
Abstract
Background
Mountains have not only provided refuge for species, but also offered dispersal corridors during the Neogene and Quaternary global climate changes. Compared with a plethora of studies on the refuge role of China’s mountain ranges, their dispersal corridor role has received little attention in plant phylogeographic studies. Using phylogeographic data of Eomecon chionantha Hance (Papaveraceae), this study explicitly tested whether the Nanling Mountains, which spans from west to east for more than 1000 km in subtropical China, could have functioned as a dispersal corridor during the late Quaternary in addition to a glacial refugium.
Results
Our analyses revealed a range-wide lack of phylogeographic structure in E. chionantha across three kinds of molecular markers [two chloroplast intergenic spacers, nuclear ribosomal internal transcribed spacer (nrITS), and six nuclear microsatellite loci]. Demographic inferences based on chloroplast and nrITS sequences indicated that E. chionantha could have experienced a strong postglacial range expansion between 6000 and 1000 years ago. Species distribution modelling showed that the Nanling Mountains and the eastern Yungui Plateau were the glacial refugia of E. chionantha. Reconstruction of dispersal corridors indicated that the Nanling Mountains also have acted as a corridor of population connectivity for E. chionantha during the late Quaternary.
Conclusions
Our results suggest that the Nanling Mountains may acted dual roles as a dispersal corridor in east-west direction and as a glacial refugium in subtropical China during the late Quaternary. The population connectivity mediated by the mountain range and a strong postglacial range expansion are the most likely reasons for the lack of phylogeographic structure in E. chionantha. The hypothesis of dual roles of the mountain range presented here sheds new insights into the phylogeographic patterns of organisms in subtropical China.
Similar content being viewed by others

Background
Given their extremely diverse topography and a wide spectrum of environmental conditions within short distances, mountains have generally provided refuge for species, especially during the Neogene and Quaternary global climate changes [1,2,3]. In addition, mountains have often offered dispersal corridors, allowing for range expansions/contractions, population connection, and gene flow between otherwise fragmented populations [1, 4, 5]. Both functions of refuge and dispersal corridor are extremely important for the persistence of organisms during periods of large climate change such as the late Cenzoic [6]. For example, many mid-elevation mountain ranges and hilly areas in southern and central Europe did not only provide shelters for the survival of Fagus sylvatica during the Quaternary glacial periods, but also facilitate the northward spread of the residual populations during inter−/postglacial periods, because beeches found their humid habitats that were absent on the great plains [7]. Such cases were also reported in the Korean Peninsula of east Asia. The uninterrupted Baekdudaegan Mountains, that runs from north to south over 1600 km, acted as a true dispersal corridor for a large assemblage of boreal and temperate elements during the Quaternary climate changes beyond serving as a refugium for many species [5].
Harboring more than 30,000 vascular plants, China is one of 17 world’s megadiversity countries [8]. Many Tertiary lineages (such as Cathaya, Ginkgo, Glyptostrobus, Metasequoia, and so forth) that were once widely distributed along the Northern Hemisphere are today endemic to China [9, 10]. An important reason is that a high percentage of the mountainous lands in the country (in China over 50% is mountainous [11]) served as refuge areas for those ancient lineages [2, 10, 12,13,14,15]. During the past decade, increasing molecular phylogeographic studies on Chinese plant species showed that climatic refugia identified by DNA-based data (reviewed in [16, 17]) are largely congruent with the centers of endemism [15], highlighting the sheltering function of China’s mountain ranges. However, compared with numerous studies on the refuge role of China’s mountain ranges, their function as dispersal corridors has received much less attention (but see [18, 19]). In addition, the mountain corridors in China have been mostly inferred subjectively according to the distribution patterns of certain taxa or genetic lineages, while very few have been tested explicitly using reproducible methods (but see [20]).
The Nanling Mountains sensu lato (i.e., including the mountains on the Guangxi-Guangdong and Hunan-Jiangxi borders) is a series of five connected mountain ranges that spans from west to east for more than 1000 km (Fig. 1c): Yuechengling, Dupangling, Mengzhuling, Qitianling and Dayuling mountains. Also known as the “Five Mountains”, and though not very high (average about 1000 m), the Nanling range is a natural dividing line in subtropical China, effectively separating the Yangtze (Changjiang) River drainage area (characterized by a central or north-subtropical climate) from the Pearl (Zhujiang) River drainage area (of south-subtropical or tropical climate). The Nanling Mountains is one of the key biodiversity hotspots in China [14, 21] and has long been viewed as a major glacial refugium [12, 16, 17] as well as an important ‘ice-free’ corridor in east-west direction [16, 18]. While its role as a glacial refugium has been demonstrated by different studies, robust evidence for the role of a corridor is still lacking.

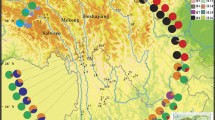
a Distribution of chloroplast haplotypes of Eomecon chionantha. b The network of 15 chloroplast haplotypes. c The distribution of Eomecon chionantha in China, the Nanling mountains is indicated with dark red color
Eomecon chionantha Hance is a perennial herb (10–30 cm in height) of the monotypic genus Eomecon from the poppy family (Papaveraceae, [22]). The Chinese name xueshuicao means ‘bleeding herb’ because its leaves and rhizomes ooze orange-red sap when bruised or cut. It is an entomophilous species (with bees and flies being the most efficient pollinators) that has mixed-mating system [23]. E. chionantha is a rhizomatous plant that also produces subterranean runners, which allows vegetative propagation. It is a potentially important economic plant given the molluscicidal activity of its alkaloids (they have shown to be active against Oncomelania hupensis , the only intermediate host of Schistosoma japonicum in China; [24]). E. chionantha always grows in shady and moist habitats along streams or trails in forests or at forest margins in mountainous areas (300–2000 m; [22]). The distribution of E. chionantha confines to subtropical China, where plenty of precipitation (800–1600 mm/year) is brought by the East Asian monsoon and Indian Monsoon. The Nanling Mountains are the southern limit of its distribution [12].
Such a distribution pattern and the close association with mountainous forests makes Eomecon a good case study to test whether the Nanling Mountains were both a major refugium and a mountain corridor for the persistence and connectivity of plant populations in subtropical China. Phylogeography, particularly in combination with species distribution modelling (SDM) and GIS-based methods, has been successfully used in locating glacial refugia and migratory corridors during the past decades [25, 26]. In this study, we investigated the phylogeographic pattern of E. chionantha using multi-loci genetic data, and then hindcasted the distribution at the Last Glacial Maximum (LGM) and Mid-Holocene (MH). At last, a recently developed method that integrates genetic and geospatial data (the categorical least cost path, CLCP [25]) was used to visualize the population connectivity of E. chionantha.
Methods
Population sampling and molecular procedure
Thirty-eight populations were collected after extensive field surveys throughout subtropical China from 2010 to 2014 (Table 1). Because this herb can spread through rhizomes to form patches, sampled individuals were spaced by ca. 20–50 m to avoid repeatedly collecting the same individual (i.e., ramets belonging to the same genet) except for a few small populations. All samples were desiccated in silica gel and stored at −20 °C until being processed. Genomic DNA was extracted using a modified CTAB procedure [27].
Two intergenic spacers (IGSs) of the chloroplast genome, rbcL-atpB [28] and trnC-rpoB [29], and nuclear ribosomal internal transcribed spacer (nrITS) [30] were amplified and sequenced. The PCR primers of the three genomic fragments are listed in Additional file 1. Amplification reactions were carried out in a volume of 20 μl, containing 10 μl 2 × Taq PCR MasterMix (Tiangen, Shanghai, China), 1 μl each forward and reverse primer (0.2 uM), 1 μl template DNA (ca. 50–100 ng) and 7 μl ddH2O. Amplification was carried out in a Bioer XP cycler (Bioer, Hangzhou, China) programmed for an initial step of 240 s at 94 °C, followed by 30 cycles of 60 s at 94 °C, 60 s at 50 °C (rbcL-atpB), 50 °C (trnC-rpoB), or 53 °C (nrITS), 60 s at 72 °C, and a final step of 600 s at 72 °C. Sequencing reactions were conducted with the corresponding forward and reverse primers commercially by Sangon Biotech Co., Ltd. (Shanghai, China). Six nuclear microsatellite (or nuclear simple sequence repeat, nSSR) loci (XSCO35, XSCO37, XSCO78, XSCQ13, XSCQ25, XSCQ60) that were specifically developed for E. chionantha were amplified and genotyped (primers and the PCR procedure were detailed in [31]). The PCR products were then separated on ABI 3730xl DNA Analyzer (Applied Biosystems, USA) to determine the precise fragment sizes at each of the six loci.
Phylogeographic and population genetic analyses
All sequences of chloroplast IGSs and nrITS were edited with Sequencher (GeneCodes Corporation, Ann Arbor, MI, USA) and were aligned using Clustal_X v1.81 [32]. Indels were discarded in subsequent analyses. Haplotype relationships were assessed using the median-joining network method [33] in Network v4.1.0.8 (http://www.fluxus-engineering.com). We used the program Haplonst [34] to calculate the total and within-population haplotype diversity (hT and hS), as well as population differentiation based on ordered (NST) and unordered (GST) haplotypes. The values of NST and GST were then compared using U-statistics to test for the presence of phylogeographic structure.
For the nSSR dataset, all six loci were checked for possible null alleles using Micro-checker v2.2.3 [35]. Deviations from Hardy-Weinberg Equilibrium (HWE) were performed with ARLEQUIN using an exact test [36] with 10,000 dememorization steps at species and population levels. Linkage disequilibrium (LD) among loci was tested using FSTAT v2.9.3 [37]. Statistical significance (α = 0.05) for inferring LD or departures from HWE was evaluated based on 1000 permutations, and corrected for multiple tests using the sequential Bonferroni method [38]. Genetic diversity was measured by allelic richness (AR, [39]), observed heterozygosity (HO), and expected heterozygosity (HE, [40]).
Genetic subgroups in the nSSR dataset were identified by a Bayesian analysis in STRUCTURE v2.3.1 [41] using the admixture model and assuming independent allele frequencies among populations. The number of clusters (K) was set to vary from one to 33. For each value of K, we performed 10 runs with a burn-in length of 10,000 and a run length of 100,000 Markov chain Monte Carlo (MCMC) replications. The mean log-likelihood for each value of K, [ln Pr(X|K)], and ΔK were employed to identify the most likely number of genetic clusters [42].
Because our STRUCTURE analysis did not included geographical information for the inference of population structure, we used a modified algorithm that includes spatially explicit prior distributions describing which sets of individuals are likely to have similar cluster membership [43]. In this approach, implemented in the program TESS v2.3 [44, 45], clusters corresponding to spatially and genetically continuous units separated by small discontinuities where genetic barriers are crossed were identified. We ran TESS with an admixture analysis for Kmax ranging from 2 to 9 (10 replicates for each value), with a burn-in period of length 20,000 cycles and estimation of length 30,000 additional cycles. For each run, we computed the Deviance Information Criterion (DIC), a model-complexity-penalized measure of how well the model fits the data. The smallest DIC values were obtained for the maximum number of clusters that best suits the data. We averaged the estimated admixture coefficients (Q matrix) over the ten runs with the smallest values of the DIC using the software Microsoft Office Excel 2007.
Demographic inference
We calculated the changes in effective population size for chloroplast and nuclear sequences separately by means of Bayesian skyline plots (BSP) in Beast v1.8.2 [46]. The combined analysis of chloroplast and nuclear sequences using the extended Bayesian skyline plot [47] was not applicable because the effective sample sizes (ESS) of the analysis were low (<100). MCMC chains were run for 10,000,000 generations for chloroplast sequences under GTR + G model and nrITS sequences under the HKY + G model chosen by jModelTest v2.1.5 [48]. The substitution rate of nrITS sequences (4.16 × 10−9 s/s/y) reported for Meconopsis of the Papaveraceae family [49] was used to date the demographic events. The synonymous substitution rate of sugarcane, maize and rice chloroplast genomes (1.52 × 10−9 s/s/y) was adopted for the two chloroplast IGSs [50].
Species distribution modelling and population connectivity
Assuming the species has not changed its climatic preference through time (i.e., niche conservatism), we reconstructed the potential range of E. chionantha at the LGM, MH and the present time according to its current distribution using a maximum entropy model (Maxent v3.1.0, [51]). The current distribution information for E. chionantha was obtained from collection records from the Chinese Virtual Herbarium (CVH, http://www.cvh.ac.cn/) combined with the sampling sites in this study. A total of 87 presence records were used for SDM construction. Current (1950–2000) and past [LGM ~ 21,000 yr. BP and MH ~6000 yr. BP] bioclimatic variables were downloaded from the WorldClim database (http://www.worldclim.org/) at 2.5-arcmin resolution. We used LGM and MH data simulated by the Community Climate System Model (CCSM; [52]). To aviod over-fitting of niche models, 6 variables with pairwise Pearson correlation coefficients r ≤ 0.5 (BIO1, Annual Mean Temperature; BIO3, Isothermality; BIO4, Temperature Seasonality; BIO7, Temperature Annual Range; BIO8, Mean Temperature of Wettest Quarter; BIO14, Precipitation of Driest Month) were retained for SDM analyses. Each model was run 10 times using the default parameters (convergence threshold of 10−5, maximum iterations of 500 and regularization multiplier of 1) and the following user-selected features: application of a random seed, duplicate presence records removal and logistic probabilities used for the output. Eighty percent of species records were used to train the model and 20% to test the model.
Putative dispersal corridors for population connectivity in E. chionantha were constructed by combining haplotype or ribotype networks and SDMs. We inverted the SDM to a “dispersal cost” layer (i.e., high probability of occurrence in the SDM has a low cost to dispersal through that region). Then, we generated a population connectivity map by summing the least cost paths (LCPs) among all shared and sister haplotypes or ribotypes from different populations in ArcGIS v9.3 (ESRI, Redlands, CA, USA) using the dispersal cost as the friction layer. The LCPs were classified into three categories: the lowest 1% LCPs, the lowest 2% LCPs, the lowest 5% LCPs and then subsequently summed each pairwise comparison [25]. Areas with the hottest colors are those hypothesized to offer the greatest ease of dispersal.
Results
Chloroplast DNA diversity and phylogeographic structure
The aligned length of E. chionantha was 796 bp for rbcL-atpB and 1142 bp for trnC-rpoB. Three and 10 substitutions were found in rbcL-atpB (GenBank accession numbers: KP658781-KP658787) and trnC-rpoB (GenBank accession numbers: KP6587811-KP658824), respectively. Six indels (4–20 bp) were discarded in subsequent analyses, resulting in a reduced length of 1876 bp of the pooled chloroplast sequences. Fifteen chloroplast haplotypes (chlorotype H1–15) were identified in 38 populations. At the species level, the cpDNA data revealed moderate haplotype diversity (hT = 0.566). Haplotype diversity at population level varied from 0 to 0.667, with the highest occurring in populations 22 and 35 (Table 1).
The chlorotype network revealed that most haplotypes were separated by one mutation to the two most common haplotypes, H1 and H2 (Fig. 1b); the only exception was H9, that was derived from H8 by one step. H1 occurred exclusively in east populations (10 populations), except for a southwest population (population 2, P2) and a west population (P9). H2 was more widespread (20 populations), but with higher frequencies in the species’ western range. Up to 10 haplotypes were exclusive to a single population, whereas one (H3) was shared by two populations (P24 and P25) and one (H8) by three populations (P33, P34 and P37) (Fig. 1a). Phylogeographic structure tested by Haplonst was not obvious in E. chionantha, because NST (0.792) was not significantly larger than GST (0.780) (U = 1.64, P < 0.05).
nrITS diversity and phylogeographic structure
A total of 301 nrITS sequences across 213 individuals from 31 populations were obtained. The aligned length was 725 bp with 25 nucleotide substitutions and two indels (that were discarded subsequently). Fifteen nuclear ribosomal ITS types (ribotypes, R1-R15) were identified among all samples surveyed (GenBank accession numbers: KP658788-KP658802).
The network of 15 ribotypes was rather complex, without any obviously differentiated clade (Fig. 2b). Three most common ribotypes (R1, R2 and R3) were successively connected by one mutation. The most common ribotype (R2) occurred across the whole range of E. chionantha, being only absent in 14 populations (45.2%). The second most common ribotype (R3) occurred mainly in central populations, whereas the third most common (R1) largely occurred in the eastern populations. The remaining ribotypes were either population-specific or regionally restricted (Fig. 2a). Phylogeographic structure was not significant at the nrITS locus (NST = 0.719, GST = 0.635, U = 0.82, P < 0.05).

a Distribution of ITS ribotypes of Eomecon chionantha. b The network of 15 ribotypes
Nuclear microsatellite diversity and population structure
Using Micro-checker, the frequency of the possible null alleles at each of the six loci surveyed was found to be lower than the threshold frequency (v = 0.12) across 389 individuals belonging to the 33 populations that were successfully genotyped. There was no evidence for LD. None of the six loci deviated from HWE significantly when all samples were treated as a single population.
A total of 117 alleles were detected at the six loci resolved for E. chionantha. The observed alleles per locus ranged from three to 15, and the alleles per locus across populations ranged from 1.17 to 8.64 (data not shown). Within-population allelic richness (AR) and expected heterozygosity (HE) ranged from 1.10 to 1.83 and from 0.083 to 0.805, respectively. Regression analyses showed that either AR or HE was dependent on latitude or longitude (P = 0.16).
Genetic differentiation at six microsatellite loci was moderate (GST = 0.367). In the Bayesian analysis of population structure, we were not able to find a meaningful K value, indicating there is no evidence of genetic cluster in E. chionantha (Additional file 2). For TESS analyses on SSR data, although DIC values were the smallest when Kmax = 6 (Additional file 2), the clusters had not geographical association. Therefore, TESS did not find any geographically meaningful genetic cluster either.
Demographic history based on cpDNA and nrITS sequence data
Both BSPs inferred from chloroplast and nrITS data revealed that E. chionantha experienced a recent population expansion (Fig. 3) although a weak decline in population size for nrITS occurred prior to the expansion. When the substitution rate (4.16 × 10−9 s/s/y) of nrITS was applied, this recent population expansion was dated to begin about 1000 years ago. While we assumed the substitution rate of the two chloroplast IGSs as 1.52 × 10−9 s/s/y, the population expansion event happened approximately at 6 ka.

Bayesian skyline analysis based on cpDNA and nITS sequence of Eomecon chionantha
Present and past species distribution models
The AUC value for the current potential distribution of E. chionantha was high (0.987), indicating good predictive model performance. The predicted distribution of the species under current conditions was generally similar to its actual distribution, with a potentially continuous range from Wuyi Mountains through Nanling Mountains to Yungui Plateau (Fig. 4a). Notably, the LGM potential range of E. chionantha was similar to the present one, thus maintaining the connectivity between the three geographic units. The only exception was a moderate contraction at the northeast and northwest tips compared to the present potential range (Fig. 4b). The MH potential range was much larger but more fragmented than the present and the LGM ones. The most suitable habitats in the Nanling Mountains obviously contracted and somewhat moved to the north at the MH (Fig. 4c).

a, b, c Potential distribution of Eomecon chionantha at present, the Last Glacial Maximum (LGM) and the Mid Holocene, respectively. The climate data at LGM and MH were simulated by the Community Climate System Model (CCSM4). d, e, f cpDNA dispersal corridor of E. chionantha at the present, the LGM and the MH, respectively. g, h, i nITS dispersal corridor of E. chionantha at the present, the LGM and the MH, respectively. Warmer color depicts higher population connectivity
Population connectivity
Both cpDNA and nrITS dispersal maps generated by the method of Chan et al. [25] showed that the most probable dispersal corridor at the LGM and the present (the areas with the hottest color) largely coincided with the Nanling Mountains, slightly extending to the Wuyi Mountains on cpDNA dispersal maps for both the present and LGM (Fig. 4d, e) or to the Luoxiao Mountains on nrITS present dispersal map (Fig. 4g). The most probable dispersal corridor at the MH slightly moved to the north edge of the Nanling Mountains (Fig. 4f, i), and the population connection in the east on nrITS dispersal map was weak (Fig. 4i).
Discussion
Genetic diversity and phylogeographic structure of Eomecon chionantha
No matter what molecular markers (chloroplast DNA sequences, nrITS sequences, nuclear microsatellites) were used, this study was not able to find obvious phylogeographic structure in E. chionantha (i.e., gene genealogies or genetic clusters not associated with geography), although genetic differentiation is quite high. This finding is at odds with the pronounced phylogeographic structure reported for most plant species in subtropical China that probably resulted from long-term survival in multiple Quaternary refugia and limited inter/postglacial expansions [15, 16, 53, 54]. According to recent studies, however, some plant species in this region may have experienced strong inter/postglacial range expansions, resulting in the lack of phylogeographic structure (Sargentodoxa cuneata [19]; Castanopsis tibetana and Machilus thunbergii [55]; Loropetalum chinense [56]; Cyclocarya paliurus, [57]). Theoretically, phylogeographic structure often arises as a consequence of restricted gene flow among populations and genetic drift within populations over relatively long periods of time [34, 58]. Although temporary genetic differentiation can arise quickly at the leading edge during range expansion due to different founders in patches [59], gene flow mediated by subsequent expanding populations may bring distantly related alleles into the same population and thus erode phylogeographic structure [34, 60]. Modelling and simulations also showed that rapid range expansion (such as leptokurtic dispersal) can produce large areas of homozygosity and these homogeneous areas may persist in space and increase with time [61]. As exemplified, Petit et al. [62] found that newly colonized populations of forest trees in north Europe displayed much weaker phylogeographic structure than those in southern glacial refugia. In this study, the demographic inferences suggest a strong (mainly northward) range expansion from putative southern refugia (located in Nanling Mountains) that could have happened at ca. 1000 to 6000 years ago, although we should caution the wide confidence in the BPS analyses. These expansions, however, could have been preceded by slight population decreases starting around the LGM (ca. 21,000 yr. BP) (Fig. 3). Therefore, it is most likely that rapid postglacial range expansion may be one of the causal factors for the lack of phylogeographic structure in E. chionantha. This expansion, however, occurred relatively late (starting about 6000 yr. BP), probably because the fully subtropical vegetation did not return to the Nanling Mountains and also the current range of E. chionantha at least until the onset of the Holocene Optimum, ca. 8000 yr. BP [63,64,65].
Even if E. chionantha have experienced an extensive northward expansion, the lack of range-wide phylogeographic structure remains elusive. In the well-studied Europe, for example, the southern range of plant species have often displayed pronounced phylogeographic structure due to long-term isolation among three peninsular glacial refugia (Iberian, Italian, and the Balkan Peninsulas) [62, 66, 67]. However, subtropical China and Europe have rather distinct physiographical characteristics that may have differential effects on phylogeographic structure. To the south of Europe, the Mediterranean Sea formed an insurmountable barrier and the three isolated peninsulas were the “last-stop” for most retreating populations during the glacial periods [68]. In contrast, subtropical China is a continuous landmass without being interrupted by seas, and has no major geographic barriers because the mountains of southern China are usually below 1500 m. The retreat to the south of many subtropical elements (such as E. chionantha) at the glacial periods would have ended at the Nanling Mountains, where they would have found suitable habitats. At the same time, the east-west orientation of such mountains would have allowed the connection of these refugial populations, thus lowering population differentiation. Our dispersal corridor maps based on both chloroplast and nuclear ITS data are rather coincident, both revealing considerable levels of connectivity across the Nanling Mountains. It can be concluded, therefore, that strong population connectivity maintained by the Nanling Mountains during the late Quaternary may have contributed, at least in part, to the lack of phylogeographic structure in E. chionantha.
The existence of cryptic refugia beyond the southern major refugia can also contribute to phylogeographic structure of plant species [62, 69]. Based on our phylogeographic data, there may be some cryptic refugia for E. chionantha beyond the Nanling Mountains, because populations P1, P2, P8 and P22 possess multiple and/or unique chloroplast or/and ITS haplotypes/ribotypes (and also show relatively high levels of microsatellite diversity; Table 1). However, as these haplotypes rarely occur in other populations, these populations may have contributed little to the overall genetic component of E. chionantha, thus having limited effects on the overall phylogeographic structure.
The dual functions of the Nanling Mountains
Mountains provide a diverse range of thermal, hydric and edaphic conditions that would have allowed mesic habitats to be present even during the coldest, most arid phases of the glacial periods of the Pleistocene (i.e. glacial maxima); mountains, therefore, are likely to have acted as refugia for many species in both temperate and tropical regions [67, 70, 71]. In two excellent reviews of plant phylogeography in China by Qiu et al. [16] and Liu et al. [17], the Nanling Mountains are regarded as one of the major refugia in China. In this study, SDMs at the LGM, MH, and the present indicate that the habitats throughout the late Quaternary within this mountain range were suitable for the survival of E. chionantha. In addition, we found two ancestral (H1 and H2) and one private chlorotypes (H15) in the mountain range, further suggesting that the mountain range was also the refugial area for E. chionantha.
However, the number of haplotypes and genetic diversity hold by the Nanling Mountains’ populations are relatively low (only two populations within the Nanling mountains, P12 and P19, have HE values above the mean for the species, 0.512; Table 1), contradicting the prediction that glacial refugia always harbor a large fraction of the intraspecific biodiversity ([67,68,69, 72]; but see [62]). Two factors may account for the relatively low genetic diversity in populations of the Nanling Mountains compared to other parts of E. chionantha’s range. First, populations in refugia are not necessarily the highest in genetic diversity because southern or ‘rear-edge’ populations may have lost diversity through processes such as genetic drift and diminished gene flow. For example, refugial southern populations in Europe, albeit genetically unique, were relatively impoverished in their intrapopulation genetic diveristy due to isolation in small populations [62, 73]. Second, the existence of cryptic refugia further north (see above) and/or the meeting of genetically divergent migratory routes (i.e., the ‘melting pot’ effect; [62, 74]) may also explain why some northern populations show very high levels of genetic diversity (e.g. populations P8, P9 and P14 show HE values even higher than 0.700; Table 1).
Compared with the refugial function, the corridor role of the Nanling Mountains has only been alluded to in early literature [16, 18]. From the results of the present study, we may conclude that the population connectivity is a major cause for the lack of phylogeographic structure in E. chionantha. At least two factors could explain why the Nanling Mountains served as a major dispersal corridor for plant species during the late Quaternary in addition to its role as refugium. First, the Nanling Mountains is the only mountain range in southern China that shows an east-west direction; With such orientation, the Nanling Mountains enabled the connection between Yungui Plateau, the main plateau in southwestern China, and Wuyi Mountains, the main mountain range of East China. Indeed, the connection of the three main orographic units of southern China is shown not only by the maps of population connectivity of E. chionantha, but also by the SDMs of our previously investigated plants (Castanopsis tibetana, Machilus thunbergii, and Schima superba, Fig. 4; [55]). This connection allowed plants to have several recolonization routes from the Nanling glacial refugium (summarized in [19], using Sargentodoxa cuneata as a case-study): (i) one towards the north-west through the Yungui Plateau; (ii) another towards the east through Wuyi Mountains, and (iii) a third route through Luoxiao Mountains.
Although plains might also be utilized by plants as dispersal corridors, mountains were much more effective for plant dispersal because the climate at low elevations was very harsh (cold and arid) during the LGM [75], causing the range fragmentation of moist forest species into residual populations within mountains where the diverse habitats may facilitate their dispersal. For example, populations of Tsuga chinensis in southeast China separated by the Middle-Lower Yangtze Plains are distinct in genetic composition; however, those within the Nanling Mountains are much more genetically homogeneous [76]. This pattern was also observed in Tapiscia sinensis [77]. In contrast, mountains were relatively moist due to orographic rainfall during the glacial periods [5] and thus facilitated plant dispersal.
In addition to the orientation, the geographic location of Nanling Mountains would have also accounted for its dual role as refugium and dispersal corridor. The mountain range is located at relatively low latitude (24–26°N), the climate in this area was warm enough (mean annual temperature only 4 °C lower than the present [78]) for the persistence of plant species. More importantly, the Nanling Mountains received almost the same annual precipitation at the LGM compared to the present (the reduction was below 10%), as occurred for most of the latitudes of China located below 28°N [78]. The amplitude of precipitation is especially important for plant species like E. chionantha that dwells in moist forests in subtropical China, because they are adapted to the humid climate dominated by the East Asian Monsoon.
Conclusions
We detected a lack of phylogeographic structure in a forest-dwelling herb, Eomecon chionantha, in subtropical China using three kinds of molecular markers. Such a phylogeographic pattern may be caused by the combined effects of a late (but rapid) postglacial expansion refugia located in the Nanling Mountains and strong population connectivity mediated by this mountain range. The hypothesis of the dual role of the Nanling Mountains provides a novel perspective for understanding the phylogeographic pattern of organisms in subtropical China. The results of this study further justify the importance of conserving the natural vegetation of the Nanling Mountains, that constitute today one of the main biodiversity hotspots in China.
Abbreviations
- BSP:
-
Bayesian skyline plots
- CCSM:
-
Community Climate System Model
- Chlorotype:
-
Chloroplast haplotype
- CVH:
-
Chinese Virtual Herbarium
- DIC:
-
Deviance Information Criterion
- ESS:
-
Effective sample sizes
- HWE:
-
Hardy-Weinberg Equilibrium
- IGS:
-
Intergenic spacers
- LCP:
-
Least cost paths
- LD:
-
Linkage disequilibrium
- LGM:
-
Last Glacial Maximum
- MH:
-
Mid-Holocene
- nrITS:
-
nuclear ribosomal internal transcribed spacer
- nSSR:
-
nuclear simple sequence repeat
- SDM:
-
Species distribution modelling
References
Spehn EM, Rudmann-Maurer K, Korner C, Maselli D, editors. Mountain biodiversity and global change. Basel: GMBA-DIVERSITAS; 2010.
López-Pujol J, Zhang FM, Sun HQ, Ying TS, Ge S. Centers of plant endemism in China: places for survival or for speciation? J Biogeogr. 2011;38:1267–80.
Feliner GN. Patterns and processes in plant phylogeography in the Mediterranean Basin. A review. Perspect Plant Ecol Evol Syst. 2014;16:265–78.
Swenson NG, Howard DJ. Clustering of contact zones, hybrid zones, and phylogeographic breaks in North America. Am Nat. 2005;166:581–91.
Chung MY, López-Pujol J, Chung MG. The role of the Baekdudaegan (Korean peninsula) as a major glacial refugium for plant species: a priority for conservation. Biol Conserv. 2017;206:236–48.
Zachos J, Pagani M, Sloan L, Thomas E, Billups K. Trends, rhythms, and aberrations in global climate 65 ma to present. Science. 2001;292:686–93.
Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gömöry D, et al. A new scenario for the quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol. 2006;171:199–221.
Mittermeier RA, Gil PR, Mittermeier CG. Megadiversity: earths biologically wealthiest nations. In: Gil PR, Wilson EO, editors. CEMEX conservation book series. México City: CEMEX; 1997.
Axelrod DI, Al-Shehbaz I, Raven PH. History of the modern flora of China. Floristic characteristics and diversity of East Asian plants. In: Zhang A, Wu S, editors. Foristic Characteristics and Diversity of East Asian Plants. Beijing: China Higher Education Press; 1996. p. 43–55.
Ying TS. Species diversity and distribution pattern of seed plants in China [in Chinese with English abstract]. Biodivers Sci. 2001;9:393–8.
Kohler T, Giger M, Hurni H, Ott C, Wiesmann U, von Dach SW, et al. Mountains and climate change: a global concern. Mt Res Dev. 2010;30:53–5.
Ying TS, Zhang YL, Boufford DE. The endemic genera of seed plants of China. Beijing: Science Press; 1993.
Wang HS, Zhang YL. The bio-diversity and characters of spermatophytic genera endemic to China [in Chinese with English abstract]. Acta Bot Yunnan. 1994;16:209–20.
Tang ZY, Wang ZH, Zheng CY, Fang JY. Biodiversity in China’s mountains. Front Ecol Environ. 2006;4:347–52.
López-Pujol J, Zhang FM, Sun HQ, Ying TS, Ge S. Mountains of southern China as “plant museums” and “plant cradles”: evolutionary and conservation insights. Mt Res Dev. 2011;31:261–9.
Qiu YX, CX F. Comes HP. plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of quaternary climate and environment change in the world’s most diverse temperate flora. Mol Phylogenet Evol. 2011;59:225–44.
Liu JQ, Sun YS, Ge XJ, Gao LM, Qiu YX. Phylogeographic studies of plants in China: advances in the past and directions in the future. J Syst Evol. 2012;50:267–75.
Wang WT. On some distribution patterns and some migration routes found in the eastern Asiatic region. Acta Phytotaxon Sin. 1992;30:1–24.
Tian S, Lei SQ, Hu W, Deng LL, Li B, Meng QL, et al. Repeated range expansions and inter−/postglacial recolonization routes of Sargentodoxa cuneata (Oliv.) Rehd. Et Wils. (Lardizabalaceae) in subtropical China revealed by chloroplast phylogeography. Mol Phylogenet Evol. 2015;85:238–46.
Yu H, Zhang Y, Liu L, Qi W, Li S, Combing HZ. The least cost path method with population genetic data and species distribution models to identify landscape connectivity during the late quaternary in Himalayan hemlock. Ecol Evol. 2015;5:5781–91.
Huang JH, Chen B, Liu C, Lai JS, Zhang JL, Ma KP. Identifying hotspots of endemic woody seed plant diversity in China. Divers Distrib. 2012;18:673–88.
Zhang M, Su Z, Lidén M, Grey-Wilson C. Papaveraceae. In: Wu ZY, Raven P, editors. Flora of China, vol. 32. Beijing: Science Press; 1999. p. 76.
Xiao Y, Chen X, XH H, Dong M. Pollination ecology of Eomecon chionantha Hance (Papaveraceae), an endemic species in China. Russ J Ecol. 2016;47:249–58.
Peng F, Liu M, Huang Q, Liu N, Yang H, Sun H, et al. Molluscicidal effect of Eomecon chionantha alkaloids against Oncomelania hupensis snails. Southeast Asian J Trop Med Public Health. 2011;42:289–96.
Chan LM, Brown JL, Yoder AD. Integrating statistical genetic and geospatial methods brings new power to phylogeography. Mol Phylogenet Evol. 2011;59:523–37.
Gavin DG, Fitzpatrick MC, Gugger PF, Heath KD, Rodríguez-Sánchez F, Dobrowski SZ, et al. Climate refugia: joint inference from fossil records, species distribution models and phylogeography. New Phytol. 2014;204:37–54.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–5.
Doi K, Kaga A, Tomooka N, Vaughan DA. Molecular phylogeny of genus Vigna subgenus Ceratotropis based on rDNA ITS and atpB-rbcL intergenic spacer region of cpDNA sequences. Genetica. 2002;114:129–45.
Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J, et al. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot. 2005;92:142–66.
Moller M, Cronk Q. Origin and relationships of Saintpaulia (Gesneriaceae) based on ribosomal DNA internal transcribed spacer (ITS) sequences. Am J Bot. 1997;84:956–65.
Wang J, Hu W, Yang Y, Xiao K, Fan DM, Zhang ZY. Isolation and characterization of microsatellite markers for Eomecon chionantha, a monotypic species endemic to China. Plant Sci J. 2015;33:855–60.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82.
Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Pons O, Petit RJ. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996;144:1237–45.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004;4:535–8.
Guo SW, Thompson EA. Performing the exact test of hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–72.
Goudet J. FSTAT, version 2.9.3.2. A program to estimate and test gene diversities and fixation indices. 2001. https://www2.unil.ch/popgen/.
Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–5.
El Mousadik A, Petit RJ. High level of genetic differentiation for allelic richness among populations of the argan tree Argania spinosa (L.) Skeels endemic to Morocco. Theor Appl Genet. 1996;92:832–9.
Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89(3):583–90.
Pritchard JK, Wen X, Falush D. Documentation for Structure software: Version 2.3. 2009. http://pritch.bsd.uchicago.edu/structure.html
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
François O, Ancelet S, Guillot G. Bayesian clustering using hidden Markov random fields in spatial population genetics. Genetics. 2006;174:805–16.
Chen C, Durand E, Forbes F, François O. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes. 2007;7:747–56.
Durand E, Chen C, François O. Comment on ‘on the inference of spatial structure from population genetics data’. Bioinformatics. 2009;25:1802–4.
Drummond AJ, Suchard MA, Xie D, Rambaut A, Rambaut A. Bayesian phylogenetics with BEAUTi and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Heled J, Drummond AJ. Bayesian inference of population size history from multiple loci. BMC Evol Biol. 2008;8:289.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772.
Yang FS, Qin AL, Li YF, Wang XQ. Great genetic differentiation among populations of Meconopsis integrifolia and its implication for plant speciation in the Qinghai-Tibetan plateau. PLoS One. 2012;7(5):e37196.
Yamane K, Yano K, Kawahara T. Pattern and rate of indel evolution inferred from whole chloroplast intergenic regions in sugarcane, maize and rice. DNA Res. 2006;13:197–204.
Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecol Model. 2006;190:231–59.
Gent PR, Danabasoglu G, Donner LJ, Holland MM, Hunke EC, Jayne SR, et al. The community climate system model version 4. J Clim. 2011;24:4973–91.
Lei M, Wang Q, ZJ W, López-Pujol J, Li DZ, Zhang ZY. Molecular phylogeography of Fagus engleriana (Fagaceae) in subtropical China: limited admixture among multiple refugia. Tree Genet Genomes. 2012;8:1203–12.
Zhang ZY, Wu R, Wang Q, Zhang ZR, Fan DM, López-Pujol J, et al. Comparative phylogeography of two sympatric beeches in subtropical China: species-specific geographic mosaic of lineages. Ecol Evol. 2013;3:4461–72.
Fan DM, Hu W, Li B, Morris AB, Zheng M, Soltis DE, et al. Idiosyncratic responses of evergreen broad-leaved forest constituents in China to the late quaternary climate changes. Sci Rep. 2016;6:31044.
Gong W, Liu W, Gu L, Kaneko S, Koch MA, Zhang D. From glacial refugia to wide distribution range: demographic expansion of Loropetalum chinense (Hamamelidaceae) in Chinese subtropical evergreen broadleaved forest. Org Divers Evol. 2016;16:23–38.
Kou YX, Cheng SM, Tian S, Li B, Fan DM, Chen Y, et al. The antiquity of Cyclocarya paliurus (Juglandaceae) provides new insights into the evolution of relict plants in subtropical China since the late early-Miocene. J Biogeogr. 2016;43:351–60.
Frankham R, Ballou JD, Briscoe DA. Introduction to conservation genetics. 2nd ed. Cambridge: Cambridge University Press; 2010.
Petit RJ, Pineau E, Demesure B, Bacilieri R, Ducousso A, Kremer A. Chloroplast DNA footprints of postglacial recolonization by oaks. Proc Natl Acad Sci U S A. 1997;94:9996–10001.
Short KH, Petren K. Fine-scale genetic structure arises during range expansion of an invasive gecko. PLoS One. 2011;6:e26258.
Ibrahim KM, Nichols RA, Hewitt GM. Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity. 1996;77:282–91.
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, et al. Glacial refugia: hotspots but not melting pots of genetic diversity. Science. 2003;300:1563–5.
Ni J, Cao X, Jeltsch F, Herzschuh U. Biome distribution over the last 22,000 yr in China. Palaeogeogr Palaeocl Palaeecol. 2014;409:33–47.
Cao X, Herzschuh U, Ni J, Zhao Y, Böhmer T. Spatial and temporal distributions of major tree taxa in eastern continental Asia during the last 22,000 years. The Holocene. 2015;25:79–91.
Zhong W, Cao J, Xue J, Ouyang J. Last deglacial and Holocene vegetation evolution and climate variability in the subalpine western Nanling Mountains in South China. The Holocene. 2015;25:1330–40.
Comes HP, Kadereit JW. The effect of quaternary climatic changes on plant distribution and evolution. Trends Plant Sci. 1998;3:432–8.
Hewitt GM. The genetic legacy of the quaternary ice ages. Nature. 2000;405:907–13.
Hewitt GM. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc. 1996;58:247–76.
Provan J, Bennett KD. Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol. 2008;23:564–71.
Hewitt GM. Genetic consequences of climatic oscillation in the quaternary. Philos Trans R Soc B. 2004;359:183–95.
Byrne M. Evidence for multiple refugia at different time scales during Pleistocene climatic oscillations in southern Australia inferred from phylogeography. Quaternary Sci Rev. 2008;27:2576–85.
Feliner GN. Southern European glacial refugia: a tale of tales. Taxon. 2011;60:365–72.
Hampe A, Petit RJ. Conserving biodiversity under climate change: the rear edge matters. Ecol Lett. 2005;8:461–7.
Stewart JR, Lister AM, Barnes I, Dalen L. Refugia revisited: individualistic responses of species in space and time. Philos T R Soc B. 2010 277: 661-671.
Harrison SP, Yu G, Takahara H, Prentice IC. Palaeovegetation (communications arising): diversity of temperate plants in east Asia. Nature. 2001;413:129–30.
Cun YZ, Wang XQ. Phylogeography and evolution of three closely related species of Tsuga (hemlock) from subtropical eastern Asia: further insights into speciation of conifers. J Biogeogr. 2015;42:315–27.
Zhang J, Li Z, Fritsch PW, Tian H, Yang A, Yao X. Phylogeography and genetic structure of a tertiary relict tree species, Tapiscia sinensis (Tapisciaceae): implications for conservation. Ann Bot. 2015;116:727–37.
Tian Z, Jiang D. Revisiting last glacial maximum climate over China and east Asian monsoon using PMIP3 simulations. Palaeogeogr Palaeocl Palaeecol. 2016;453:115–26.
Acknowledgements
We are grateful to the people who provided help with the field work. Special thanks go to Shupeng Dong, Bo Li, and Yi Luo for their participation in sample collection. We are also indebted to Wan Hu and Min Zheng for their laboratory assistance.
Funding
This study was supported by grants from the National Natural Science Foundation of China (grant nos. 41,061,006, 31,560,064).
Availability of data and materials
Genome sequences of Eomecon chionantha generated during the current study are available in the NCBI GenBank, under the accession numbers: KP658781-KP658787, KP6587811-KP658824, KP658788-KP658802. The data sets supporting the results and conclusions of this article are available in the Additional files 1 and 2.
Author information
Authors and Affiliations
Contributions
ZyZ, DF and ST designed the study. ZyZ, DF and DL collected the samples. ZrZ and LY performed the experiments. ST and YK performed the analyses. ZyZ, DF, ST, YK and JLP wrote and/or critically reviewed the manuscript. All authors read, provided revisions and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
PCR primers of two chloroplast intergenic spacers and nuclear ribosomal internal transcribed spacer in Eomecon chionantha. (PDF 73 kb)
Additional file 2:
a. The mean log-likelihood for each value of K, [ln Pr(X|K)], and ΔK in STRUCTURE analysis on nSSR data of Eomecon chionantha. b. DIC as a function of Kmax for nSSR in TESS analysis. (PDF 110 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tian, S., Kou, Y., Zhang, Z. et al. Phylogeography of Eomecon chionantha in subtropical China: the dual roles of the Nanling Mountains as a glacial refugium and a dispersal corridor. BMC Evol Biol 18, 20 (2018). https://doi.org/10.1186/s12862-017-1093-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-017-1093-x