
Abstract
This review examines the landscape genetics literature from 2011–2015 and summarises the genetic evidence for the roles of habitat amount (i.e. total area of habitat), configuration (i.e. spatial arrangement of habitat), and matrix (i.e. nature of the intervening landscape between habitat patches) in shaping genetic differentiation and diversity of populations. We found that the vast majority of landscape genetic studies focused on the effects of habitat configuration and intervening matrix permeability on genetic differentiation of populations, and very few consider the consequences of habitat loss (i.e. change in habitat amount) versus fragmentation per se (i.e. change in habitat configuration). In addition, disproportionately few studies consider genetic diversity as a response variable in landscape genetic models. We argue that by ignoring the effects of habitat amount, landscape geneticists are missing an important component of how landscape structure shapes patterns of genetic variation. On the other hand, landscape ecologists may need to consider the confounding role of the matrix to resolve the ongoing debate about the relative importance of habitat loss versus fragmentation per se in determining biological diversity.
Similar content being viewed by others

Introduction
Landscape genetics is an interdisciplinary field combining tools and concepts from both landscape ecology and population genetics to relate landscape structure to patterns of genetic variation [1, 2]. The field has evolved tremendously since Manel’s landmark paper in 2003 [1], moving from descriptive assignment tests used to define population boundaries to a more explicit analytical framework including landscape variables as predictors in genetic models [3]. A recent review categorized the types of questions and methods used in landscape genetic studies compared to papers published in its predecessor fields and found that most self-identifying landscape genetic studies fall more into the realm of population genetics (e.g. using terms such as “genetic”, “gene”, and “barrier” [4]) than landscape ecology (e.g. using terms such as “vegetation”, “resource”, “properties” [4]). This begs the question: what can genetics contribute to the field of landscape ecology? Could this trend reflect a lack of initiative from the landscape ecology community to drive the agenda for landscape genetics?
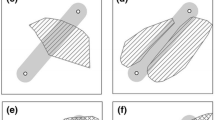
As a tool, molecular genetics can make hard-to-observe processes visible and thus should be useful for landscape ecologists working on species whose movement is hard to track. For example, genetic markers have been used to estimate both contemporary and historical effective population size [5], assess sex-biased dispersal [6–8], identify population bottlenecks [9], and characterize meta-population dynamics [10]. Genetics can be used to quantify actual functional connectivity either directly or indirectly, and thus provides the means to test hypotheses about how aspects of the intervening landscape matrix support or inhibit dispersal and gene flow [11–13]. Most studies in landscape genetics focus on this aspect, i.e. testing if the permeability of the intervening landscape matrix between populations matters beyond the effects of the spatial configuration of populations in explaining among-population genetic differentiation. Such studies typically use link-level analysis (Fig. 1; [14]), where each data point, or row in a data table, refers to a pair of sampling units and quantifies their pairwise genetic, geographic, or ecological distance. Far fewer studies have tested the effects of habitat amount and habitat configuration (i.e. spatial arrangement of habitat) on genetic diversity within populations [15]. Such studies typically use node-level analysis (Fig. 1; [14]), where each data point refers to a focal patch or population. Few papers combine node- and link-based analysis in gravity models [16, 17]. Note that another set of landscape genetic studies, landscape genomics, focuses on adaptive genetic variation [18, 19], which is beyond the scope of this review.

Approaches for quantifying the role of landscape structure on genetic variation. Letters represent focal patches or populations, and grey shapes represent habitat in a matrix of non-habitat (white background). In link-based approaches and gravity models, the genetic response (genetic differentiation) is measured in a pairwise fashion between focal patches/populations. In node-based approaches, the genetic response is measured per focal patch/population. Link-based approaches model genetic differentiation as a function of the distance (e.g. isolation-by-distance, IBD) or matrix permeability (e.g. isolation-by-resistance, IBR) between populations or individuals. For node-based approaches, genetic diversity or population-specific genetic differentiation is modeled as a function of landscape structure measured in buffered areas around focal patches/populations (dashed circles). Gravity models combine the two approaches, where genetic differentiation is modeled as a function of landscape predictors measured at both nodes (e.g. in buffered areas around each patch/population) and links (e.g. pairwise distance or matrix resistance). Note: incorporating the effects of habitat amount on genetic differentiation in link-based approaches is possible, but requires sampling of multiple landscapes
There is an ongoing and lively debate in ecology about the relative importance of habitat loss (i.e. change in habitat amount) versus fragmentation per se (i.e. change in habitat configuration independently of habitat amount) in shaping populations and communities. The two processes can occur independently, where the same amount of habitat can be in the shape of one large patch or several smaller patches. Thus, fragmentation reduces patch size and increases the proportion of edge to interior habitat. Studies in landscape ecology have produced strong theoretical and empirical evidence that patch occupancy (e.g. [20–23]), species diversity (e.g. [24, 25]), and abundance (e.g. [26, 27]) are often more influenced by the amount of habitat in a landscape rather than the degree of fragmentation [28]. These findings have culminated into a provocative new hypothesis, Fahrig’s “habitat amount hypothesis”, which posits that the amount of habitat in a local landscape determines species richness and that metrics of configuration such as patch size (e.g. size of focal patch) and isolation (e.g. distance to neighbouring patches) can largely be ignored [29]. In contrast, existing evidence from metapopulation ecology indicates a strong role of habitat configuration in explaining species richness (e.g. [30, 31]) and population persistence (e.g. [32]). In response to Fahrig, Hanski argues that the habitat amount hypothesis may only be valid at small spatial scales and when overall habitat amount is large [33]. Landscape configuration should be important in real landscapes where the amount of remaining habitat is often quite low [33]. Evidence from population and landscape genetics seems to support the metapopulation view that configuration and isolation (or its opposite, connectivity) are key determinants of population outcomes. For example, gene flow among populations decreases as a function of population isolation (i.e. isolation-by-distance; [34]), and this relationship can be modified by the permeability of the intervening landscape matrix (i.e. isolation-by-resistance; [35]). These patterns have been demonstrated in a variety of plant (e.g. [36–38]) and animal (e.g. [39, 40]) systems. However, the amount of habitat at the local or landscape scale should also play an important role in shaping within-population genetic variation, as larger amounts of habitat can accommodate higher effective populations sizes (Ne), and consequently, populations will experience lower levels of genetic drift and retain higher levels of diversity [41]. Ultimately, it is the balance of genetic drift and gene flow that determines neutral genetic diversity within, and genetic differentiation among, populations [41], and only by considering the two together can we fully appreciate the role of landscape structure on genetic variation.
Habitat loss and fragmentation tend to be highly correlated in nature, and as a consequence, their independent effects are often difficult to tease apart. Beyond this analytical challenge, there are inconsistencies in terminology and metrics used to measure fragmentation across disciplines [42]. For example, metapopulation studies tend to operate at the patch scale, measuring patch size and isolation individually for each focal patch. However, in landscape ecology, patch size is measured at the landscape scale (e.g. mean patch size in landscape) and is not considered a measure of habitat amount, but as a metric of configuration (i.e., fragmentation). At the patch scale, patch size is an ambiguous metric of fragmentation as it does not account for local landscape context [28]. Fragmentation occurs at the landscape level and thus can only be measured per se after accounting for effects of habitat amount. This mismatch in approach and terminology across disciplines has led to a large number of publications that measure the effects of certain aspects of fragmentation on a variety of population and genetic processes (e.g. [43, 44]), but very few actually quantify fragmentation per se (i.e. control for habitat amount [28, 45]).
Here we examine the last five years of the landscape genetic and relevant population genetic literature and summarise the genetic evidence for the roles of habitat amount, configuration, and matrix resistance in shaping connectivity and diversity of populations. We identify key gaps in our current knowledge and ask: (1) are landscape geneticists considering the independent effects of habitat amount versus configuration on genetic variation? And (2), are the effects of landscape structure on genetic diversity versus genetic differentiation equally considered by landscape geneticists? We end with a discussion of how researchers can address the identified gaps to help resolve a key debate in ecology.
Literature Review
We conducted a final literature search on 26 October 2015 of articles published between 2011–2015 in the ISI Web of Science Core Collection and BIOSIS Citation Index using the following search terms in the TOPIC field: landscape AND genet* AND (frag* OR “habitat loss” OR connectivity OR “gene flow” OR isolation). We refined results to include only the following research areas: ecology, genetics and heredity, evolutionary biology, biodiversity, conservation, plant science, zoology, environmental sciences, multidisciplinary sciences, biology, and ornithology. This search returned 1346 papers. As a first pass, we looked at titles and abstracts to remove papers that did not incorporate genetic data, were reviews or opinions, or were mainly methodological in nature and did not use a new empirical dataset. We also removed papers that did not explicitly test the influence of landscape structure (habitat amount, configuration, or matrix, as described below and in Fig. 1) on genetic variation (i.e. did not include them as predictors). This resulted in the exclusion of papers that investigated the genetic consequences of certain aspects of fragmentation (e.g. studies that compare genetic diversity between a reference and fragmented landscape), but these types of studies have been reviewed elsewhere (e.g. [43, 44]). Studies comparing genetic variation amongst islands were only included if habitat amount was defined and included as a predictor in genetic models (i.e. not just island size as predictor), and we excluded studies conducted in marine systems or seascapes. We placed the remaining 541 articles in one of three categories based on the landscape process investigated and whether the genetic response was measured at the link (i.e. pairwise genetic differences among populations or individuals; Fig. 1) or node (i.e. genetic variation measured per population; Fig. 1) level: 1) influence of habitat configuration on genetic differentiation measured at the link level (e.g. isolation-by-distance, IBD), 2) influence of matrix permeability on genetic differentiation measured at the link level (e.g. isolation-by-resistance, IBR), or 3) influence of habitat amount and/or configuration on within-population genetic variation (e.g. genetic diversity or relatedness) or genetic differentiation (e.g. population-specific Fst), measured at the node level. Many papers considered two or more of the above processes, and we categorized them hierarchically: papers that tested IBR in addition to IBD were placed in the second category, and papers that considered IBD or IBR and the influence of landscape structure on genetic variation measured at the node level were placed in the third category. For each paper that fell into the third category, we recorded the genetic response variable and information about the landscape predictors measured. Each landscape predictor was categorized as one of the following: 1) habitat amount, measured at the landscape or local-landscape scale (i.e. percent habitat in buffered area around focal patches or populations), 2) patch size measured at the patch scale (i.e. size of the focal patch), or 3) habitat configuration. Metrics of habitat configuration included patch-level measures of isolation (e.g. nearest neighbour distances, connectivity index), patch number and density metrics measured at the landscape scale (e.g. mean patch size, number of patches), and landscape-scale aggregation indices (e.g. clumping/dispersion of patches). We considered a study to have controlled for the independent effects of habitat loss versus fragmentation per se [28] if they included habitat amount and at least one (patch or landscape level) metric of habitat configuration as predictors in genetic models, or used an experimental approach. We did not consider patch size measured at the patch scale to be a metric of habitat configuration as it lacks local landscape context, and is often used instead as a proxy of population size in the metapopulation literature. We further summarized results of the retained node-level studies by recording the direction and statistical significance of the effects of habitat amount, patch size, and configuration on genetic variation. We did not carry out formal meta-analysis because few studies reported sufficient information.
Characterizing Gaps in the Literature
Out of the 541 retained studies, 485 were conducted at the link level (IBD: n = 298; IBR: n = 187) and 56 at the node level (Fig. 2a). We found similar biases in study taxa as previous reviews (Fig. 2a; [15]). Given the over-representation of link-level studies, it was thus not surprising to find that the vast majority of retained studies used genetic differentiation as the response variable in genetic models and that the most common landscape predictors were habitat configuration and matrix permeability (Fig. 2b). In contrast, only 23 studies—all of which were conducted at the node level—included habitat amount as a predictor, with either genetic differentiation (n = 5) or genetic diversity (n = 18), as the response variable. Only 11 of these controlled for the confounding effects of habitat amount versus fragmentation per se (Table 1). We have thus uncovered two key gaps in our knowledge of the effects of landscape structure on genetic variation: 1) most studies on landscape genetics are conducted at the link level and thus only consider effects of configuration and/or matrix on genetic variation, ignoring possible effects of habitat amount; and 2) genetic diversity is severely under-represented as a response variable in landscape genetic models.

Mosaic plot showing the proportion of studies conducted at the link and node level across taxa (a), and barplots showing the number times configuration only, matrix, or habitat amount were used as predictors in genetic models with either genetic differentiation or diversity as the response variable (b). One link-based IBR study included three separate taxa, and one node-based study included two taxa and are represented more than once in the mosaic plot. Non-insect invertebrates were included in the insect category and studies on fungi were included in the plant category
Habitat Amount Versus Fragmentation
Current evidence from landscape genetics would suggest a strong and almost singular effect of landscape configuration on functional connectivity and genetic structure across a variety of taxa. This goes against considerable evidence from landscape ecology, which suggests that habitat amount is the most important determinant of demographic outcomes (e.g. patch occupancy, abundance, species richness [28]), and that configuration only becomes important below a critical threshold of habitat loss [57]. However, our search of the recent literature suggests that this discordance among disciplines likely reflects a bias in the type of questions being asked and approaches used in landscape genetics compared to landscape ecology. For example, the vast majority of the studies that we identified were conducted at the link level, i.e. they quantified the relationship between pairwise genetic differentiation among populations and their spatial configuration (IBD; Fig. 2a) or aspects of the intervening landscape matrix (IBR; Fig. 2a). A much smaller proportion incorporated approaches at the node level, where aspects of the local landscape around focal patches were related to genetic variation. It is in this second scenario where the best opportunity exists to quantify the effects of habitat amount on genetic variation. This can also be accomplished at the link level, but it requires sampling of multiple landscapes where overall genetic differentiation among populations within landscapes can be related to landscape-level habitat amount, which is often not feasible. It may be possible to conduct such analysis within landscapes (i.e. per population pair); however, it would be difficult to create meaningful buffers around pairs of populations in which to measure local habitat amount. A third approach, using network-based gravity models, has the potential to integrate both node- and link-based data, but to our knowledge has not been used to include the effects of local habitat amount in genetic models. This approach will be discussed in a later section of this review.
Landscape Effects on Gene Flow and Genetic Drift
To understand the importance of including habitat amount in genetic models, we must consider the landscape determinants of gene flow and drift (Fig. 3). The balance between these two opposing forces ultimately defines both within- and among-population genetic variation [58]. When the relative strength of drift is higher than gene flow, populations are expected to exhibit low genetic diversity and high among-population differentiation. When the strength of gene flow outweighs that of drift, populations are expected to exhibit the opposite pattern, where genetic diversity within population is high and differentiation among them is low [58]. Whereas gene flow is the product of migration and thus should be influenced heavily by habitat configuration and matrix permeability, the strength of drift is determined by the effective size of populations (Ne) and directly influenced by habitat amount (Fig. 3). A strong link between genetic variation and Ne has been empirically demonstrated in a large number of species [59–61], yet the potential landscape determinants of Ne , and thus genetic drift, are largely ignored in landscape genetics in favour of landscape hypotheses relating to gene flow. This is despite the fact that measures of both genetic diversity and differentiation will contain signals of both processes. Including Ne as a covariate can potentially increase the strength of correlations between landscape structure and both genetic diversity [62, 63] and genetic differentiation [64, 65]. Including Ne is expected to be most important for species with low dispersal ability, but should not be ignored for vagile organisms. For example, Prunier et al. [65] showed through simulations that including a proxy of Ne explained up to 50 % of the variation in pairwise population genetic differentiation when migration rates were low, and still explained up to 20 % of the variation for high simulated migration rates.

Predicted relationships between landscape structure, genetic drift and gene flow, and genetic diversity and differentiation. Habitat amount and quality are expected to strongly determine local effective population size (Ne) and thus the local pool of alleles (i.e. genetic diversity), whereas habitat configuration and matrix will affect the probability of migration and gene flow among populations and thus leave a stronger signal in measures of genetic differentiation. Habitat configuration and matrix can indirectly influence local Ne by promoting migration among nearby populations. Habitat amount and quality can directly impact migration by promoting philopatry when local resources are high or indirectly through density-dependent processes (e.g. emigration when a population approaches carrying capacity)
Although a variety of methods and programs exist to estimate Ne from genetic markers, effective population sizes are difficult to quantify for real landscapes because these estimates can be biased when populations are spatially structured [66, 67]. A landscape proxy for Ne may thus be preferred. But what is the appropriate metric? Patch size measured at the patch-scale is often used as a proxy of population size in metapopulation models, but multiple meta-analyses have revealed highly inconsistent relationships between patch size and abundance across species [68, 69]. Measured at the patch scale, patch size lacks local landscape context and may only be an appropriate measure of habitat amount, and thus population size, when patches are quite isolated [28]. In our search of the recent literature, only 11 out of the 37 models that included patch size found it to be a significant predictor of genetic variation, and in two instances the results showed the opposite of the expected relationship (Table 2). Alternatively, habitat amount, measured at the local landscape scale (i.e. in buffered area around focal patches) may be a more appropriate proxy for population size [29]. However, of the 34 instances where local-landscape habitat amount was included as a predictor in genetic models, just half (n = 17) found the predicted relationship, while 15 found no significant effect and two found an opposite effect (Table 2). The small number of studies retained in our literature search precluded an in-depth meta-analysis, but there are a number of potential reasons for these inconsistencies. First, the relevant scale for measuring a landscape proxy of Ne is likely species-specific and will depend on how species perceive patch boundaries [70, 71]. Second, Ne is not only determined by habitat amount, but is also indirectly impacted by habitat configuration and matrix permeability by allowing migration among nearby patches (Fig. 3). We expect these effects to be most important when local habitat amount is small, where small patches will only be able to maintain a viable population if they are connected via gene flow (i.e. genetic rescue). Third, in addition to its impact on genetic drift, habitat amount can have contrasting effects on gene flow, which may obscure predicted relationships. For example, Robinson et al. [52] and Dharmarajan et al. [72] found that high local habitat availability was associated with higher levels of average relatedness among individuals within populations of white-tailed deer (Odocoileus virginianus) and racoons (Procyon lotor), respectively. This suggests that these species exhibit higher natal philopatry when local resources and habitat are plentiful, a process that can lead to a pattern of low genetic diversity in populations surrounded by large amounts of habitat. Similarly, density-dependent migration can lead to the movement of individuals from large populations that have surpassed carrying capacity to smaller populations [73]. In this case accounting for the differences in Ne among pairwise populations may produce stronger models [64, 65].
Habitat quality, shaped by local environmental conditions, resource availability, and species interactions, can also limit the size of populations and impact rates of migration. It is increasingly recognized that these effects can shape genetic outcomes (e.g. [74–77]) and thus should be incorporated into landscape genetic models (see [78] for a review on the subject). A binary definition of habitat and non-habitat is unlikely to capture the full range of these effects, and ideally, variation in habitat quality should be integrated directly into measures of habitat amount. For example, habitat suitability models are often used as a starting point for parameterizing resistance surfaces in landscape genetics, and these same models can be translated to measure the amount of high quality habitat in buffered areas around focal patches or populations.
Genetic Diversity Versus Differentiation
Our literature search revealed another important gap; very few studies consider the effects of landscape structure on genetic diversity within populations, as opposed to genetic differentiation among them. Genetic diversity is a strong predictor of individual and population fitness, extinction risk, and the ability to respond to future environmental change, and thus is an important indicator for landscape-level conservation planning [79, 80]. Understanding the causes and consequences of patterns of genetic diversity has been a longstanding goal of biogeography, population and conservation genetics. Theoretical and empirical evidence from these fields have given us a strong understanding of the effects of population size and population isolation on genetic diversity. More recently, the field of landscape genomics has made great strides relating local environmental conditions to adaptive genetic diversity (reviewed in [18, 19]). However, our knowledge of the landscape determinants of neutral genetic diversity in complex landscapes is limited and has largely been ignored in favour of studies on genetic differentiation in landscape genetics.
Recent simulations have demonstrated that within-population and among-population genetic variation can respond quite strongly to different aspects of landscape structure. For example, using individual-based simulations of gene flow for five hypothetical organisms that varied in dispersal ability, Jackson and Fahrig [81] found that habitat amount explained 88 % of variation in genetic diversity compared to 7 % explained by fragmentation. Likewise, Ezard and Travis [82] found that the fixation time of neutral alleles within simulated populations was predicted best by the amount of habitat. In contrast, simulations by both Bruggeman et al. [83] and Cushman et al. [84] demonstrated a stronger role of habitat configuration compared to habitat amount in explaining patterns of genetic differentiation among populations. These results appear to be reflected in a subset of the empirical studies that we retained in our literature search: Barr et al. [47], Taylor and Hoffman [49], and Capurucho et al. [50] (Table 1) found correlations between habitat configuration and genetic differentiation among populations, but habitat amount and not fragmentation correlated best with within-population genetic diversity. These results are again supported by population genetic theory. While habitat amount is expected to play a stronger role in determining population size and thus the local pool of alleles (i.e. genetic diversity; a measure of alpha diversity), the configuration of habitat determines rates of migration and thus leaves a stronger signal in measures of differentiation among populations (a measure of beta diversity; Fig. 3).
Genetic diversity and differentiation reach migration-drift equilibrium at different rates and are thus expected to respond to landscape structure at different spatiotemporal scales [85]. When gene flow or population size is reduced by a change in landscape structure, populations will become genetically differentiated from each other faster than genetic diversity is lost within populations. This difference in lag time is important to consider when quantifying the impact of landscape structure on genetic variation [86]. For example, measures of genetic differentiation might correlate with current landscape structure while genetic diversity might be better explained by a historical landscape (e.g. [48, 87]).
Both genetic diversity and differentiation are the products of many generations of dispersal and mating. As the lag time in the ability to detect a signal of landscape change increases, the more important spatially distant individuals and their landscape context will be for explaining genetic variation. For example, using simulations, Jackson and Fahrig [81] found that habitat amount explained genetic diversity best when it was measured at a broad spatial scale. In contrast, demographic outcomes (patch occupancy and abundance) correlated best with habitat amount measured at smaller spatial scales. Similarly, Millette and Keyghobadi [53] found that fine-scale genetic structure of a pitcher-plant insect (Metriocnemus knabi) was best explained by broad-scale habitat configuration. These findings are significant from a conservation perspective as they may lead to different decisions about how to conserve the landscape depending on the outcome measured, further emphasizing the need for both demographic and genetic approaches.
If it takes longer for a signal of landscape change to emerge in measures of genetic diversity compared to differentiation, we might also expect the relevant spatial scale to be larger for within-population diversity. However, it appears that this is not always the case. For example, Taylor and Hoffman [49] tested the influence of habitat amount and fragmentation on the white footed mouse (Peromycus leucopus) at three spatial scales and found that habitat amount measured at the smallest radius (500 m) around focal populations was the single best predictor of genetic diversity. Balkenhol et al. [88] found that landscape-level habitat amount influenced the contribution of local-landscape and patch-scale measures of connectivity to both genetic diversity and differentiation in the forest dwelling marsupial Marmosops incanus. Similarly, Millette and Keyghobadi [53] found an interactive effect of habitat amount and patch connectivity; genetic differentiation of M. knabi populations increased with patch isolation when habitat amount was low, but decreased with patch isolation when habitat amount was high. Together these results suggest that both habitat amount and configuration can impact within- and among-population measures of genetic variation at multiple spatial and temporal scales and underlines the importance of multi-scale approaches in landscape genetics.
Importance of the Matrix
The last decade of landscape genetics has produced overwhelming evidence that functional connectivity is affected by the permeability of the intervening landscape matrix beyond the effects of the spatial configuration of populations (e.g. [89–91]) and that different species can respond to the same landscape in vastly different ways [92–97]. Thus, using a binary classification of habitat versus non-habitat when quantifying landscape structure is unrealistic and can lead to misleading conclusions about the impact of habitat loss and fragmentation on populations [70, 98]. However, our knowledge of how habitat amount, configuration, and the matrix interact to determine gene flow is quite limited, as it requires sampling across replicate landscapes, or the use of node-based or gravity model approaches (Fig. 1), which are quite rare in landscape genetics. The few studies that have considered multiple landscapes have found that different aspects of the landscape matrix predicted gene flow for the same species in different regions (e.g. [40, 99]). However, we know of only one study with sufficient replication to determine if these differences in landscape effects on gene flow could be explained by landscape-level differences in habitat amount and configuration: Short Bull et al. [100] contrasted patterns of genetic differentiation in black bears (Ursus americanus) across 12 landscapes and found that landscape features were only identified as significant predictors of population differentiation if they were highly variable or fragmented at the landscape scale. For example, forest (considered suitable habitat for black bears) was more likely to be identified as a predictor of genetic differentiation in landscapes where forest was more fragmented [100]. This study did not control for differences in overall habitat amount among the landscapes, so it is unclear if fragmentation per se or if the amount of different features in each landscape was driving the effect. This suggests that broad-scale habitat amount and configuration can alter species response to the matrix or, alternatively, alter our ability to detect landscape genetic correlations. Simulations by Cushman et al. [84] suggests that the reverse can also be true; the relative resistance of the intervening landscape matrix can modify the relative contribution of landscape-level habitat amount versus fragmentation per se to patterns of genetic differentiation. Although they identified habitat configuration as the best overall predictor of genetic differentiation in simulated landscapes, the amount of variance explained by habitat amount (alone, and interactively with configuration) increased with relative matrix resistance. At the highest level of matrix resistance simulated, habitat amount outperformed three of the four tested fragmentation metrics. This result suggests that landscape ecologists need to take into account the intervening matrix in order to resolve the ongoing debate on the relative importance of habitat loss vs. fragmentation. However, more evidence from empirical studies and further simulations are urgently needed.
The Way Forward
How can we test the relative importance of the effects of habitat amount versus fragmentation per se on genetic variation? We see three possible approaches: 1) Link-based analysis in multiple landscapes (Fig 1a): by quantifying gene flow across multiple landscapes that vary in overall habitat amount and degree of fragmentation, we can start to understand the interactive effects of habitat amount, configuration, and matrix. Link-based analysis is by far the most common approach in landscape genetics, but usually only a single landscape is considered. We recognize that replication at the landscape scale is not always feasible, but as shown by Short Bull et al. [100], it is desperately needed to build a comprehensive understanding of how landscape structure influences gene flow; 2) Node-based analysis (Fig. 1): habitat amount and fragmentation are measured either at the landscape-scale or locally within a given radius around focal patches and are related to patch-specific measures of genetic variation. The genetic response variable can either be a measure of genetic diversity, or population-specific Fst that represents the relative genetic differentiation of a local population from all others in a metapopulation [101]. This focal patch approach is commonly used in landscape ecology and can be adapted to directly test Fahrig’s habitat amount hypothesis [29], but can also incorporate matrix effects by measuring connectivity of the focal patch to all others in the local landscape as a function of matrix permeability (e.g. using an incidence function; [102]); 3) Network-based gravity models (Fig. 1): this approach models genetic differentiation among populations as a function of both local-landscape variables that are measured at the node level and matrix permeability measured at the link level. This method has been used to model the effects of local environment and habitat quality on gene flow (e.g. [16, 17]), but to the best of our knowledge has not yet been employed to quantify the relative contribution of local habitat amount, fragmentation, and matrix.
Ideally, future research in landscape genetics should employ all three approaches, as each captures the potential effects of landscape structure on genetic variation at different spatio-temporal scales. Certain approaches will be more appropriate or feasible in particular systems or for particular species. For example, the use of the individual as the study unit is common in link-based studies for continuously distributed species (i.e. genetic differentiation is measured among individuals rather than among populations); however, it is less clear if individual-based methods can be translated to node or gravity model frameworks. It has been suggested that landscape features could be measured around sampling locations of focal individuals at radii relevant to the species’ dispersal ability or home-range size (see [78]); however, more theoretical work is needed to demonstrate the validity and strength of this approach.
Node-based analysis is most intuitively applicable when habitat is discrete. For more continuously distributed species, binary habitat classification may not be appropriate, and this may explain why we found an under-representation of node-based studies. In comparison, link-based analysis of either IBD or IBR does not necessarily require the quantification of landscape pattern with a strict definition of patch boundaries. For example, matrix permeability can be modeled as a continuous variable such as habitat suitability or outputs from species distribution models. Translating these continuous representations of landscapes to categorical ones that can be used to quantify the amount and configuration of habitat is possible, but not always straightforward [70, 103].
Conclusions
Here we have identified three key gaps in our knowledge of the effects of landscape structure on genetic variation. (1) Very few empirical studies have included habitat amount as a covariate in genetic models, and as a result, empirical evidence of the relative influence of habitat loss versus fragmentation per se on genetic diversity and differentiation is limited. (2) In addition, disproportionately few studies in landscape genetics consider genetic diversity as a response variable. Habitat amount can have important impacts on both genetic drift and gene flow at multiple spatial scales. Moreover, the effects of drift and gene flow are unequally represented in the signals of genetic diversity and differentiation and thus may correlate with different aspects of landscape structure. (3) The effects of habitat amount, configuration, and matrix (and ideally also habitat quality) should be considered simultaneously to help resolve the debate about the relative importance of habitat loss versus fragmentation in determining biological diversity. This will require a more balanced analytical framework that incorporates node-based and landscape-level approaches commonly used in landscape ecology. We thus encourage landscape ecologists take a more active role in setting the agenda for landscape genetics.
References
Manel S, Schwartz MK, Luikart G, Taberlet P. Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol. 2003;18(4):189–97.
Holderegger R, Wagner HH. Landscape genetics. Bioscience. 2008;58(3):199–207.
Storfer A, Murphy MA, Evans JS, et al. Putting the ‘landscape’ in landscape genetics. Heredity. 2007;98(3):128–42.
Dyer RJ. Is there such a thing as landscape genetics? Mol Ecol. 2015;24(14):3518–28.
Chiucchi JE, Gibbs HL. Similarity of contemporary and historical gene flow among highly fragmented populations of an endangered rattlesnake. Mol Ecol. 2010;19(24):5345–58.
Vangestel C, Callens T, Vandomme V, Lens L. Sex-biased dispersal at different geographical scales in a cooperative breeder from fragmented rainforest. Plos One. 2013;8(8):e71624.
Goudet J, Perrin N, Waser P. Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol Ecol. 2002;11(6):1103–14.
Wang Y, Lane A, Ding P. Sex-biased dispersal of a frog (odorrana schmackeri) is affected by patch isolation and resource limitation in a fragmented landscape. Plos One. 2012;7(10):e47683.
Tucker JM, Schwartz MK, Truex RL, Pilgrim KL, Allendorf FW. Historical and contemporary DNA indicate fisher decline and isolation occurred prior to the European settlement of California. Plos One. 2012;7(12):e52803.
Andreasen AM, Stewart KM, Longland WS, Beckmann JP, Forister ML. Identification of source-sink dynamics in mountain lions of the Great Basin. Mol Ecol. 2012;21(23):5689–701.
Jaquiery J, Broquet T, Hirzel AH, Yearsley J, Perrin N. Inferring landscape effects on dispersal from genetic distances: how far can we go? Mol Ecol. 2011;20(4):692–705.
Wang IJ, Savage WK, Shaffer HB. Landscape genetics and least-cost path analysis reveal unexpected dispersal routes in the California tiger salamander (Ambystoma californiense). Mol Ecol. 2009;18(7):1365–74.
Lowe WH, Allendorf FW. What can genetics tell us about population connectivity? (vol 19, pg 3038, 2010). Mol Ecol. 2010;19(23):5320.
Wagner HH, Fortin MJ. A conceptual framework for the spatial analysis of landscape genetic data. Conserv Genet. 2013;14(2):253–61.
Storfer A, Murphy MA, Spear SF, Holderegger R, Waits LP. Landscape genetics: where are we now? Mol Ecol. 2010;19(17):3496–514.
Murphy MA, Dezzani R, Pilliod DS, Storfer A. Landscape genetics of high mountain frog metapopulations. Mol Ecol. 2010;19(17):3634–49.
Dileo MF, Siu JC, Rhodes MK, et al. The gravity of pollination: integrating at-site features into spatial analysis of contemporary pollen movement. Mol Ecol. 2014;23(16):3973–82.
Manel S, Joost S, Epperson BK, et al. Perspectives on the use of landscape genetics to detect genetic adaptive variation in the field. Mol Ecol. 2010;19(17):3760–72.
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R. A practical guide to environmental association analysis in landscape genomics. Mol Ecol. 2015;24(17):4348–70.
Scherer RD, Muths E, Noon BR. The importance of local and landscape-scale processes to the occupancy of wetlands by pond-breeding amphibians. Popul Ecol. 2012;54(4):487–98.
Betts MG, Forbes GJ, Diamond AW. Thresholds in songbird occurrence in relation to landscape structure. Conserv Biol. 2007;21(4):1046–58.
Hornseth ML, Walpole AA, Walton LR, et al. Habitat loss, not fragmentation, drives occurrence patterns of Canada Lynx at the southern range periphery. Plos One. 2014;9(11):e113511.
Mortelliti A, Amori G, Capizzi D, et al. Independent effects of habitat loss, habitat fragmentation and structural connectivity on the distribution of two arboreal rodents. J Appl Ecol. 2011;48(1):153–62.
Fischer J, Lindenmayer DB, Barry S, Flowers E. Lizard distribution patterns in the Tumut Fragmentation “natural experiment” in south-eastern Australia. Biol Conserv. 2005;123(3):301–15.
Flick T, Feagan S, Fahrig L. Effects of landscape structure on butterfly species richness and abundance in agricultural landscapes in eastern Ontario, Canada. Agric Ecosyst Environ. 2012;156:123–33.
Holbrook SJ, Forrester GE, Schmitt RJ. Spatial patterns in abundance of a damselfish reflect availability of suitable habitat. Oecologia. 2000;122(1):109–20.
McGarigal K, McComb WC. Relationships between landscape structure and breeding birds in the oregon coast range. Ecol Monogr. 1995;65(3):235–60.
Fahrig L. Effects of habitat fragmentation on biodiversity. Annu Rev Ecol Evol Syst. 2003;34:487–515.
Fahrig L. Rethinking patch size and isolation effects: the habitat amount hypothesis. J Biogeogr. 2013;40(9):1649–63.
Hanski I, Zurita GA, Bellocq MI, Rybicki J. Species-fragmented area relationship. Proc Natl Acad Sci U S A. 2013;110(31):12715–20.
Rybicki J, Hanski I. Species-area relationships and extinctions caused by habitat loss and fragmentation. Ecol Lett. 2013;16:27–38.
Hanski I, Pakkala T, Kuussaari M, Lei GC. Metapopulation persistence of an endangered butterfly in a fragmented landscape. Oikos. 1995;72(1):21–8.
Hanski I. Habitat fragmentation and species richness. J Biogeogr. 2015;42(5):989–93.
Wright S. Isolation by distance. Genetics. 1943;28(2):114–38.
McRae BH. Isolation by resistance. Evolution. 2006;60(8):1551–61.
Dyer RJ, Chan DM, Gardiakos VA, Meadows CA. Pollination graphs: quantifying pollen pool covariance networks and the influence of intervening landscape on genetic connectivity in the North American understory tree, Cornus florida L. Landsc Ecol. 2012;27(2):239–51.
Rico Y, Holderegger R, Boehmer HJ, Wagner HH. Directed dispersal by rotational shepherding supports landscape genetic connectivity in a calcareous grassland plant. Mol Ecol. 2014;23(4):832–42.
Kamm U, Gugerli F, Rotach P, Edwards P, Holderegger R. Open areas in a landscape enhance pollen-mediated gene flow of a tree species: evidence from northern Switzerland. Landsc Ecol. 2010;25(6):903–11.
Row JR, Blouin-Demers G, Lougheed SC. Habitat distribution influences dispersal and fine-scale genetic population structure of eastern foxsnakes (Mintonius gloydi) across a fragmented landscape. Mol Ecol. 2010;19(23):5157–71.
Trumbo DR, Spear SF, Baumsteiger J, Storfer A. Rangewide landscape genetics of an endemic Pacific northwestern salamander. Mol Ecol. 2013;22(5):1250–66.
Wright S. Evolution in Mendelian populations. Genetics. 1931;16(2):0097–159.
Lindenmayer DB, Fischer J. Tackling the habitat fragmentation panchreston. Trends Ecol Evol. 2007;22(3):127–32.
Keyghobadi N. The genetic implications of habitat fragmentation for animals. Can J Zool. 2007;85(10):1049–64.
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J. Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol. 2008;17(24):5177–88.
Hadley AS, Betts MG. The effects of landscape fragmentation on pollination dynamics: absence of evidence not evidence of absence. Biol Rev. 2012;87(3):526–44.
Toma Y, Imanishi J, Yokogawa M, et al. Factors affecting the genetic diversity of a perennial herb Viola grypoceras A. Gray var. grypoceras in urban fragmented forests. Landsc Ecol. 2015;30(8):1435–47.
Barr KR, Kus BE, Preston KL, Howell S, Perkins E, Vandergast AG. Habitat fragmentation in coastal southern California disrupts genetic connectivity in the cactus wren (Campylorhynchus brunneicapillus). Mol Ecol. 2015;24(10):2349–63.
Flavenot T, Fellous S, Abdelkrim J, Baguette M, Coulon A. Impact of quarrying on genetic diversity: an approach across landscapes and over time. Conserv Genet. 2015;16(1):181–94.
Taylor ZS, Hoffman SMG. Landscape models for nuclear genetic diversity and genetic structure in white-footed mice (Peromyscus leucopus). Heredity. 2014;112(6):588–95.
Capurucho JMG, Cornelius C, Borges SH, et al. Combining phylogeography and landscape genetics of Xenopipo atronitens (Aves: Pipridae), a white sand campina specialist, to understand Pleistocene landscape evolution in Amazonia. Biol J Linn Soc. 2013;110(1):60–76.
Levy E, Tomkins JL, LeBas NR, Kennington WJ. Contrasting effects of landscape features on genetic structure in different geographic regions in the ornate dragon lizard, Ctenophorus ornatus. Mol Ecol. 2013;22(15):3904–15.
Robinson SJ, Samuel MD, Lopez DL, Shelton P. The walk is never random: subtle landscape effects shape gene flow in a continuous white-tailed deer population in the Midwestern United States. Mol Ecol. 2012;21(17):4190–205.
Millette KL, Keyghobadi N. The relative influence of habitat amount and configuration on genetic structure across multiple spatial scales. Ecol Evol. 2015;5(1):73–86.
Coster SS, Babbitt KJ, Cooper A, Kovach AI. Limited influence of local and landscape factors on finescale gene flow in two pond-breeding amphibians. Mol Ecol. 2015;24(4):742–58.
Peterman WE, Anderson TL, Ousterhout BH, Drake DL, Semlitsch RD, Eggert LS. Differential dispersal shapes population structure and patterns of genetic differentiation in two sympatric pond breeding salamanders. Conserv Genet. 2015;16(1):59–69.
Hahn T, Kettle CJ, Ghazoul J, Hennig EI, Pluess AR. Landscape composition has limited impact on local genetic structure in mountain clover, Trifolium montanum L. J Hered. 2013;104(6):842–52.
Fahrig L. When does fragmentation of breeding habitat affect population survival? Ecol Model. 1998;105(2–3):273–92.
Wright S. On the roles of directed and random changes in gene frequency in the genetics of populations. Evolution. 1948;2(4):279–94.
Frankham R. Relationship of genetic variation to population size in wildlife. Conserv Biol. 1996;10(6):1500–8.
Ellstrand NC, Elam DR. Population genetic consequences of small population-size - implications for plant conservation. Annu Rev Ecol Syst. 1993;24:217–42.
Leimu R, Mutikainen P, Koricheva J, Fischer M. How general are positive relationships between plant population size, fitness and genetic variation? J Ecol. 2006;94(5):942–52.
Mendez M, Vogeli M, Tella JL, Godoy JA. Joint effects of population size and isolation on genetic erosion in fragmented populations: finding fragmentation thresholds for management. Evol Appl. 2014;7(4):506–18.
Carvalho CD, Ribeiro MC, Cortes MC, Galetti M, Collevatti RG. Contemporary and historic factors influence differently genetic differentiation and diversity in a tropical palm. Heredity. 2015;115(3):216–24.
Weckworth BV, Musiani M, DeCesare NJ, McDevitt AD, Hebblewhite M, Mariani S. Preferred habitat and effective population size drive landscape genetic patterns in an endangered species. Proc R Soc B Biol Sci. 2013;280(1769):20131756.
Prunier J, Dubut V, Chikhi L, Blanchet S: Isolation-by-drift: quantifying the respective contributions of genetic drift and gene flow in shaping spatial patterns of genetic differentiation. bioRxiv 2015.
Neel MC, McKelvey K, Ryman N, et al. Estimation of effective population size in continuously distributed populations: there goes the neighborhood. Heredity. 2013;111(3):189–99.
Ryman N, Allendorf FW, Jorde PE, Laikre L, Hossjer O. Samples from subdivided populations yield biased estimates of effective size that overestimate the rate of loss of genetic variation. Mol Ecol Resour. 2014;14(1):87–99.
Bowers MA, Matter SF. Landscape ecology of mammals: Relationships between density and patch size. J Mammal. 1997;78(4):999–1013.
Thornton DH, Branch LC, Sunquist ME. The influence of landscape, patch, and within-patch factors on species presence and abundance: a review of focal patch studies. Landsc Ecol. 2011;26(1):7–18.
Betts MG, Fahrig L, Hadley AS, et al. A species-centered approach for uncovering generalities in organism responses to habitat loss and fragmentation. Ecography. 2014;37(6):517–27.
Bender DJ, Contreras TA, Fahrig L. Habitat loss and population decline: a meta-analysis of the patch size effect. Ecology. 1998;79(2):517–33.
Dharmarajan G, Beasley JC, Fike JA, Rhodes OE. Effects of landscape, demographic and behavioral factors on kin structure: testing ecological predictions in a mesopredator with high dispersal capability. Anim Conserv. 2014;17(3):225–34.
Matthysen E. Density-dependent dispersal in birds and mammals. Ecography. 2005;28(3):403–16.
Wang IJ. Environmental and topographic variables shape genetic structure and effective population sizes in the endangered Yosemite toad. Divers Distrib. 2012;18(10):1033–41.
Pitra C, Suarez-Seoane S, Martin CA, Streich WJ, Alonso JC. Linking habitat quality with genetic diversity: a lesson from great bustards in Spain. Eur J Wildl Res. 2011;57(3):411–9.
Kahilainen A, Keranen I, Kuitunen K, Kotiaho JS, Knott KE. Interspecific interactions influence contrasting spatial genetic structures in two closely related damselfly species. Mol Ecol. 2014;23(20):4976–88.
Koen EL, Bowman J, Wilson PJ. Node-based measures of connectivity in genetic networks. Mol Ecol Resour. 2016;16(1):69–79.
Pflueger FJ, Balkenhol N. A plea for simultaneously considering matrix quality and local environmental conditions when analysing landscape impacts on effective dispersal. Mol Ecol. 2014;23(9):2146–56.
Frankham R. Genetics and extinction. Biol Conserv. 2005;126(2):131–40.
Reed DH, Frankham R. Correlation between fitness and genetic diversity. Conserv Biol. 2003;17(1):230–7.
Jackson ND, Fahrig L. Landscape context affects genetic diversity at a much larger spatial extent than population abundance. Ecology. 2014;95(4):871–81.
Ezard THG, Travis JMJ. The impact of habitat loss and fragmentation on genetic drift and fixation time. Oikos. 2006;114(2):367–75.
Bruggeman DJ, Wiegand T, Fernandez N. The relative effects of habitat loss and fragmentation on population genetic variation in the red-cockaded woodpecker (Picoides borealis). Mol Ecol. 2010;19(17):3679–91.
Cushman SA, Shirk A, Landguth EL. Separating the effects of habitat area, fragmentation and matrix resistance on genetic differentiation in complex landscapes. Landsc Ecol. 2012;27(3):369–80.
Varvio SL, Chakraborty R, Nei M. Genetic-variation in subdivided populations and conservation genetics. Heredity. 1986;57:189–98.
Anderson CD, Epperson BK, Fortin M-J, et al. Considering spatial and temporal scale in landscape-genetic studies of gene flow. Mol Ecol. 2010;19(17):3565–75.
Keyghobadi N, Roland J, Matter SF, Strobeck C. Among- and within-patch components of genetic diversity respond at different rates to habitat fragmentation: an empirical demonstration. Proc R Soc B Biol Sci. 2005;272(1562):553–60.
Balkenhol N, Pardini R, Cornelius C, Fernandes F, Sommer S. Landscape-level comparison of genetic diversity and differentiation in a small mammal inhabiting different fragmented landscapes of the Brazilian Atlantic Forest. Conserv Genet. 2013;14(2):355–67.
Lange R, Diekotter T, Schiffmann LA, Wolters V, Durka W. Matrix quality and habitat configuration interactively determine functional connectivity in a widespread bush cricket at a small spatial scale. Landsc Ecol. 2012;27(3):381–92.
Perez-Espona S, Perez-Barberia FJ, McLeod JE, Jiggins CD, Gordon IJ, Pemberton JM. Landscape features affect gene flow of Scottish Highland red deer (Cervus elaphus). Mol Ecol. 2008;17(4):981–96.
Stevens VM, Verkenne C, Vandewoestijne S, Wesselingh RA, Baguette M. Gene flow and functional connectivity in the natterjack toad. Mol Ecol. 2006;15(9):2333–44.
Paquette SR, Talbot B, Garant D, Mainguy J, Pelletier F. Modelling the dispersal of the two main hosts of the raccoon rabies variant in heterogeneous environments with landscape genetics. Evol Appl. 2014;7(7):734–49.
Engler JO, Balkenhol N, Filz KJ, Habel JC, Rodder D. Comparative landscape genetics of three closely related sympatric hesperid butterflies with diverging ecological traits. Plos One. 2014;9(9):e106526.
Amos JN, Harrisson KA, Radford JQ, et al. Species- and sex-specific connectivity effects of habitat fragmentation in a suite of woodland birds. Ecology. 2014;95(6):1556–68.
Poelchau MF, Hamrick JL. Differential effects of landscape-level environmental features on genetic structure in three codistributed tree species in Central America. Mol Ecol. 2012;21(20):4970–82.
Richardson JL. Divergent landscape effects on population connectivity in two co-occurring amphibian species. Mol Ecol. 2012;21(18):4437–51.
Shanahan DF, Possingham HP, Riginos C. Models based on individual level movement predict spatial patterns of genetic relatedness for two Australian forest birds. Landsc Ecol. 2011;26(1):137–48.
Prugh LR, Hodges KE, Sinclair ARE, Brashares JS. Effect of habitat area and isolation on fragmented animal populations. Proc Natl Acad Sci U S A. 2008;105(52):20770–5.
Moore JA, Tallmon DA, Nielsen J, Pyare S. Effects of the landscape on boreal toad gene flow: does the pattern-process relationship hold true across distinct landscapes at the northern range margin? Mol Ecol. 2011;20(23):4858–69.
Bull RAS, Cushman SA, Mace R, et al. Why replication is important in landscape genetics: American black bear in the Rocky Mountains. Mol Ecol. 2011;20(6):1092–107.
Gaggiotti OE, Foll M. Quantifying population structure using the F-model. Mol Ecol Resour. 2010;10(5):821–30.
Hanski I. A practical model of metapopulation dynamics. J Anim Ecol. 1994;63(1):151–62.
Lausch A, Blaschke T, Haase D, et al. Understanding and quantifying landscape structure - a review on relevant process characteristics, data models and landscape metrics. Ecol Model. 2015;295:31–41.
Acknowledgments
We thank A. Hadley for helpful discussion during the preparation of this manuscript, and M. Betts and two anonymous reviewers for their comments. This research was funded by the Natural Sciences and Engineering Research Council of Canada Discovery grant to HHW an Ontario Graduate Scholarship to MFD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Effects of Landscape Structure on Conservation of Species and Biodiversity
Rights and permissions
About this article

Cite this article
DiLeo, M.F., Wagner, H.H. A Landscape Ecologist’s Agenda for Landscape Genetics. Curr Landscape Ecol Rep 1, 115–126 (2016). https://doi.org/10.1007/s40823-016-0013-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40823-016-0013-x