
Abstract
Background
Due to the environmental heterogeneity along elevation gradients, alpine ecosystems are ideal study objects for investigating how ecological variables shape the genetic patterns of natural species. The highest region in the world, the Qinghai-Tibetan Plateau, is a hotspot for the studies of evolutionary processes in plants. Many large rivers spring from the plateau, providing abundant habitats for aquatic and amphibious organisms. In the present study, we examined the genetic diversity of 13 Ranunculus subrigidus populations distributed throughout the plateau in order to elucidate the relative contribution of geographic distance and environmental dissimilarity to the spatial genetic pattern.
Results
A relatively low level of genetic diversity within populations was found. No spatial genetic structure was suggested by the analyses of molecular variance, Bayesian clustering analysis and Mantel tests. Partial Mantel tests and multiple matrix regression analysis showed a significant influence of the environment on the genetic divergence of the species. Both climatic and water quality variables contribute to the habitat heterogeneity of R. subrigidus populations.
Conclusions
Our results suggest that historical processes involving long-distance dispersal and local adaptation may account for the genetic patterns of R. subrigidus and current environmental factors play an important role in the genetic differentiation and local adaptation of aquatic plants in alpine landscapes.
Similar content being viewed by others
Background
By limiting gene flow within species, both spatial distance and environmental heterogeneity influence evolutionary as well as ecological processes [1, 2]. Gene flow among populations may be reduced due to the geographic isolation through the combined force of dispersal limitation and genetic drift [3]. A linear relationship between genetic differentiation and geographic distance (isolation-by-distance, IBD) indicates a general tendency for speciation or intraspecific differentiation [3]. The pattern will be disrupted when migration, colonization or mating are under the influence of ecological factors [1, 4, 5]. Recently, genetic isolation-by-environment (IBE) was discovered as another important driver of adaptive divergence processes, related to both small-scale heterogeneity and short-term changes of the environment [6, 7].
In alpine ecosystem, physical barriers and a complex topography can significantly reduce the dispersal of organisms [8, 9]. On the other hand, elevation gradients in alpine regions lead to increased habitat heterogeneity as to photoperiod, radiation, soil characteristics or biotic factors in addition to the climatic gradient [10]. Thus, plant communities in mountains provide an important context for studying mechanisms of genetic structuring related to patterns of genetic isolation among populations at different scales. Manel et al. [11] have shown that alpine environments may facilitate rapid adaptation in plants. Several subsequent studies supported these findings [12, 13]. However, more studies on representative taxa are necessary for a comprehensive understanding of the issue [2, 14, 15]. The highest region in the world, the Qinghai-Tibetan Plateau (QTP), is highly vulnerable to global climate change [16, 17]. It is characterized by wide variations in atmospheric pressure, solar radiation, topographies, wind regimes and mesoclimates [18, 19]. The plateau is considered to be an ideal ‘natural laboratory’ for investigations on adaptive evolution [20,21,22].
The QTP gives rise to many large rivers of eastern and southern Asia. Therefore, river channels, lakes and other wetlands are found in great abundance [23]. The QTP provides a great wealth of habitats for vascular aquatic plants [24], an important functional group in freshwater [25, 26]. Aquatic plants show a limited taxonomic differentiation and low intraspecific genetic variation, due to the broad tolerance ranges and plasticity of genotypes, as well as preferential clonal reproduction [27]. The way aquatic plants react to the heterogeneous and harsh environments at multiple scales is poorly understood [28, 29]. Phylogeographic studies suggest an early natural establishment of aquatic plants on the QTP in mid-Pleistocene [30, 31]. The complexity of environments may have promoted specialization and divergence of aquatic plants [32]. To date, landscape genetic studies on aquatic plants of the QTP are rare. A previous study showed that geographic isolation could be a main factor influencing the spatial genetic structure of Myriophyllum spicatum L. on southeastern edge of the QTP [33].
Ranunculus section Batrachium (Ranunculaceae) comprises about 30 species with aquatic or semiaquatic habitat requirements [34, 35]. Ranunculus sect. Batrachium species show worldwide distributions from arctic to meridional zones. They are characterized by extreme phenotypic plasticity, diverse breeding systems, polyploidization, and network evolution by means of frequent hybridization [34, 36,37,38]. Ranunculus sect. Batrachium species are frequently found across the QTP [24]. We regard them as representative taxa to understand how the environment is influencing genetic variation of aquatic plants at a large geographic scale. Ranunculus sect. Batrachium on the QTP comprises three distinct species (Ranunculus subrigidus W. B. Drew, Ranunculus flavidus (Hand.-Mazz.) C.D.K. Cook and Ranunculus trichophyllus Chaix) based on our field identifications and herbarium studies. The species differ in flower characteristics, life cycle and genetics [35, 39]. In present study, our aim was estimating the extent and spatial pattern of genetic variation of R. subrigidus. The species has an Amphipacific distribution [35]. R. subrigidus tends to colonize hard water habitats, sometimes even brackish waters [35], which makes the species one of most common aquatic species on the QTP. Absence of IBD pattern has been revealed in R. sect. Batrachium on the QTP, whereas the effects of environment were not concerned [30, 40]. Severely diverse habitats might promote the divergence in aquatic plants [41,42,43]. We therefore conducted further landscape genetic analysis to evaluate the hypothesis that the environmental variables on the QTP might influence the population genetic structure of R. subrigidus.
Results
A total of 164 genets were identified in the 13 populations of R. subrigidus. The number of multilocus genotypes per population ranged from 5 to 20, all ramets of MQ, NM and CM were from different genets (Table 1). The number of alleles per locus (NA), the expected heterozygosity (HE) and the observed heterozygosity (HO) ranged from 1.294 to 2.471, from 0.035 to 0.122 and from 0.078 to 0.24, respectively (Table 1).
An analysis of molecular variance (AMOVA) suggested only 4% of genetic variation was partitioned between subregions, and about half of the genetic variation was distributed among populations (Table 2). Two genetic clusters were suggested, inferred from STRUCTURE analysis (Fig. 2a and b). Cluster A consists of GE1, GE2, GJ, ZB, NM (the southern and western QTP, SWQTP) and DT (the northeast QTP, NEQTP), when Cluster B consists of SG, DR, CM (SWQTP) and QM, DL (NEQTP) (Fig. 2). Genetic admixture was found in a lake population MD and an adjacent downstream population MQ (Figs. 1 and 2c).

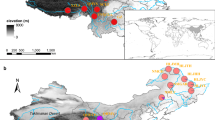
The sampling sites of Ranunculus subrigidus on the Qinghai-Tibetan Plateau are mapped with relative code using ArcGIS. The elevation range, main rivers, lakes and mountains are visualized, and the sources are supported by National Earth System Science Data Center, National Science & Technology Infrastructure of China (http://www.geodata.cn)

Modelling of the number of genetic clusters in Ranunculus subrigidus based on the Estimated Ln Prob. (a) and ΔK (b) in STRUCTURE. (c) The bar plot depicts the genetic assignment of Ranunculus subrigidus when K = 2, a single vertical bar displays the membership coefficient of each genet. The codes of sample site are labeled
The pairwise genetic distance among populations was calculated on the individual and population level respectively. The results showed that the matrices of genetic distance estimated with different approaches were significantly correlated (r = 0.644, p < 0.001). The DT and DL, DT and SG, DL and CM were the most divergent populations suggested by both approaches (Additional file 1: Tables S1 & S2).
Multiple MIGRATE runs show consistent result for direct estimation of gene flow. Asymmetric migration rates (M) varied from 0.678 (DL - > NM) to 11.357 (QM - > SG) (Additional file 1: Table S3), suggesting limited migration between populations. Small effective population sizes (θ < 50) were also indicated by MIGRATE (Table 1). The number of migrants per generation (Nm) was calculated using the Nm = (M * θ)/4, the migrants between R. subrigidus populations on the QTP ranged from 0.623 to 25.506 per generation (Additional file 1: Table S3). Immigration to the lake population MD and high-elevation population QM was higher than to others (Additional file 1: Table S3). For the populations at the northeast edge of the QTP (DT and DL), gene flow to other populations was relatively low (Additional file 1: Table S3).
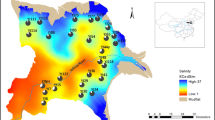
The environmental space of 13 populations was quantified using principle component analysis (PCA) on environmental variables, including elevation, climate and water quality of the sample sites (Table 3, Fig. 3, Additional file 1: Table S4). The first two axes explain 62.5% of environmental variation. The first axis of the environmental space is closely associated with temperature seasonality, precipitation and water quality, and the second axis is closely associated with elevation and temperature (Table 3). The populations assigned to different genetic clusters occupied distinct environment space (Fig. 3). The environment dissimilarity among populations were calculated with Euclidean distance based on the coordinates of first two PC axes (the results of subsequent analyses were consistent when first three PC axes were used, not showed). We found no significant relationship between the genetic distance/migration rate and geographic distance within all genetic similarity matrices (Table 4). The relationship between genetic distance/gene flow rate and environmental dissimilarity was significant (population-based: r = 0.409, p = 0.023; individual-based: r = 0.496, p = 0.008; migration rate: r = 0.553, p = 0.001, Table 4). The relationship was maintained when we set geographic distance as a control (population-based: r = 0.351, p = 0.027; individual-based: r = 0.471, p = 0.011; migration rate: r = 0.542, p = 0.001, Table 4). The Multiple matrix regression with randomization (MMRR) analysis also showed a high relationship between ecology and genetic differentiation (population-based: β = 0.356, p = 0.031; individual-based: β = 0.489, p = 0.019; migration rate: β = 0.568, p = 0.001, Table 5), whereas the effects of geographic isolation were not significant (population-based: β = 0.167, p = 0.173; individual-based: β = 0.023, p = 0.795; migration rate: β = 0.046, p = 0.616, Table 5). Distance-based redundancy analysis (dbRDA) revealed the first two principal component variables could explain one-third of the variations in genetic differentiation, and 58.7% of the variations in migration rates between populations (Additional file 1: Tables S5). Significant effect of PC1 on the genetic pattern was suggested by regressions on different genetic similarity matrices (Additional file 1: Tables S5).

The environmental space of the 13 Ranunculus subrigidus populations quantified with PCA axes. Coordinates of the first two axes are presented. The genetic cluster of each population identified by STRUCTURE are labeled
Discussion
In the present study, low genetic variation was revealed in R. subrigidus on the QTP with polymorphic microsatellite markers (Table 1). Most ramets present distinct multilocus genotypes, implying the clonal reproduction was not predominant in the species (Table 1). The value of Ho (0.139, in average) and FST (0.508, globally) in R. subrigidus resembled a species with a selfing breeding system (HO = 0.05, FST = 0.42, [44]). The mean HE (0.095) is much lower than that of a species with a regional distribution [44]. A lack of genetic variation has been revealed in population genetics studies on R. sect. Batrachium by ISSR and cpDNA markers [45, 46]. It has also been found for Russian populations of R. subrigidus [47]. Self-compatibility and vegetative reproduction are common in R. sect. Batrachium species, which may influence the level of genetic variation in different habitats [48,49,50]. The small population and effective population size in R. subrigidus caused by severe isolation and aquatic habitat fragmentation on the QTP make the plants more vulnerable to inbreeding, genetic drift/founder effects and ecological selection (Table 1) [24, 51], all of which mainly influence the level of genetic variation in R. subrigidus.
Absence of spatial genetic structure in R. subrigidus on the QTP is indicated by the following results. The genetic clusters identified in the present study are not corresponding to the two distant subregions, as suggested by STURCTURE (Fig. 2). The hierarchical analyses showed a significant genetic variation component among and within populations, but no genetic differentiation between the subregions (Table 2). No significant linear relationship between genetic differentiation and geographic distance was found (Table 4). IBD is regarded as the general pattern of neutral genetic differentiation in plants [1,2,3], especially in alpine landscapes due to the greater impact of historical processes such as glaciation and post-glaciation colonization [52]. Most available studies on aquatic plants also showed that geographic isolation played an essential role on their evolutionary processes [27, 53]. Nevertheless, the current genetic structure of alpine plants could be shaped by repeated range changes. It more likely occurs in some cold tolerant taxa, which might have survived the glaciation in situ on the plateau platform [20, 54]. According to previous studies on phylogeography, R. subrigidus might have experienced expansions and retreats in mid-to-late Pleistocene [30], undergoing a similar evolutionary history as the aquatic herb Hippuris vulgaris L. [31]. On the other hand, compared to the terrestrial plants on the QTP, the dispersal of aquatic plants was facilitated by rivers or streams, while dispersal by waterfowl may have allowed crossing watersheds and physical barriers such as high mountains [55,56,57]. The dispersal of propagules in Ranunculus species through duck guts has been proved [56, 58]. The potential of long distance dispersal in R. subrigidus contributes to the genetic similarity between distant populations. Previous studies suggested the important role of passive dispersal on the genetic structuring in aquatic plants in different scales [59,60,61]. MD (Zhaling-Eling Lake) is one of largest lakes for the breeding of migration waterfowls on the QTP [62]. We therefore consider the random movements of R. subrigidus via transport by waterfowl or humans might be a viable explanation for the current distribution (Fig. 2). The assumption is supported by the relatively high gene flow rates between MD and some distant populations (e.g. GE1, GJ) (Additional file 1: Table S3). More empirical studies are necessary on how waterfowl or human activities contribute to the dispersal and divergence of aquatic plant populations [63].
IBE may be another important cause for the absence of IBD, when the influence of the contemporary environmental conditions is strong [2, 7]. In R. subrigidus, IBE was confirmed by both partial Mantel test and multiple regression under the effect of geography as a covariate (Tables 4 and 5). We also found that the two genetic clusters had different niche requirements (Fig. 3). It implies that the effective dispersal among R. subrigidus populations on the QTP was significantly influenced by environmental factors. For instance, near-by populations in significantly different environments (e.g. DT and DL, Figs. 1 and 3) were highly divergent despite relatively low rates of movement (Additional file 1: Tables S1, S2 and S3). Gene flow among adjacent populations also benefited from environmental similarity (e.g. SG/DR vs ZB/NM, Fig. 1, Additional file 1: Tables S1, S2 and S3). Hostile environments on the QTP, especially short growing seasons and low temperature, negatively influence both photosynthetic rates and reproduction of plants [18, 64]. It would limit the gene flow when natural selection operates on the immigrates or their offspring with local individuals [4, 7, 65]. Climate was considered as one of the best predictors for the differentiation among alpine populations [22, 66]. It was identified as the primary driver for the IBE pattern in landscape genetic studies [67, 68]. Temperature, precipitation and water quality mainly contributed to environmental heterogeneity of R. subrigidus (Table 3), suggesting that they might induce the differentiation and potential adaptation of R. subrigidus populations. The results of multivariate regression also support these variables significantly influence the genetic pattern of R. subrigidus (Additional file 1: Table S5). Temperature seasonality greatly varied among the sites of R. subrigidus on the QTP, and significantly differed between the two genetic clusters (Additional file 1: Table S4). The factor is generally known to shaping life history strategies [69]. It may elicit adaptive traits on the growth and reproduction of plants [69], which could promote the divergence of the populations from different seasonal environments. The precipitation regime might decide the geographic pattern and adaptive strategies of aquatic plants as well [70]. On the QTP, precipitation is significantly reduced with the increasing altitude. Insufficient precipitation might promote the plants to be tolerant to droughts and competitive at low water levels [71]. It also affects habitat quality such as the mineralization (water salinity, pH) of isolated lakes [71, 72]. The habitat specificity could shape the spatial pattern of species richness and community [43] and enhance intraspecies genetic differentiation of aquatic macrophytes [73, 74], due to the combined effect of water qualities. These variables might simultaneously drive the local adaptation of R. subrigidus in alpine environments. Although it was generally expected that local adaptation served as a week barrier for gene flows in aquatic macrophytes due to the insignificance of sexual recombination [41], adaptations to broad climatic gradients or extreme water environments has been proved in some widespread species [75, 76]. We therefore assume that large-scaled environmental heterogeneities of climatic and habitat features might drive the local adaptation, simultaneously maintaining the genetic divergence of R. subrigidus on the QTP.
Conclusions
R. subrigidus is a representative taxon to study the influence of alpine environments on the genetic structuring of wetland plants. Similar as in many other species of R. sect. Batrachium, a lack of genetic variation was revealed in R. subrigidus. Severe isolation and habitat fragmentation on the QTP make the wetland plants more vulnerable to genetic drift, inbreeding and selection caused by heterogeneous and harsh environments. An IBE pattern, but no IBD pattern was found in R. subrigidus, implying a significant influence of the environment (especially climatic seasonality) on the genetic divergence within the species. Long-distance dispersals and historical processes may also contribute to the patterns. The results would further our understanding about how aquatic plants react and adapt to the heterogeneous and harsh environments.
Methods
Field sampling
Field sampling was carried out in the August of 2013 and 2016, respectively. All samples were collected from small water bodies in wetlands and from lakeshores. Idenfication of specimens in the field followed [35]. Water quality (salinity and pH) were measured three times at different places of the sites for an average before sampling the plant material (Additional file 1: Table S4), using a handheld multiparameter meter (PROPLUS, YSI, USA). We sampled a total of 228 individuals at thirteen sites throughout the QTP. Size ranged from 13 to 22 individuals per site (Table 1). As R. subrigidus can extend clonally via rhizomes, a genet (genetic individual) may consist of individual shoots (ramets) covering several m2. All samples were therefore collected at an interval of at least 20 m. Fresh leaves were dried in silica gel in the field and frozen at − 20 °C in the laboratory.
Clone identification and genetic diversity
Genomic DNA was extracted using the Novel DNA Plant Kit (Kangwei Biotech, Beijing, China). Thirteen self-developed EST-SSRs and four feasible SSRs isolated in Ranunculus species were used in R. subrigidus [39]. All PCR amplifications were performed in 20 μL reaction mixtures containing 1.5 μL genomic DNA (~ 30 ng/μL), 0.5 μL of each primer (10 μM), and 10 μL 1x master PCR Mix (Tiangen Biotech, Beijing, China). The PCR conditions comprised an 5 min initial denaturation step at 95 °C; followed by 35 cycles of 30 s at 95 °C, 30 s at suggested annealing temperature according to Wu et al. [39], 1 min at 72 °C; and a final extension at 72 °C for 7 min. PCR products were analyzed on the ABI 3730XL analyzer (Applied Biosystems, Foster City, CA, USA). The microsatellite genotyping was performed using the GeneMarker V.1.5 software (SoftGenetics, State College, Pennsylvania, PA, USA).
As the sampling distance of 20 m does not guarantee that the samples do not belong to the same genet (e.g. in case of short-distance propagation by fragments), we carried out an analysis of clone identification using RClone [77] in R 3.3.3 (R Development Core Team 2017). The probability-based method can identify identical multilocus genotypes (MLGs) having arisen via sexual reproduction (psex) [78]. Duplicate MLGs were removed for the following analyses. Number of alleles and observed/expected heterozygosity were estimated to evaluate the genetic diversity of each population using GenALEx 6.501 [79].
Genetic structure and gene flow
Ranunculus subrigidus populations on the QTP were divided into two geographic subregions: the southern and western QTP (SWQTP, comprising both Shiquan River and Brahmaputra River basins) and the northeast QTP (NEQTP, comprising the upper reaches of Yangtze River and Yellow River basins). The two subregions are separated by the great mountains in the central QTP such as Tanggula Mountains and Nyenchen Tanglha Mountains (Fig. 1). We calculated the distribution of genetic variation between subregions, and among and within populations based on an AMOVA using Arlequin ver. 3.5 [80]. A Bayesian clustering method approach was also used for genetic cluster assignment for individuals, implemented in STRUCTURE 2.3.4 [81, 82]. Twenty independent runs were performed for each K value (K = 1 to 10) with a burn-in period of 200,000 iterations and 1,000,000 MCMC iterations under the admixture model. The best-fit number of clusters was determined using STRUCTUREHARVEST [83]. Visualization of the Bayesian clustering was conducted in DISTRUCT 1.3 [84].
The genetic distance was measured based on the individual and population level respectively. Genetic relatedness between individuals was presented by Rousset’s â [85], using SPAGeDi 1.3 [86]. The index was suggested to be one of the most accurate individual-based genetic distance metrics for landscape genetic analysis [87]. Considering the individuals from the same site can not represent the populations independently, we randomly picked one genet per sampling site to calculate pairwise relatedness and repeated the calculations 100 times. The averaged value of genetic distance for each pair of localities was subsequently obtained. The pairwise inter-population FST was also calculated using Genelax 6.501 [88]. Given the accuracy of genetic estimation could be influenced by the small sample size (the number of genets per site ≥5, see results), we gave 100 replicates of sub-population random sampling of five individuals from all sites and calculated the respective FST estimates [68]. We found genetic distance matrix across different replicates were highly correlated (Mantel r = 0.841 ± 0.065, all P values < 0.001), which indicated a genet number of no less than five can provide reliable estimation of pairwise FST values. The matrices of genetic distance measured with individual-based and population-based approach were used for the subsequent analysis respectively.
We used the program MIGRATE 3.7.2 [89] to calculate historical mutation-scaled asymmetric immigration rates (M) for direct measurement of gene flow. The effective population size (θ) was also generated in the program. The data were analyzed with Bayesian inference strategy and followed a Brownian motion model. The starting values of both θ and M were generated using FST calculation method. We specified uniform priors for θ with a minimum of 0, maximum of 200, and a delta of 20, and for M with a minimum of 0, maximum of 100, and a delta of 10. We set static heating with four chains (temperatures: 1, 1.5, 3.0, 100,000.0), a sampling increment of 100, 50,000 recorded steps, and a burn-in of 50,000.
The pattern of isolation by distance was tested by the Mantel tests with 10,000 permutations based on pairwise genetic similarity index and geographic distance among all populations, using the VEGAN package in R [90].
Landscape genetics
Growing season temperature, extreme low temperate, temperature seasonality and annual precipitation were introduced as climate variables (Table 3, Additional file 1: Table S4), which were derived from data collected at meteorological stations across China based on the geographic coordinates [91]. The environmental space of all sample sites was quantified and visualized using PCA, with standardized variables of the climate and water quality (pH and salinity) in R software. The environmental space was constructed by the first two PCA axes (Table 3). The principal component variables of the studied sites were used to calculate environmental (Euclidean) distances between populations.
All index of genetic distance and gene flow were suitable for the landscape genetic studies [67]. Because MIGRATE produced asymmetrical pairwise migration rates, we made averages on immigrates and emigrates to obtain single values between populations for subsequent analyses [92]. A combination of partial Mantel test and matrix regression analysis was employed to evaluate the independent contribution of environmental heterogeneity and geographic isolation to the genetic variation of R. subrigidus on the QTP. Partial Mantel tests with 10,000 permutations were performed between genetic distance and geographic/environmental distance under the influence of the other factor, using R package VEGAN. MMRR was implemented with 10,000 permutations in R with the MMRR function script [93]. We also used distance-based redundancy analysis (dbRDA) to determine the contributions of environmental principal component variables in driving the genetic patterns. The dbRDA performed a multivariate regression on the response variable (genetic distance/gene flow) using the VEGAN package.
Availability of data and materials
The data set supporting the results of this article is available in the Dryad repository, in https://doi.org/10.5061/dryad.j6q573n8v.
Abbreviations
- AMOVA:
-
An analysis of molecular variance
- dbRDA:
-
distance-based redundancy analysis
- HE:
-
Expected heterozygosity
- HO:
-
Observed heterozygosity
- IBD:
-
Isolation-by-distance
- IBE:
-
Isolation-by-environment
- MLGs:
-
Multilocus genotypes
- MMRR:
-
Multiple matrix regression with randomization
- NA:
-
Number of alleles per locus
- NEQTP:
-
Northeast Qinghai-Tibetan Plateau
- PCA:
-
Principle component analysis
- QTP:
-
Qinghai-Tibetan Plateau
- SWQTP:
-
Southern and western Qinghai-Tibetan Plateau
References
Orsini L, Vanoverbeke J, Swillen I, Mergeay J, De Meester L. Drivers of population genetic differentiation in the wild: isolation by dispersal limitation, isolation by adaptation and isolation by colonization. Mol Ecol. 2013;22:5983–99.
Sexton JP, Hangartner SB, Hoffmann AA. Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution. 2014;68:1–15.
Slatkin M. Isolation by distance in equilibrium and non-equilibrium populations. Evolution. 1993;47:264–79.
Lee C-R, Mitchell-Olds T. Quantifying effects of environmental and geographical factors on patterns of genetic differentiation. Mol Ecol. 2011;20:4631–42.
Shafer ABA, Wolf JBW. Widespread evidence for incipient ecological speciation: a meta-analysis of isolation-by-ecology. Ecol Lett. 2013;16:940–50.
Wang IJ, Summers K. Genetic structure is correlated with phenotypic divergence rather than geographic isolation in the highly polymorphic strawberry poison-dart frog. Mol Ecol. 2010;19:447–58.
Wang IJ, Bradburd GS. Isolation by environment. Mol Ecol. 2014;23:5649–62.
Manel S, Schwartz MK, Luikart G, Taberlet P. Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol. 2003;18:189–97.
Murphy MA, Dezzani R, Pilliod DS, Storfer A. Landscape genetics of high mountain frog metapopulations. Mol Ecol. 2010;19:3634–49.
Normand S, Treier UA, Randin C, Vittoz P, Guisan A, Svenning J-C. Importance of abiotic stress as a range-limit determinant for European plants: insights from species responses to climatic gradients. Glob Ecol Biogeogr. 2009;18:437–49.
Manel S, Gugerli F, Thuiller W, Alvarez N, Legendre P, Holderegger R, et al. Broad-scale adaptive genetic variation in alpine plants is driven by temperature and precipitation. Mol Ecol. 2012;21:3729–38.
Frei ER, Ghazoul J, Matter P, Heggli M, Pluess AR. Plant population differentiation and climate change: responses of grassland species along an elevational gradient. Glob Chang Biol. 2014;20:441–55.
Hämälä T, Mattila TM, Savolainen O. Local adaptation and ecological differentiation under selection, migration, and drift in Arabidopsis lyrata. Evolution. 2018;72:1373–86.
DiLeo MF, Wagner HH. A landscape ecologist’s agenda for landscape genetics. Curr Landscape Ecol Rep. 2016;1:115–26.
Morente-López J, García C, Lara-Romero C, García-Fernández A, Draper D, Iriondo JM. Geography and environment shape landscape genetics of Mediterranean alpine species Silene ciliata Poiret. (Caryophyllaceae). Front. Plant Sci. 2018;9:1698.
Liu X, Chen B. Climatic warming in the Tibetan plateau during recent decades. Int J Climatol. 2000;20:1729–42.
Zhao L, Ping C-L, Yang D, Cheng G, Ding Y, Liu S. Changes of climate and seasonally frozen ground over the past 30 years in Qinghai–Xizang (Tibetan) plateau, China. Glob Planet Chang. 2004;43:19–31.
Körner C. Alpine plant life: functional plant ecology of high mountain ecosystems. 2nd ed. Berlin: Springer; 2003.
Shen M, Zhang G, Cong N, Wang S, Kong W, Piao S. Increasing altitudinal gradient of spring vegetation phenology during the last decade on the Qinghai–Tibetan Plateau. Agr forest Meteorol. 2014;189–190:71–80.
Qiu Y-X, Fu C-X, Comes HP. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogenet Evol. 2011;59:225–44.
Wen J, Zhang JQ, Nie ZL, Zhong Y, Sun H. Evolutionary diversifications of plants on the Qinghai-Tibetan plateau. Front Genet. 2014;5:1–16.
Woolbright SA, Whitham TG, Gehring CA, Allan GJ, Bailey JK. Climate relicts and their associated communities as natural ecology and evolution laboratories. Trends Ecol Evol. 2014;29:406–16.
Wang SM, Dou HS. Lakes in China. Beijing: Science Press; 1998.
Wang D. The geography of aquatic vascular plants of Qinghai-Xizang (Tibet) plateau. Ph.D. dissertation. Wuhan: Wuhan University; 2003.
Carpenter SR, Lodge DM. Effects of submersed macrophytes on ecosystem processes. Aquat Bot. 1986;26:341–70.
Engelhardt KAM, Ritchie ME. Effects of macrophyte species richness on wetland ecosystem functioning and services. Nature. 2001;411:687–9.
Santamaría L. Why are most aquatic plants widely distributed? Dispersal, clonal growth and small-scale heterogeneity in a stressful environment. Acta Oecol. 2002;23:137–54.
Grace JB. The adaptive significance of clonal reproduction in angiosperms: an aquatic perspective. Aquat Bot. 1993;44:159–80.
Kliber A, Eckert CG. Interaction between founder effect and selection during biological invasion in an aquatic plant. Evolution. 2005;59:1900–13.
Chen J-M, Du Z-Y, Yuan Y-Y, Wang Q-F. Phylogeography of an alpine aquatic herb Ranunculus bungei (Ranunculaceae) on the Qinghai–Tibet plateau. J Syst Evol. 2014;52:313–25.
Lu Q, Zhu J, Yu D, Xu X. Genetic and geographical structure of boreal plants in their southern range: phylogeography of Hippuris vulgaris in China. BMC Evol Biol. 2016;16:34.
Wang Z, Xia C, Yu D, Wu Z. Low-temperature induced leaf elements accumulation in aquatic macrophytes across Tibetan plateau. Ecol Eng. 2015;75:1–8.
Wu Z, Yu D, Wang Z, Li X, Xu X. Great influence of geographic isolation on the genetic differentiation of Myriophyllum spicatum under a steep environmental gradient. Sci Rep. 2015;5:15618.
Cook CDK. A monographic study of Ranunculus subgenus Batrachium (DC.) A. Gray. Mitt Bot Staatssamml München. 1966;6:47–237.
Wiegleb G, Bobrov AA, Zalewska-Gałosz J. A taxonomic account of Ranunculus section Batrachium (Ranunculaceae). Phytotaxa. 2017;319:1–55.
Webster SD. Ranunculus penicillatus (Dumort.) Bab. In Great Britain and Ireland. Watsonia. 1988;17:1–22.
Dahlgren G. Differentiation patterns in Ranunculus subgenus Batrachium (Ranunculaceae). Pl Syst Evol. 1995; [Suppl.];9:305–17.
Zalewska-Gałosz J, Jopek M, Ilnicki T. Hybridization in Batrachium group: controversial delimitation between heterophyllous Ranunculus penicillatus and the hybrid Ranunculus fluitans × R. peltatus. Aquat Bot. 2015;120:160–8.
Wu Z, Wu J, Wang Y, Hou H. Development of EST-derived microsatellite markers in the aquatic macrophyte Ranunculus bungei (Ranunculaceae). Appl Plant Sci. 2017;5:1700022.
Wang Y, Chen J, Xu C, Liu X, Wang Q, Motley TJ. Population genetic structure of an aquatic herb Batrachium bungei (Ranuculaceae) in the Hengduan Mountains of China. Aquat Bot. 2010;92:221–5.
De Meester L, Gómez A, Okamura B, Schwenk K. The monopolization hypothesis and the dispersal–gene flow paradox in aquatic organisms. Acta Oecol. 2002;23:121–35.
Nies G, Reusch TBH. Evolutionary divergence and possible incipient speciation in post-glacial populations of a cosmopolitan aquatic plant. J Evol Biol. 2005;18:19–26.
Viana DS, Santamaría L, Schwenk K, Manca M, Hobæk A, Mjelde M, Preston CD, Gornall RJ, Croft JM, King RA, Green AJ, Figuerola J. Environment and biogeography drive aquatic plant and cladoceran species richness across Europe. Freshw Biol. 2014;59:2096–106.
Nybom H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol. 2004;13:1143–55.
Koga K, Kadono Y, Setoguchi H. Phylogeography of Japanese water crowfoot based on chloroplast DNA haplotypes. Aquat Bot. 2008;89:1–8.
Butkuvienė J, Sinkevičienė Z, Naugžemys D, Patamsytė J, Žvingila D. Genetic diversity of Batrachium (Ranunculaceae) species reveals the necessity of their protection in Lithuanian rivers. Aquat Bot. 2017;142:61–70.
Bobrov AA, Maltseva SY, Movergoz EA, Wiegleb G, Zalewska-Gałosz J. A revision of rigid-leaved water crowfoots of Ranunculus circinatus group (Batrachium, Ranunculaceae). In: Sokoloff DD, et al., editors. Taxonomy and evolutionary morphology of plants, materials of the conference dedicated to 85th anniversary of V. N. Tikhomirov. Мoscow: MAKS Press; 2017. p. 106–9.
Hong D-Y. A biosystematic study on Ranunculus subgenus Batrachium in S Sweden. Nord J Bot. 1991;11:41–59.
Lambertini C, Gustafsson MHG, Baattrup-Pedersen A, Riis T. Genetic structure of the submersed Ranunculus baudotii (sect. Batrachium) population in a lowland stream in Denmark. Aquat Bot. 2017;136:186–96.
Wang Y-H. Studies on reproductive characters and population genetic differentiation of Batrachium bungei in Hengduan Mountains region. Ph.D. dissertation. Wuhan: Wuhan University; 2009.
Wu Y, Colwell RK, Han N, Zhang R, Wang W, Quan Q, et al. Understanding historical and current patterns of species richness of babblers along a 5000-m subtropical elevational gradient. Glob Ecol Biogeogr. 2014;23:1167–76.
Frei ES, Scheepens JF, Armbruster GFJ, Stöcklin J. Phenotypic differentiation in a common garden reflects the phylogeography of a widespread alpine plant. J Ecol. 2012;100:297–308.
Barrett SCH, Eckert CG, Husband BC. Evolutionary processes in aquatic plant populations. Aquat Bot. 1993;44:105–45.
Wu ZY, Wu SG. A proposal for a new floristic kingdom (realm) – the E. Asiatic kingdom, its delimitation and characteristics. In: Zhang AL, Wu SG, editors. Proceedings of the first international symposium on floristic characteristics and diversity of East Asian plants. Beijing, China/Berlin: China Higher Education Press/Springer-Verlag; 1996. p. 3–42.
Lacoul P, Freedman B. Relationships between aquatic plants and environmental factors along a steep Himalayan altitudinal gradient. Aquat Bot. 2006;84:3–16.
Brochet A-L, Guillemain M, Fritz H, Gauthier-Clerc M, Green AJ. The role of migratory ducks in the long-distance dispersal of native plants and the spread of exotic plants in Europe. Ecography. 2009;32:919–28.
Reynolds C, Miranda NAF, Cumming GS. The role of waterbirds in the dispersal of aquatic alien and invasive species. Diversity Distrib. 2015;21:744–54.
Brochet AL, Guillemain M, Fritz H, Gauthier-Clerc M, Green AJ. Plant dispersal by teal (Anas crecca) in the Camargue: duck guts are more important than their feet. Freshw Biol. 2010;55:1262–73.
Mader E, Van Vierssen W, Schwenk K. Clonal diversity in the submerged macrophyte Potamogeton pectinatus L. inferred from nuclear and cytoplasmic variation. Aquat Bot. 1998;62:147–60.
Pollux BJA, Santamaría L, Ouborg NJ. Differences in endozoochorous dispersal between aquatic plant species, with reference to plant population persistence in rivers. Freshw Biol. 2005;50:232–42.
Wu Z, Yu D, Li X, Xu X. Influence of geography and environment on patterns of genetic differentiation in a widespread submerged macrophyte, Eurasian watermilfoil (Myriophyllum spicatum L., Haloragaceae). Ecol Evol. 2016;6:460–8.
Zhang Y. Comparative investigation of birds diversity of 6 nature Reserve in Sanjiangyuan National Nature Reserve of Qinghai Province. Master dissertation. Xining: Qinghai Normal University; 2015.
Liu W, Wu Y, DuBay SG, Zhao C, Wang B, Ran J. Dung-associated arthropods influence foraging ecology and habitat selection in black-necked cranes (Grus nigricollis) on the Qinghai–Tibet plateau. Ecol Evol. 2019;9:2096–105.
Sun H, Niu Y, Chen Y-S, Song B, Liu C-Q, Peng D-L, Chen J-G, Yang Y. Survival and reproduction of plant species in the Qinghai–Tibet plateau. J Syst Evol. 2014;52:378–96.
Nosil P, Vines TH, Funk DJ. Reproductive isolation caused by natural selection against immigrants from divergent habitats. Evolution. 2005;59:705–19.
Weinig C, Ewers BE, Welch SM. Ecological genomics and process modeling of local adaptation to climate. Curr Opin Plant Biol. 2014;18:66–72.
Epps CW, Keyghobadi N. Landscape genetics in a changing world: disentangling historical and contemporary influences and inferring change. Mol Ecol. 2015;24:6021–40.
Ortego J, Gugger PF, Sork VL. Climatically stable landscapes predict patterns of genetic structure and admixture in the Californian canyon live oak. J Biogeogr. 2015;42:328–38.
Varpe Ø. Life history adaptations to seasonality. Integr Comp Biol. 2017;57:943–60.
Xia C, Yu D, Wang Z, Xie D. Stoichiometry patterns of leaf carbon, nitrogen and phosphorous in aquatic macrophytes in eastern China. Ecol Eng. 2014;70:406–13.
Lacoul P, Freedman B. Environmental influences on aquatic plants in freshwater ecosystems. Environ Rev. 2006;14:89–136.
Zheng M, Liu X. Hydrochemistry of salt lakes of the Qinghai-Tibet plateau. China Aquat Geochem. 2009;15:293–320.
Gao L, Tang S, Zhuge L, Nie M, Zhu Z, Li B, Yang J. Spatial genetic structure in natural populations of Phragmites australis in a mosaic of saline habitats in the Yellow River delta, China. PLoS ONE. 2012;7:e43334.
Katayama N, Kato M, Imaichi R. Habitat specificity enhances genetic differentiation in two species of aquatic Podostemaceae in Japan. Amer J Bot. 2016;103:317–24.
Santamaría L, Figuerola J, Pilon JJ, Mjelde M, Green AJ, Boer TD, et al. Plant performance across latitude: the role of plasticity and local adaptation in an aquatic plant. Ecology. 2003;84:2454–61.
Zhou W, Wang Z, Davy AJ, Liu G. Geographic variation and local adaptation in Oryza rufipogon across its climatic range in China. J Ecol. 2013;101:1498–508.
Bailleul D, Stoeckel S, Arnaud-Haond S. RClone: a package to identify multiLocus clonal lineages and handle clonal data sets in R. Methods Ecol Evol. 2016;7:966–70.
Arnaud-Haond S, Duarte CM, Alberto F, SerrÃO EA. Standardizing methods to address clonality in population studies. Mol Ecol. 2007;16:5115–39.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research—an update. Bioinformatics. 2012;28:2537–9.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform. 2005;1:47–50.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–87.
Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–61.
Rosenberg NA. Distruct: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–8.
Rousset F. Genetic differentiation between individuals. J Evol Biol. 2000;13:58–62.
Hardy OJ, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002;2:618–20.
Shirk AJ, Landguth EL, Cushman SA. A comparison of individual-based genetic distance metrics for landscape genetics. Mol Ecol Resour. 2017;17:1308–17.
Rousset F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997;145:1219–28.
Beerli P, Felsenstein J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. P Natl Acad Sci USA. 2001;98:4563–8.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Package ‘vegan’. Community ecology package. R package version 2.2–1. 2013.
Fang J, Piao S, Tang Z, Peng C, Ji W. Interannual variability in net primary production and precipitation. Science. 2001;293:1723.
Blair C, Jiménez Arcos VH, de la Cruz FRM, Murphy RW. Historical and contemporary demography of leaf-toed geckos (Phyllodactylidae: Phyllodactylus tuberculosus saxatilis) in the Mexican dry forest. Conserv Genet. 2015;16:419–29.
Wang IJ. Examining the full effects of landscape heterogeneity on spatial genetic variation: a multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution. 2013;67:3403–11.
Acknowledgements
We thank Zhong Wang for the field assistance and Ian Wang for help in the data analysis. We also thank the two anonymous reviewers for the many valuable suggestions on the revision of manuscript. Studies of G. Wiegleb at herbarium PE were supported by German Academic Exchange service (DAAD). Xian-Chun Zhang (Beijing) gave valuable support during the stay. Alexander A. Bobrov (Yaroslavl) gave valuable hints on taxonomy and distribution of R. subrigidus. The geographic source in Figures was supported from National Earth System Science Data Center, National Science & Technology Infrastructure of China (http://www.geodata.cn).
Funding
This research was supported by the National Natural Science Foundation of China (NSFC, 31700190) and National Key R & D Program of China (2018YFD0900801). Any opinion, finding and conclusion or recommendation expressed in this publication is that of the authors and the NSFC does not accept liability in this regard.
Author information
Authors and Affiliations
Contributions
ZW, XX and HH conceived and designed the study. ZW and ZJ performed experimental work and analyses. GW performed the herbarium studies. ZW wrote the draft and GW provided support and revision. All authors gave their approval for this publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1 Pairwise individual-based genetic distance (Rousset’s â) between 13 Ranunculus subrigidus populations. Table S2 Pairwise population-based genetic distance (FST) between 13 Ranunculus subrigidus populations. Table S3 Estimates of historical asymmetric migration rates (M)/ number of migrants per generation (Nm) between populations of Ranunculus subrigidus. Table S4 Measures of environmental variables of 13 Ranunculus subrigidus populations. Table S5 Results of dbRDA on the contributions of environmental principal component variables on the genetic pattern.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wu, Z., Xu, X., Zhang, J. et al. Influence of environmental factors on the genetic variation of the aquatic macrophyte Ranunculus subrigidus on the Qinghai-Tibetan Plateau. BMC Evol Biol 19, 228 (2019). https://doi.org/10.1186/s12862-019-1559-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-019-1559-0