https://doi.org/10.1093/aob/mcl129.
Li Q, Zhan Y, Wang EP, et al. Research progress of medicinal value and cultivation technology of Allium Macrostemon. Specialty Res. 2022;44(01):111–5.
Hamrick JL. Factors influencing levels of genetic diversity in woody plant species.New forests, 1992, 6.https://doi.org/10.1007/BF00120641
Weimin LI, Sifeng LI. Genetic diversity of natural populations of Abies chinling was analyzed using SSR molecular markers. Acta Bot Sinica. 2012;47(04):413–21. https://doi.org/10.3724/SP.J.1259.2012.00413.
Correa Ribeiro P, Lemos-Filho JP, de Oliveira Buzatti RS, et al. Species-specific phylogeographical patterns and Pleistocene east–west divergence in Annona (Annonaceae) in the Brazilian Cerrado. Bot J Linn Soc. 2016;181(1):21–36.
Fiorini CF, Peres EA, da Silva MJ, et al. Phylogeography of the specialist plant Mandirola Hirsuta (Gesneriaceae) suggests ancient habitat fragmentation due to savanna expansion. Flora. 2020;262:151522. https://doi.org/10.1016/j.flora.2019.151522.
Sun M, Wong KC. Genetic structure of three orchid species with contrasting breeding systems using RAPD and allozyme markers. Am J Bot. 2001;88(12):2180–8.
Balloux F, Lugon-Moulin N. The estimation of population differentiation with microsatellite markers. Mol Ecol. 2010;11(2). https://doi.org/10.1046/j.0962-1083.2001.01436.x.
Wright S. Evolution and the genetics of populations, volume 3: experimental results and evolutionary deductions. Volume 3. University of Chicago Press; 1977.
Bai WN, Liao WJ, Zhang DY. Nuclear and chloroplast DNA phylogeography reveal two refuge areas with asymmetrical gene flow in a temperate walnut tree from East Asia. New Phytol. 2010;188(3):892–90. https://doi.org/10.1111/j.1469-8137.2010.03407.x.
Dong Y, Shi X, Sun S et al. Co-evolution of the Cenozoic tectonics, geomorphology, environment and ecosystem in the Qinling Mountains and adjacent areas, Central China. Geosystems and Geoenvironment.2022.
Zhang F, Cawood PA, Dong Y, Wang Y. Petrogenesis and tectonic implications of early cretaceous andesitic–dacitic rocks, western Qinling (Central China): Geochronological and geochemical constraints. Geosci Front. 2018;10(4). https://doi.org/10.1016/j.gsf.2018.09.013.
Zhou B-F, et al. Linked selection, ancient polymorphism, and ecological adaptation shape the genomic landscape of divergence in Quercus dentata. J Syst Evol. 2022;60. https://doi.org/10.1111/jse.12817.
Shi BP. DaSun, YufangLiu, XiaoruiLv, XiulinCumberlidge, NeilSun, Hongying.Regional climates shape the biogeographic history of a broadly distributed freshwater crab species complex. J Biogeogr, 2021, 48(6).
Yan J, Wang Q, Chang Q, Ji X, Zhou K. The divergence of two independent lineages of an endemic Chinese gecko, Gekko swinhonis, launched by the Qinling orogenic belt. Mol Ecol. 2010;19(12):2490–500. https://doi.org/10.1111/j.1365-294X.2010.04660.x.
Wan DS, Feng JJ, Jiang DC, et al. The quaternary evolutionary history, potential distribution dynamics, and conservation implications for a Qinghai-Tibet Plateau endemic herbaceous perennial, Anisodus tanguticus (Solanaceae). Ecol Evol. 2016;6(7):1977–95. https://doi.org/10.1002/ece3.2019.
Zhao YP, Fan G, Yin PP, et al. Resequencing 545 ginkgo genomes across the world reveals the evolutionary history of the living fossil. Nat Commun. 2019;10(1):4201. https://doi.org/10.1038/s41467-019-12133-5.
Liu ZG, Shu QY, Wang L, Yu MF, Hu YP, Zhang HG, et al. Genetic diversity of the endangered and medically important Lycium Ruthenicum Murr. Revealed by sequence-related amplified polymorphism (SRAP) markers. Biochem Syst Ecol. 2012a;45:86–97. https://doi.org/10.1016/j.bse.2012.07.017.
Wu LL, Cui XK, Milne RI, Sun YS, Liu JQ. Multiple autopolyploidizations and range expansion of Allium przewalskianum regel. (Alliaceae) in the Qinghai-Tibetan plateau. Mol Ecol. 2010;19:1691–704. https://doi.org/10.1111/j.1365-294X.2010.04613.x.
Li XH, Shao JW, Lu C, Zhang XP, Qiu YX. Chloroplast phylogeography of a temperate tree Pteroceltis tatarinowii (Ulmaceae) in China. J Syst Evol. 2012;50:325–33. https://doi.org/10.1111/j.1759-6831.2012.00203.x.
Muellner-Riehl AN. Mountains as evolutionary arenas: patterns, emerging approaches, paradigm shifts, and their implications for Plant Phylogeographic Research in the Tibeto-Himalayan Region. Front Plant Sci. 2019;10:195. https://doi.org/10.3389/fpls.2019.00195.
Gong W, Chen C, Dobes C, Fu CX, Koch MA. Phylogeography of a living fossil: pleistocene glaciations forced Ginkgo biloba L. (Ginkgoaceae) into two refuge areas in China with limited subsequent postglacial expansion. Mol Phylogenet Evol. 2008;48(3):1094–105. https://doi.org/10.1016/j.ympev.2008.05.003.
Chen F, Song YF, Li XJ, Chen JH, Mo L, Zhang XT. Genome sequences of horticultural plants: past, present, and future. Hortic Res. 2019;6(1):23. https://doi.org/10.1038/s41438-019-0195-6.
Wang MN, Duan L, Qiao Q, et al. Phylogeography and conservation genetics of the rare and relict Bretschneidera sinensis (Akaniaceae). PLoS ONE. 2018;13(1):e0189034. https://doi.org/10.1371/journal.pone.0189034.
Hussein AA, Eid M, Rahimi M, Filimban M, Abd El-Moneim FZ. Comparative Assessment of SSR and RAPD markers for genetic diversity in some mango cultivars. PeerJ. 2023;11:e15722. https://doi.org/10.7717/peerj.15722.
Ying LX, Zhang TT, Chiu CA, et al. The phylogeography of Fagus hayatae (Fagaceae): genetic isolation among populations. Ecol Evol. 2016;6(9):2805–16. https://doi.org/10.1002/ece3.2042.
Nybom H, Carlson-Nilsson, et al. Different levels of morphometric variation in three heterogamous dogrose species (Rosa sect. Caninae. Rosaceae).Plant Systematics & Evolution; 1997.
Chen C, Lu RS, Zhu SS, Tamaki I, Qiu YX. Population structure and historical demography of Dipteronia dyeriana (Sapindaceae), an extremely narrow palaeoendemic plant from China: implications for conservation in a biodiversity hot spot. Heredity (Edinb). 2017;119(2):95–106. https://doi.org/10.1038/hdy.2017.19.
Tan X, Qi J, Liu Z, et al. Phylogenomics reveals high levels of Incomplete Lineage sorting at the ancestral nodes of the Macaque Radiation. Mol Biol Evol. 2023;40(11):msad229. https://doi.org/10.1093/molbev/msad229.
Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 2006;55(1):21–30. https://doi.org/10.1080/10635150500354928.
Avise JC, Robinson TJ. Hemiplasy: a new term in the lexicon of phylogenetics. Syst Biol. 2008;57(3):503–7. https://doi.org/10.1080/10635150802164587.
Liu M, Hu X, Wang X, Zhang J, Peng X, Hu Z, Liu Y. (2020). Constructing a Core Collection of the Medicinal Plant Angelica biserrata Using Genetic and Metabolic Data. Frontiers in Plant Science, 11, 600249. Liu M, Hu X, Wang X,. Constructing a Core Collection of the Medicinal Plant Angelica biserrata Using Genetic and Metabolic Data. Front Plant Sci. 2020;11:600249. https://doi.org/10.3389/fpls.2020.600249
Acknowledgements
This work was supported by the projects of Guizhou Science and Technology Support Plan Project (Guizhou Science and Technology Cooperation Support ([2019] 2451-2); National Natural Science Foundation of China (NSFC, No. 32260252); Construction Program of Biology First-class Discipline in Guizhou (GNYL [2017] 009); Guizhou Province Chinese Herbal Medicine Modern Industrial Technology System Construction Project (GZYCCYJSTX); Key Technologies of Mountainous Agriculture in Guizhou Province (GZNYGJHX-2023011). Thanks very much for those above founders.
Funding
Guizhou Science and Technology Support Plan Project (Guizhou Science and Technology Cooperation Support ([2019] 2451-2); National Natural Science Foundation of China (NSFC, No. 32260252); Construction Program of Biology First-class Discipline in Guizhou (GNYL [2017] 009); Guizhou Province Chinese Herbal Medicine Modern Industrial Technology System Construction Project (GZYCCYJSTX); Key Technologies of Mountainous Agriculture in Guizhou Province (GZNYGJHX-2023011).
Author information
Authors and Affiliations
Contributions
Shi came up with the original concept. Shi, Mo, Jiang and Zhao designed the study, selected species and conducted field investigations. Shi and Jiang did the lab work, analyzed the genetic data and wrote the manuscript. Zhao supervised the laboratory work and genetic analysis. All authors contributed to the final draft.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Voucher sample
Allium macrostemon specimens were preserved in the Department of Botany, Guizhou University, deposition number is zhaocX1-ZhaocX50 (Table S10).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, C., Shi, T., Mo, Z. et al. Across a phylogeographic break in the Qinling Mountains-Huaihe River Line: Quaternary evolutionary history of a medicinal and edible homologous plant (Allium macrostemon) in China. BMC Ecol Evo 24, 107 (2024). https://doi.org/10.1186/s12862-024-02297-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-024-02297-0