
Abstract
Background
Chemolithoautotrophic primary production sustains dense invertebrate communities at deep-sea hydrothermal vents and hydrocarbon seeps. Symbiotic bacteria that oxidize dissolved sulfur, methane, and hydrogen gases nourish bathymodiolin mussels that thrive in these environments worldwide. The mussel symbionts are newly acquired in each generation via infection by free-living forms. This study examined geographical subdivision of the thiotrophic endosymbionts hosted by Bathymodiolus mussels living along the eastern Pacific hydrothermal vents. High-throughput sequencing data of 16S ribosomal RNA encoding gene and fragments of six protein-coding genes of symbionts were examined in the samples collected from nine vent localities at the East Pacific Rise, Galápagos Rift, and Pacific-Antarctic Ridge.
Results
Both of the parapatric sister-species, B. thermophilus and B. antarcticus, hosted the same numerically dominant phylotype of thiotrophic Gammaproteobacteria. However, sequences from six protein-coding genes revealed highly divergent symbiont lineages living north and south of the Easter Microplate and hosted by these two Bathymodiolus mussel species. High heterogeneity of symbiont haplotypes among host individuals sampled from the same location suggested that stochasticity associated with initial infections was amplified as symbionts proliferated within the host individuals. The mussel species presently contact one another and hybridize along the Easter Microplate, but the northern and southern symbionts appear to be completely isolated. Vicariance associated with orogeny of the Easter Microplate region, 2.5–5.3 million years ago, may have initiated isolation of the symbiont and host populations. Estimates of synonymous substitution rates for the protein-coding bacterial genes examined in this study were 0.77–1.62%/nucleotide/million years.
Conclusions
Our present study reports the most comprehensive population genetic analyses of the chemosynthetic endosymbiotic bacteria based on high-throughput genetic data and extensive geographical sampling to date, and demonstrates the role of the geographical features, the Easter Microplate and geographical distance, in the intraspecific divergence of this bacterial species along the mid-ocean ridge axes in the eastern Pacific. Altogether, our results provide insights into extrinsic and intrinsic factors affecting the dispersal and evolution of chemosynthetic symbiotic partners in the hydrothermal vents along the eastern Pacific Ocean.
Similar content being viewed by others


Background
Symbiosis involving chemoautolithotrophic bacteria plays a predominant role supporting the diverse invertebrate fauna that flourishes at deep-sea hydrothermal vents, hydrocarbon seeps, whale and wood falls, and organically enriched shallow water sediments [1]. The invertebrate animals use three different means to acquire the symbionts. For example, vent clams in the family Vesicomyidae, transmit their bacteria vertically through an ovarial pathway [2,3,4], though some species might acquire bacteria laterally from unrelated hosts or from the environment, a process known as leaky vertical transmission [5]. Most chemosymbiotic taxa, such as the iconic vent tubeworm Riftia pachyptila, acquire their symbionts horizontally via infections by a free-living stage of microbe [6]. Various transmission modes pose different trade-offs (reviewed in [7]). Vertical transmission provides ‘symbiont assurance’ resulting in the joint dispersal of hosts and symbionts, but it might constrain host species to a narrow ecological niche. Horizontal acquisition risks the failure of dispersing propagules acquiring the ‘right kind’ of bacteria when they settle in new habitats, but it creates opportunities to adopt diverse locally adapted strains of symbiotic bacteria wherever they settle [8, 9]. Because leaky vertical transmission involves vertical and horizontal components, it allows hosts to replace their symbionts with locally optimal strains, but it also engenders a risk of infections by “cheater” strains and pathogens [7]. Although numerous studies have examined the population genetics and geographical connectivity of various invertebrate hosts (reviewed in [10]), only a few studies have examined their bacterial partners [11,12,13,14,15,16]. Often the genetic markers employed to study the symbionts were limited in number and too conservative to adequately discern fine-scale geographical structure.
Herein, we attempt to assess geographical structure and dispersal of horizontally transmitted chemosynthetic bacteria hosted by parapatric sister-species of Bathymodiolus mussels. Various members of the mytilid subfamily Bathymodiolinae that commonly dominate vent and seep communities worldwide host a diverse suite of chemoautotrophic eubacteria capable of oxidizing H2S, CH4 or H2 gases [17,18,19,20,21,22,23,24]. Apparently, their exceptional capacity for adopting phylogenetically diverse and locally adapted strains of bacteria has contributed to their rapid global radiation during the Middle Eocene and Early Oligocene Epochs [9, 25].
A combination of microscopy and molecular evidence indicate that bathymodiolin larvae acquire their endosymbionts from the environments in which they settle [8, 9, 26,27,28,29]. Initial infections of larvae occur in a range of epithelial tissues and then shift to the developing gills, where the bacteria proliferate [30,31,32]. Researchers hypothesized that dispersing mussel larvae might carry hitchiking symbionts from natal sites to new habitats in which they settle [8], but the genetic evidence was insufficient and inconclusive. Regardless, environmental symbiont acquisition renders bathymodiolins susceptible to metabolic cheaters and pathogens [7], but successful infections appear to be highly specific to potential symbiont species [31]. Unlike the experimentally tractable legume/rhizobium system [33,34,35], signalling pathways and metabolic interactions that control specificity have not been investigated in the mussel/symbiont mutualism.
The goal of this study was to determine whether geographic barriers known to act on subdivision of the Bathymodiolus host species also govern genetic structure of the thiotrophic endosymbionts. Closely related sister-species of mussels are abundant at southeastern Pacific vents (Fig. 1). Bathymodiolus thermophilus Kenk & Wilson 1985, occupies the Galápagos Rift (GAR, at 0° latitude) and East Pacific Rise (EPR, between 13°N latitude and 18°S), whereas Bathymodiolus antarcticus Johnson, Won, Harvey, and Vrijenhoek 2013, occupies a northeastern extension of Pacific-Antarctic Ridge (PAR, between 32and 38°S latitude). The host species contact one another and hybridize at 23°S along the northwestern margin of the Easter Microplate [36]. Strong cross-axis currents intersect the ridge axes in this uplifted region, creating dispersal barriers for a number of vent-restricted animals (reviewed in [10]). However, a gap exists in our knowledge about the symbiotic bacteria hosted by these Bathymodiolus sister-species, particularly with respect to their host specificity and biogeographic population structure. Although the northern species, B. thermophilus, is known to harbor a single thiotrophic symbiont species [37], symbionts associated with the southern species, B. antarcticus are poorly understood. To ascertain whether the southern mussel is infected by the same symbiont species, and if physical barriers to dispersal similarly impede the northern and southern symbiont populations, we used high-throughput DNA sequencing of the 16S rRNA encoding gene and six protein-coding genes isolated from symbiotic bacteria sampled throughout the known ranges of their mussel hosts. We applied anlaysis of molecular variance (AMOVA) methods to estimate the proportions of symbiont genetic diversity: (1) contained between the northern and southern regions; (2) contained within population samples within regions; and (3) housed within individual host mussels.

Bathymodiolus sample localities along the East Pacific Rise (EPR), Galápagos Rift (GAR), and Pacific-Antarctic Ridge (PAR). Numbers in parenthesis indicate tectonic spreading rates in mm/year. Lines perpendicular to ridge axes indicate fracture zones. Inset illustrates a B. thermophilus bed at 9°N latitude (Photo credit: Stephen Low Productions, Courtesy of R. A. Lutz, Rutgers University)
Methods
Sampling
Oceanographic expeditions conducted between 1990 and 2005 sampled Bathymodiolus mussels from hydrothermal vent fields distributed along the Galápagos Rift (GAR), the East Pacific Rise (EPR), and the Pacific-Antarctic Ridge (PAR) (Fig. 1). Details regarding geographical coordinates, bathymetric depths, and sampling methods were previously reported [36]. Following recovery of the research submarine HOV Alvin, mussels were immediately transferred to buckets containing 2 °C filtered seawater. Gill tissue samples were dissected from adult mussels and preserved directly in cryovials containing 95% ethanol or frozen at −80 °C. The DNeasy Blood and Tissue Kit (QIAGEN Inc., Valencia, CA) were used to extract total DNA from approximately 25 mg of gill tissue from each specimen.
Community analysis of bacteria from Bathymodiolus gill tissues
A subsample of 45 mussels from seven vent fields was screened for eubacteria-specific 16S rRNA sequences (Table 1A). We designed new PCR primers (with barcodes and linkers) to span the 27F and 516R region of 16S rRNA encoding gene conventionally used for eubacterial ribotyping (Additional file 1: Table S1, barcodes in Additional file 1: Table S2A). Amplicons from 45 independent reactions were pooled and sequenced using 454 GS-FLX titanium sequencing machine (Roche, Branford, CT). We used PyroTrimmer [38] to remove the barcode, linker and primer sequences, and trim the 3′-ends of sequences with low-quality reads (i.e. average score < 20 nucleotides per 5-bp sliding window). Ambiguous sequences and those shorter than 300 bp were discarded. The de novo chimera detection algorithm incorporated in UCHIME program [39] was used to detect and discard chimeric reads. To detect different phylotypes, sequence reads were trimmed to have 300 bp and identical or different only by insertion/deletion of bases were grouped together. The most frequent representative sequence of 16S rRNA encoding gene was deposited in GenBank [GenBank: KX987235] and the other rest variant sequences have been deposited in the Dryad data repository as a polymorphic table. To see if the size selection (300 bp) effects on the final result, the similar trimming and same downstream analyses were also applied to the original 454-pyrosequencing data with different criteria for DNA lengths (250, 350, 400, and 450 bp). The predominant sequence in each phylotype group was used to represent the group in subsequent phylogenetic analyses. Taxonomic affiliation of each representative sequence was determined by RDP classifier [40] against 16S rRNA training set 16 of RDP project. We initially analyzed the forward and reverse sequences, but ultimately used the reverse sequences alone as they provided improved recognition of phylotypes.
Symbiont protein-coding genes
To further characterize the thiotrophic endosymbionts, we examined six protein-coding genes from 72 mussels sampled from nine vent fields, which included the subset of individuals examined for 16S rRNA sequences (Table 1B). For each individual, the 16S rRNA-coding and six protein-coding DNA sequences were amplified from a whole genomic DNA extract obtained from a single gill tissue sample. Symbiont-specific PCR primers and barcodes were designed to span at least 400 bp of each gene (Additional file 1: Table S1, barcodes in Additional file 1: Table S2B). These six genes were chosen from our unpublished genomic sequence (~2.7 Mb) of the thiotrophic endosymbionts (Won Y-J, unpublished data assembled by PacBio sequencing method from a host gill tissue collected at the latitude of 9°N, East Pacific Rise) under the following conditions: (1) existence as a single copy; (2) even representation of genomic positions (Additional file 2: Figure S1); and (3) diverse coverage of the functional category of bacteria such as transcription (rpoD), chaperones (COI and dnaK), carbohydrate transport and metabolism (pgi and pykF), and sulfur metabolism (soxA). Nested PCRs were conducted with two sets of primers for each gene. Primers used for the second PCR attached unique barcodes for eight individuals from each sample location (Additional file 1: Table S2B) to 5′-ends of the forward and reverse sequences.
We used a nested PCR protocol to preferentially amplify sequences known to mark Bathymodiolus thiotrophic symbionts and minimize potential contamination by sequences from environmental bacteria. The nested thermal cycling was performed in 20 μl of reaction with initial denaturation for 1 min at 94 °C; denaturation for 40 s at 92 °C, 60 s at 58 °C (35 cycles), extension for 1 min at 72 °C; and final extension for 7 min at 72 °C. All PCR products were then pooled according to sampling locations (nine vent localities as shown in Fig. 1). The next step involved ligation of MID adapters containing the recognition sequence for nine sampling sites (Table 1B). Ligation was performed according to the GS FLX short-gun DNA library preparation quick guide (Roche, Branford, CT). Once MID adapters were ligated, all amplicons were pooled into a single tube and sequenced using 454 GS-FLX titanium sequencing machine (Roche, Branford, CT).
We used UCHIME [39] to remove erroneous chimeric reads from the protein-coding sequences. A two-step algorithm was developed to correct sequencing errors (Additional file 2: Figure S2). First, we imported raw 454-pyrosequencing reads into Geneious v.6.1.5, and aligned them with the MUSCLE algorithm [41]. The barcodes and primers were removed and the remaining sequences were scanned for stop-codons or frame-shift errors. Second, we developed a Python script to correct probable sequencing errors (Additional file 3). Although the pyrosequencing error rate was unknown, a range of conservative error-correction criteria (1–10%) was applied to the data. For example, a 1% criterion treated nucleotide substitutions occurring at 1% within a population sample as sequencing errors and corrected them to the most frequent base at that position. The process was repeated for the 2–10% criteria and the effects of these criteria on estimates of F-statistics were compared (Additional file 2: Figure S3). Generally, pairwise F ST estimates and hierarchical AMOVAs were robust to these criteria; so, we only report results from the 1% criterion.
Comparisons with partial genomic data from Bathymodiolus thiotrophs (Won Y-J, unpublished data), revealed that all of the 454-pyrosequencing reads of pgi exhibited complete deletion of a single ‘T’ in a poly ‘T’ track that would lead to premature termination the polypeptide. To restore the normal reading-frame we manually inserting a ‘T’ at the deletion site in all the pgi sequences. We also compared our COI and dnaK pyrosequences with Sanger-sequencing data from B. thermophilus thiotrophs [42] to confirm that our primer sets were specific for the bacteria genes (Additional file 2: Figure S4). Unfortunately, the remaining protein-coding genes could not be evaluated in this way due to an absence of published sequence data. The most frequent representative sequences of the six protein-coding genes were deposited in GenBank [GenBank: KX987236-KX987241], and the other rest variant sequences of each gene have been deposited in the Dryad data repository as polymorphic tables.
Population genetics
We used Splitstree v.4.12 [43] to generate unrooted sequence networks for the six protein-coding genes. Genetic diversity indices, pairwise F ST estimates, and the analysis of molecular variance (AMOVA) were obtained with Arlequin v. 3.5 [44]. We used the NUVEL-1A model option implemented in a web-based application (http://www.ldeo.columbia.edu/~menke/plates 2.html) to estimate seafloor spreading rates. Correlations between average sequence diversity (H) and seafloor-spreading rates were estimated with SPSS v.21 (IBM, Armonk, NY). Pairwise geographical distances between sample localities were estimated in Google Earth Pro v.7.1.2. Correlations between pairwise F ST estimates and geographical distances were examined. Because hierarchical subdivision can also generate patterns resembling IBD signals [45], we used the stratified Mantel test implemented in GenoDive [46] to remove the effects of subdivision.
We used a vicariant event to estimate synonymous and nonsynonymous substitution rates for the protein-coding genes. The sequences were sorted into groups located north and south of the Easter Microplate, a geomorphological feature that is estimated to be 2.5–5.3 million years (MY) old [47, 48]. Assuming that no symbiont exchanges crossed this geographic barrier following the initial separation, we used the Nei and Gojobori [49] method to estimate corresponding substitution rates for each gene. A Python script developed for use with high-throughput genetic data can be provided upon request from Y-J Won.
Results
Community analysis of bacteria from Bathymodiolus gill tissues
The 16S sequences revealed a single dominant eubacterial species occupying gill tissues of mussels sampled throughout the known ranges of the two hosts (Additional file 4: Table S3). After quality-control measures were applied to delete questionable sequences, we were left with 43,421 sequence reads of ≥300 bp length. Gammaproteobacteria overwhelmingly predominated, constituting 93.4 to 100% of the sequence reads from 45 mussels. Most of these sequences matched previously published phylotypes for thiotrophic endosymbionts hosted by Bathymodiolus mussels. Minor eubacterial classes found in these mussels included Betaproteobacteria (0–5.9%), Epsilonproteobacteria (0–4.1%), and Flavobacteriia (0–1%) (Additional file 4: Table S3). Further discussion of these minor eubacterial classes is premature, however. Their potential roles as symbionts, environmental contaminants, or pathogens, must be verified with fluorescence in situ hybridization (FISH) methods to determine their location internally or externally on gill tissues [21, 22]. Those analyses are beyond the scope of this population genetic study.
Small subunit rRNA diversity in Bathymodiolus symbionts
Altogether, 94% of the 43,421 16S sequence reads could be grouped into seven relatively frequent phylotypes that differed by only one nucleotide to each other (Additional file 1: Table S4). Phylotypes HT1–HT7 were nearly identical to previously published Sanger sequences from endosymbionts hosted by EPR B. thermophilus (DQ321716) and PAR B. antarcticus (DQ321717) [27]. Phylotype HT1, which dominated all seven samples and constituted 93% of the total sequence reads (Additional file 1: Table S4), was 100% identical with a 300-bp segment from the published sequences. Sequence diversity of the seven-most frequent phylotypes was extremely low (mean π population = 0.0001, and mean H population = 0.043, Table 2 and Additional file 5: Table S5). Differentiation among the sampled populations was very small (mean pairwise F ST = 0.048 ± 0.008 SD; Table 3) with one exception. Elevated pairwise F ST’s involving the small 7°S sample (mean F ST = 0.106 ± 0.011 SD; n = 3) probably resulted from sampling error.
Hierarchical AMOVA (Table 4A) revealed that none of the bacterial ribotype diversity resided in differences between host samples from the EPR versus PAR regions and only 3.13% resided in differences among samples within the regions. Remarkably, the remaining 98.05% of the ribotype variation was contained in the differences among symbionts within (89.52%) and among host individuals (9.8%) from vent localities (Table 4B).
Diversity of protein-coding genes in Bathymodiolus symbionts
Unlike the 16S rRNA encoding gene, DNA sequence networks for six protein-coding gene segments (Fig. 2) all exhibited bifurcating patterns (northern and southern symbiont types in Fig. 1) corresponding with the northern (n = 56; EPR + GAR) and southern host species (n = 16; PAR). All but one of the nucleotide substitutions between the northern and southern symbiont lineages constituted synonymous substitutions (Additional file 1: Table S6). The pgi fragment alone showed a single fixed nonsynonymous substitution (Thr ↔ Ala). All other substitutions (fixed or polymorphic) in the six protein-coding genes were synonymous. Both pgi and pykF exhibited the greatest number of distinct sequences (h) (Table 2 and Additional file 6: Table S7). Except for soxA, haplotypic diversities (H) were relatively homogeneous across the sample locations. SoxA diversity was lowest at three southern locations. Pairwise F ST estimates of the six protein-coding genes were much greater between the host groups (mean F ST = 0.83 ± 0.095 SD) than among populations within groups (mean F ST = 0.15 ± 0.042 SD) (Table 3).

Sequence networks of six protein-coding genes of Bathymodiolus bacterial symbionts. Two representative geographical regions are overlaid on the networks with two different colors corresponding to the regional sampling sites as in Fig. 1: blue for northern symbiont type (EPR + GAR) and orange for southern symbiont type (PAR) harbored by B. thermophilus and B. antarcticus host mussels, respectively
Hierarchical AMOVA revealed that most of the diversity in protein-coding genes (61.97–90.65%) resided in differences between endosymbiont populations hosted by two mussel species (Table 4A). Only 1.43–4.34% resided among population samples within the host groups, and 7.92–34.07% resided among sequence reads within population samples. We further partitioned the within-sample latter component into within- and among-host individual components (Table 4B). An unexpectedly large proportion of variation resided in symbiont compositions among host individuals (27.48–40.76%), and the balance resided within hosts (33.73–66.31%). Frequencies of the most abundant protein-coding sequences varied greatly among host individuals across all nine of the population samples (Additional file 6: Table S7), resulting in the unexpectedly large among-host individual variability.
Symbiont isolation-by-distance
We used a stratified Mantel procedure to test for an Isolation-by-Distance (IBD) pattern in the protein-coding genes. Because geographic subdivision can generate spurious IBD patterns [45], samples were partitioned into regions based on distributions of the mussel hosts. The averaged pairwise F ST estimates within the partitions still presented significant positive correlations with distance (r = 0.439; P = 0.021). Consequently, an Isolation-by-Distance (IBD) pattern holds for Bathymodiolus symbiont populations.
Mussels and symbionts in the hybrid zone
The hybrid zone population at 23°S latitude includes individuals with varying proportions of B. thermophilus and B. antarcticus genes [36]. To examine possible correspondence between the northern and southern symbiont strains and their respective hosts, we used the results of Johnson et al.’s [36] NEWHYBRIDS analysis that assigned individual mussels to putative parental, F1, F2, or backcross categories. The symbiont sequences we obtained from a random subsample of eight hybrid zone mussels only revealed the presence of the northern symbiont strain, regardless of the hosts’ genotypes (Fig. 3). It should be noted that two of the mussels had relatively high proportions of southern (B. antarcticus) genes, but they harbored the northern symbiont strain. The symbiont DNA sequences from this sample of eight mussels provided no evidence that the southern symbiont strain occurred in the 23°S mussels, but we cannot exclude the possibility that they might exist in the ambient environment.

Genetic relationship between host mussels and their thiotrophic endosymbionts from the hybrid zone at 23°S. The left panel represents genetic assignments of eight individual mussels to five categories: blue = northern parental (B. thermophilus); red = southern parental (B. antarcticus), black = F1 hybrid; white = F2 hybrid, light blue = northern backcross hybrid, and pink = southern backcross hybrid [36]. Vertical lines mark 0.75 and 0.90 values of posterior probabilities for the assignments. The right panel represents the corresponding symbiont type of each host mussel. Dark blue color-codes for northern symbiont type (EPR) and orange color codes for southern symbiont type (PAR)
Nucleotide substitution rates
We estimated nucleotide substitution rates for the six protein-coding genes based on a vicariance associated with orogeny of the Easter Microplate, about 2.5–5.3 million years ago (MYA) (Table 5). Based on this range of dates, mean synonymous substitution rates were estimated as 0.77–1.62% per nucleotide per million years (MY). Likewise, nonsynonymous rates were estimated as 0.01–0.023% per nucleotide per MY, roughly 70 times slower than synonymous substitutions.
Discussion
The realization that B. thermophilus hosts a single thiotrophic symbiont species was identified by reverse transcription sequencing of small subunit rRNA with oligonucleotide primers [37]. Subsequent molecular studies confirmed this result [27], but the PCR/direct sequencing methods used in these studies have limited power to detect rare strains or species. The present high-throughput metagenomics analysis revealed that B. antarcticus and B. thermophilus both host a single overwhelmingly predominant ‘ribospecies’ of thiotrophic Gammaproteobacteria. We loosely use term ‘ribospecies’ [50, 51] to denote a grouping of 16S rRNA phylotypes that share ≥97% sequence similarity.
Nonetheless, extensive sequence variation in six protein-coding genes revealed that this symbiont ‘ribospecies’ comprised two highly divergent evolutionary lineages that were geographically separated by the Easter Microplate, corresponding with parapatric distributions of the B. thermophilus and B. antarcticus hosts. On average, 80% of the protein-coding sequence diversity was contained in differences between these symbiont lineages and 17.2% resided in the differences within vent fields. Only 2.9% of the total diversity occurred among vent fields within geographical regions, but this small variance component manifested an Isolation-by-Distance (IBD) signal that was significant even after hierarchical subdivision was taken into account. Remarkably, an IBD pattern was not found in the Bathymodiolus hosts [36, 52]. Therefore, the bacteria appeared to exhibit more limited “realized” dispersal than the mussel hosts, which produce relatively long-lived planktotrophic larvae [53]. Johnson et al. [36] reported that B. antarcticus and B. thermophilus mussels contact one another and hybridize at 23°S, but we found no evidence that corresponding symbiont lineages were mixed at this locality. They also reported evidence for asymmetrical introgression of southern B. antarcticus alleles into northern B. thermophilus, but the northern and southern symbionts appear to be completely isolated. Our examination of host and symbiont genotypes in a sample of eight mussels from the hybrid zone provided no evidence for a host/symbiont specificity. Instead, we only found the northern symbiont lineage, despite the existence of some mussels with a large proportion of B. antarcticus genes (Fig. 3). The apparent absence of southern symbionts at this locality suggests the possibility of complete geographical isolation of the symbiont stains across the Easter Microplate region. Although, we do not know how these bacteria disperse, the present evidence does not support a hypothesis that the symbionts might be transported with dispersing mussel larvae [8].
The Easter Microplate boundary acts as a variable dispersal filter for a number of vent-restricted taxa. It separates sister-species pairs of bythograeid crabs [54, 55], and lepetodrilid limpets [56]. The boundary also separates genetically differentiated metapopulation segments of the siboglinid tubeworm Tevnia jerichonana, and the alvinellid palmworm Alvinella pompejana, but the degrees of differentiation do not warrant species recognition [57, 58]. In contrast, it is not associated with differentiation in Branchipolynoe symmytilida, a polynoid annelid that resides in the mantle cavities of mussels [59], or Riftia pachyptila, the giant siboglinid tubeworm that is emblematic of vents [60]. The isolating potential of this boundary is taxon-specific, reflecting complex interactions between the unique life histories of species, their historical distributions throughout southeast Pacific, and metapopulation processes related to regional extinctions, recolonization events, range expansions, and dispersal modes (reviewed in [10]).
Little is known, however, about the life history of these mussel symbionts. A free-living stage has been identified in vent habitats [42], and the early stages of infection was identified in juvenile mussels [31, 32, 61]. However, it is unknown if the symbiotic stages recycle to the free-living demographic component, as occurs in a siboglinid tubeworm symbiont [62]. Although the symbionts and mussel hosts might experience independent demographic processes, they appear to have experienced similar biogeographical histories. Orogeny of Easter Microplate region probably played a common role in vicariance of their northern and southern populations. This small tectonic plate is estimated to have formed 2.5 to 5.3 million years ago [47, 48] (Fig. 1). Its east and west rifts are connected by northern and southern transform faults [63]. Topographically elevated seamount chains extending east and west of the Microplate are believed to interrupt deep-ocean circulation, creating strong cross-ridge axis currents in the Easter Microplate region [64]. Empirical evidence based on the oceanic distribution of unique vent gases (i.e. Helium-3) supports the ocean circulation models [65]. Consequently, Won et al. [66] hypothesized that the strong cross-axis currents in this region create a contemporary barrier to dispersal for many vent-restricted animals, and particularly for species like mussels that produce planktotrophic larvae. Furthermore, the southern EPR and PAR exhibit superfast tectonic spreading rate of 141–151 mm/yr [67] that are believed to control the rate of habitat turnover in these regions [10, 68]. Regional differences in tectonic and volcanic activities could alter local geochemical conditions that, in turn, might affect the reduced allelic diversity of soxA in the SEPR and PAR endosymbionts (Additional file 6: Table S7). Further research on vent geochemistry in this region might shed some light on factors affecting the physiological ecology of these symbionts and their mussel hosts.
Assuming a 2.5–5.3 million-year (MY) time to the most recent common ancestor of the northern and southern symbiont lineages, we estimated synonymous substitution rates for the six protein-coding genes. The estimated range of rates, 0.77–1.62% per site per MY (Table 5), is comparable to estimates for synonymous substitution in other bacteria: 0.45% for genomic and 0.6–0.8% for protein-coding in E. coli [69, 70]; 0.82% for genomic and 0.39–0.8% for elongation factor Tu (tuf) in Buchnera [69, 71]. The estimated nonsynonymous substitution rates for Bathymodiolus endosymbionts (0.01–0.023% per site per MY, Table 5) were almost identical to rates for the tuf gene in Buchnera (0.013–0.025%, [71]).
Divergence of the northern and southern symbiont lineages was characterized by numerous fixed and polymorphic substitutions at synonymous sites. Only one fixed nonsynonymous substitution was found among the six nuclear gene fragments (Additional file 1: Table S6). The data provided insufficient statistical power to test for adaptive differentiation between the northern and southern symbiont alleles at these loci. Nonetheless, an absence of evidence for natural selection acting on these alleles does not preclude adaptive divergence between the northern and southern symbionts. The lineages might be differentially adapted to EPR and PAR environments or genetically co-adapted with their B. thermophilus and B. antarcticus hosts. Bathymetric variation probably does not contribute to divergence, as all the sampled sites fell within a narrow depth range of 2236–2747 m (Table 1C), but detailed comparative information about geochemical conditions in the two regions is lacking (e.g., [72,73,74]). Differentiation in the communities of Gamma- and Epsilon-proteobacteria hosted by western Pacific vent gastropods of the genus Alviniconcha to be triggered by local patchiness and regional scale differences in vent geochemistry [14]. Yet, Alviniconcha snails differ greatly from B. thermophilus and B. antarcticus mussels, which host a single predominant ‘ribospecies’ of Gammaproteobacteria. Perhaps the excess of synonymous substitutions observed in the present sample of genes from this bacterium only represents neutral differentiation between historically isolated populations. Although several physiological studies have been conducted for these uncultivable symbionts [75,76,77], comparative studies of temporal and spatial variation in vent biogeochemistry have not been undertaken. Examining a larger sample of protein-coding loci in these mussels or a very large number of genome-wide single nucleotide polymorphisms (SNPs) might provide the statistical power needed to conduct tests for adaptive differentiation (e.g., [78, 79]).
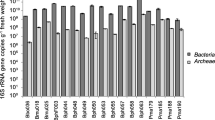
As previously noted, Bathymodiolus mussels acquire thiotrophic endosymbionts via infection by free-living stages that occur in the local environment. High levels of differentiation among host individuals within vent samples (35.28% of total variation; Table 4B) versus low differentiation among vent samples within a region (2.89% of total variation; Table 4A) appears to be anomalous, unless infections are a stochastic consequence of small-scale temporal and spatial genetic heterogeneity of the free-living bacterial strains (Fig. 4). Poisson sampling of the free-living bacterial population seems likely if they are relatively scarce. Fontanez and Cavanaugh [42] reported densities of ~1.8 × 106 free-living bacteria with the appropriate ribotypes living in biofilms on basaltic blocks experimentally deployed at western Pacific hydrothermal vents. Ambient seawater sampled near adult mussels contained lower densities (~1.74 × 105/l) that declined with distances from the Bathymodiolus patch. Once settling mussel larvae are infected, rapid proliferation of the bacteria would greatly enrich the abundance of a small number founding strains (e.g., [31, 32]). Bathymodiolus adults from the Mid-Atlantic Ridge were estimated to host ~2.5 × 1012 symbionts per individual [61]; thus, the high variance among host individuals probably reflects initial sampling bias associated with infections and secondary biases that accrue during enrichment. A random sample of host individuals from a given locality averages these individual sampling biases and provides a better estimate of strain frequencies at that locality. As long as the mussels were not sampled from a single potentially aberrant patch, these averages would explain the low among-locality variation along a ridge axis.

Model for the hierarchical differentiation of Bathymodiolus symbionts. The differently colored symbiotic bacteria represent different strains of the conspecific bacteria in both free-living and endosymbiotic phases. The large arrows represent environmental acquisition of free-living symbionts by the mussel hosts. An intervening physical barrier, in this case the Easter Microplate region, isolates EPR + GAR and PAR populations of the symbiotic bacteria
The adage, “Everything is everywhere, but the environment selects” ([80], p. 15), provides a useful and potentially falsifiable starting hypothesis for studies of microbial diversity [81]. Are the pre-infectious thiotroph strains identified in this study distributed evenly throughout a vent field, or does small-scale environmental heterogeneity in geochemical conditions favor different strains? The genetic composition of pre-infectious stages remains unknown, but small-scale heterogeneity in the chemistry of Bathymodiolus habitats, diffuse-flow low-temperature vents, does influence the distribution of free-living bacteria [82]. Habitat patchiness might contribute to the symbiont variance among host individuals in most of the samples. Conversely, the low among-host variance observed at 17and 38°S might have resulted from sampling of mussels from a single patch (Additional file 6: Table S7). Multiple sample chambers have been used with ROVs to characterize the small-scale patchiness in the symbionts hosted by siboglinid tubeworms and Alviniconcha snails [14, 15]. Unfortunately, they were not available on HOV Alvin during our 1999 through 2005 expeditions. Consequently, the present study represents a starting point for addressing these difficult questions. More directed efforts must be made to isolate biological subsamples and obtain corresponding biogeochemical data from discrete environmental patches, a goal for future studies.
Conclusions
The previous understanding of extrinsic and intrinsic factors affecting the dispersal and evolution of chemosynthetic symbiotic partners has been mostly limited to invertebrate hosts due to the difficulties in sampling and culturing of the bacteria. Here, we attempted to overcome these methodological challenges through the combination of parallel DNA pyrosequencing, highly variable genetic markers, and appropriate geographical sampling of horizontally transmitted thiotrophic endosymbiotic bacteria of deep-sea hydrothermal vent invertebrate hosts, Bathymodiolus mussels (Mollusca: Mytilidae), in the eastern Pacific Ocean. The community analysis based on sequences of slowly evolving 16S rRNA encoding gene confirmed that all the host individuals belonging to two allopatric host species, northern B. thermophilus and southern B. antarcticus, harbor the same numerically dominant thiotrophic Gammaproteobacteria. However, anlaysis of molecular variance of the variable sequences of six protein-coding genes of the endosymbionts revealed a strong genetic disconnection due to the formation of the Easter Microplate, which is also responsible for the subdivision of allopatric host species. We found no evidence for adaptive differentiation between the northern and southern symbiont groups but Isolation-by-Distance in the protein-coding genes. The age of Easter Microplate, 2.5–5.3 million years ago, enabled us to estimate synonymous substitution rates of the protein-coding genes, 0.77–1.62%/nucleotide/million years, which turned out to be remarkably similar to those of E. coli and endosymbionts of aphids. Finally, the unexpected high heterogeneity of symbiont sequences among host individuals sampled from the same location suggested that stochasticity associated with initial infections was amplified as symbionts proliferated within the host individuals.
Abbreviations
- 16S rRNA:
-
16S ribosomal RNA
- AMOVA:
-
Analysis of molecular variance
- COI :
-
Cytochrome C oxidase subunit I
- dnaK :
-
Chaperone protein
- EPR:
-
East Pacific Rise
- GAR:
-
Galápagos Rift
- HOV:
-
Human occupied vehicle
- IBD:
-
Isolation-By-Distance
- MID:
-
Multiplex Identifier sequences
- MY:
-
million years
- PAR:
-
Pacific-Antarctic Ridge
- pgi :
-
Glucose-6-phosphate isomerase
- pykF :
-
Pyruvate kinase
- rpoD :
-
RNA polymerase, sigma D factor
- SEPR:
-
southern East Pacific Rise
- soxA :
-
Sulfur oxidation protein A
References
Dubilier N, Bergin C, Lott C. Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat Rev Microbiol. 2008;6(10):725–40.
Cary SC, Giovannoni SJ. Transovarial inheritance of endosymbiotic bacteria in clams inhabiting deep-sea hydrothermal vents and cold seeps. Proc Natl Acad Sci U S A. 1993;90(12):5695–9.
Szafranski KM, Gaudron SM, Duperron S. Direct evidence for maternal inheritance of bacterial symbionts in small deep-sea clams (Bivalvia: Vesicomyidae). Naturwissenschaften. 2014;101:373–83.
Ikuta T, Igawa K, Tame A, Kuroiwa T, Kuroiwa H, Aoki Y, Takaki Y, Nagai Y, Ozawa G, Yamamoto M, et al. Surfing the vegetal pole in a small population: extracellular vertical transmission of an 'intracellular' deep-sea clam symbiont. R Soc Open Sci. 2016;3(5):160130.
Decker C, Olu K, Arnaud-Haond S, Duperron S. Physical proximity may promote lateral Acquisition of Bacterial Symbionts in Vesicomyid clams. PLoS One. 2013;8(7):e64830.
Nussbaumer AD, Fisher CR, Bright M. Horizontal endosymbiont transmission in hydrothermal vent tubeworms. Nature. 2006;441(7091):345–8.
Vrijenhoek RC. Genetics and evolution of deep-sea chemosynthetic bacteria and their invertebrate hosts. In: Kiel S, editor. The vent and seep biota, topics in Geobiology 33. Dordrecht: Springer Netherlands; 2010. p. 15–50.
Won Y-J, Hallam SJ, O'Mullan GD, Pan IL, Buck KR, Vrijenhoek RC. Environmental acquisition of thiotrophic endosymbionts by deep-sea mussels of the genus Bathymodiolus. Appl Environ Microbiol. 2003;69(11):6785–92.
Duperron S. The diversity of deep sea mussels and thier bacterial symbioses. In: Kiel S, editor. The vent and seep biota: aspect from microbes to ecosystems. Dordrecht: Springer Netherlands; 2010. p. 485.
Vrijenhoek RC. Genetic diversity and connectivity of deep-sea hydrothermal vent metapopulations. Mol Ecol. 2010;19(20):4391–411.
Di Meo CA, Wilbur AE, Holben WE, Feldman RA, Vrijenhoek RC, Cary SC. Genetic variation among endosymbionts of widely distributed vestimentiferan tubeworms. Appl Environ Microbiol. 2000;66(2):651–8.
van der Heijden K, Petersen JM, Dubilier N, Borowski C. Genetic connectivity between north and south mid-Atlantic ridge chemosynthetic bivalves and their symbionts. PLoS One. 2012;7(7):e39994.
Hurtado LA, Mateos M, Lutz RA, Vrijenhoek RC. Coupling of bacterial endosymbiont and host mitochondrial genomes in the hydro- thermal vent clam Calyptogena magnific. Appl Environ Microbiol. 2003;69(4):2058–64.
Beinart RA, Sanders JG, Faure B, Sylva SP, Lee RW, Becker EL, Gartman A, Luther GW, Seewald JS, Fisher CR, et al. Evidence for the role of endosymbionts in regional-scale habitat partitioning by hydrothermal vent symbioses. Proc Natl Acad Sci U S A. 2012;109(47):E3241–50.
Vrijenhoek RC, Duhaime M, Jones WJ. Subtype variation among bacterial endosymbionts of tubeworms (Annelida: Siboglinidae) from the Gulf of California. Biol Bull. 2007;212:180–4.
Laming SR, Szafranski KM, Rodrigues CF, Gaudron SM, Cunha MR, Hilário A, Le Bris N, Duperron S. Fickle or faithful: the roles of host and environmental context in determining symbiont composition in two Bathymodioline mussels. PLoS One. 2016;10(12):e0144307.
Fontanez K, Cavanaugh C. Phylogenetic relationships of hydrothermal vent mussels (Bathymodiolinae) and their symbionts. Mar Ecol Prog Ser. 2013;474:147–54.
Lorion J, Kiel S, Faure B, Kawato M, Ho SYW, Marshall B, Tsuchida S, Miyazaki JI, Fujiwara Y. Adaptive radiation of chemosymbiotic deep-sea mussels. Proc R Soc Biol Sci Ser B. 2013;280(1770):20131243.
Cavanaugh CM. Microbial symbiosis: patterns of diversity in the marine environment. Am Zool. 1994;34(1):79–89.
Childress JJ, Fisher CR. The biology of hydrothermal vent animals: physiology, biochemistry and autotrophic symbioses. Oceanogr Mar Biol Annu Rev. 1992;30:337–441.
Duperron S, Nadalig T, Caprais JC, Sibuet M, Médioni AF, Amann E, Dubilier N. Dual symbiosis in a Bathymodiolus sp. mussel from a methane seep on the Gabon continental margin (Southeast Atlantic): 16S rRNA phylogeny and distribution of the symbionts in gills. Appl Environ Microbiol. 2005;71(4):1694–700.
Duperron S, Halary S, Lorion J, Sibuet M, Gaill F. Unexpected co-occurrence of six bacterial symbionts in the gills of the cold seep mussel Idas sp. (Bivalvia: Mytilidae). Environ Microbiol. 2008;10(2):433–45.
Petersen JM, Zielinski FU, Pape T, Seifert R, Moraru C, Amann R, Hourdez S, Girguis PR, Wankel SD, Barbe V, et al. Hydrogen is an energy source for hydrothermal vent symbioses. Nature. 2011;476(7359):176–80.
Ikuta T, Takaki Y, Nagai Y, Shimamura S, Tsuda M, Kawagucci S, Aoki Y, Inoue K, Teruya M, Satou K, et al. Heterogeneous composition of key metabolic gene clusters in a vent mussel symbiont population. ISME J. 2015;10:990–1001.
Won Y-J, Hallam SJ, O'Mullan GD, Vrijenhoek RC. Cytonuclear disequilibrium in a hybrid zone involving deep-sea hydrothermal vent mussels of the genus Bathymodiolus. Mol Ecol. 2003;12(11):3185–90.
Le Pennec M, Diouris M, Herry A. Endocytosis and lysis of bacteria in gill epithelium of Bathymodiolus thermophilus, Thyasira flexuosa and Lucinella divaricata (bivalve, molluscs). J Shellfish Res. 1988;7(3):483–9.
Won Y-J, Jones WJ, Vrijenhoek RC. Absence of cospeciation between deep-sea mytilids and their thiotrophic endosymbionts. J Shellfish Res. 2008;27(1):129–38.
DeChaine EG, Bates AE, Shank TM, Cavanaugh CM. Off-axis symbiosis found: characterization and biogeography of bacterial symbionts of Bathymodiolus mussels from lost City hydrothermal vents. Environ Microbiol. 2006;8(11):1902–12.
Laming SR, Duperron S, Cunha MR, Gaudron SM. Settled, symbiotic, then sexually mature: adaptive developmental anatomy in the deep-sea, chemosymbiotic mussel Idas modiolaeformis. Mar Biol. 2014;161(6):1319–33.
Salerno JL, Macko SA, Hallam SJ, Bright M, Won YJ, McKiness ZP, Van Dover CL. Characterization of symbiont populations in life-history stages of mussels from chemosynthetic environments. Biol Bull. 2005;208(2):145–55.
Wentrup C, Wendeberg A, Huang JY, Borowski C, Dubilier N. Shift from widespread symbiont infection of host tissues to specific colonization of gills in juvenile deep-sea mussels. ISME J. 2013;7(6):1244–7.
Wentrup C, Wendeberg A, Schimak M, Borowski C, Dubilier N. Forever competent: deep-sea bivalves are colonized by their chemosynthetic symbionts throughout their lifetime. Environ Microbiol. 2014;16(12):3699–713.
Kiers ET, Rousseau RA, West SA, Denison RF. Host sanctions and the legume–rhizobium mutualism. Nature. 2003;425(6953):78–81.
Desbrosses Guilhem J, Stougaard J. Root nodulation: a paradigm for how plant-microbe symbiosis influences host developmental pathways. Cell Host Microbe. 2011;10(4):348–58.
Ferguson BJ, Mathesius U. Phytohormone regulation of legume-rhizobia interactions. J Chem Ecol. 2014;40(7):770–90.
Johnson SB, Won Y-J, Harvey JB, Vrijenhoek RC. A hybrid zone between Bathymodiolus mussel lineages from eastern Pacific hydrothermal vents. BMC Evol Biol. 2013;13(1):21.
Distel DL, Lane DJ, Olsen GJ, Giovannoni SJ, Pace B, Pace NR, Stahl DA, Felbeck H. Sulfur-oxidizing bacterial endosymbionts: analysis of phylogeny and specificity by 16S rRNA sequences. J Bacteriol. 1988;170(6):2506–10.
Oh J, Kim BK, Cho WS, Hong SG, Kim KM. PyroTrimmer: a software with GUI for pre-processing 454 amplicon sequences. J Microbiol. 2012;50(5):766–9.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–000.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9.
Fontanez KM, Cavanaugh CM. Evidence for horizontal transmission from multi-locus phylogeny of deep-sea mussel (Mytilidae) symbionts. Environ Microbiol. 2014;16(12):3608–21.
Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2005;23(2):254–67.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under linux and windows. Mol Ecol Resour. 2010;10(3):564–7.
Meirmans PG. The trouble with isolation by distance. Mol Ecol. 2012;21(12):2839–46.
Meirmans PG, van Tienderen PH. GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes. 2004;4(4):792–4.
Cogné J, Francheteau J, Courtillot V, Team PS. Large rotation of the Easter microplate as evidenced by oriented paleomagnetic samples from the ocean floor. Earth Planet Sci Lett. 1995;136(3):213–22.
Naar DF, Hey R. Tectonic evolution of the Easter microplate. J Geophys Res Solid Earth. 1991;96(B5):7961–93.
Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3(5):418–26.
Stackebrandt E, Goebel B. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Evol Microbiol. 1994;44(4):846–9.
Day MD, Beck D, Foster JA. Microbial communities as experimental units. Bioscience. 2011;61(5):398–406.
Craddock C, Hoeh WR, Lutz RA, Vrijenhoek RC. Extensive gene flow among mytilid (Bathymodiolus thermophilus) populations from hydrothermal vents of the eastern Pacific. Mar Biol. 1995;124(1):137–46.
Arellano SM, Young CM. Spawning, development, and the duration of larval life in a deep-sea cold-seep mussel. Biol Bull. 2009;216(2):149–62.
Guinot D, Hurtado LA. Two new species of hydrothermal vent crabs of the genus Bythograea from the southern East Pacific Rise and from the Galapagos Rift (Crustacea Decapoda Brachyura Bythograeidae). C R Biol. 2003;326(4):423–39.
Mateos M, Hurtado LA, Santamaria CA, Leignel V, Guinot D. Molecular systematics of the deep-sea hydrothermal vent endemic brachyuran family Bythograeidae: a comparison of three Bayesian species tree methods. PLoS One. 2012;7(3):e32066.
Johnson S, Warén A, Vrijenhoek RC. DNA barcoding of Lepetodrilus limpets reveals cryptic species. J Shellfish Res. 2008;27(1):43–51.
Zhang H, Johnson SB, Flores VR, Vrijenhoek RC. Intergradation between discrete lineages of Tevnia jerichonana, a deep-sea hydrothermal vent tubeworm. Deep-Sea Res II Top Stud Oceanogr. 2015;121:53–61.
Jang S-J, Lee W-K, Park E, Johnson SB, Vrijenhoek RC, Won Y-J. Population subdivision of hydrothermal vent polychaete Alvinella pompejana across equatorial and Easter microplate boundaries. BMC Evol Biol. 2016;16:235.
Hurtado LA, Lutz RA, Vrijenhoek RC. Distinct patterns of genetic differentiation among annelids of eastern Pacific hydrothermal vents. Mol Ecol. 2004;13(9):2603–15.
Coykendall DK, Johnson SB, Karl SA, Lutz RA, Vrijenhoek RC. Genetic diversity and demographic instability in Riftia pachyptila tubeworms from eastern Pacific hydrothermal vents. BMC Evol Biol. 2011;11:96.
Duperron S, Quiles A, Szafranski KM, Léger N, Shillito B. Estimating symbiont abundances and gill surface areas in specimens of the hydrothermal vent mussel Bathymodiolus puteoserpentis maintained in pressure vessels. Front Mar Sci. 2016;3:Aticle 16.
Klose J, Polz MF, Wagner M, Schimak MP, Gollner S, Bright M. Endosymbionts escape dead hydrothermal vent tubeworms to enrich the free-living population. Proc Natl Acad Sci U S A Biol Sci. 2015;112(36):11300–5.
Searle R, Rusby R, Engeln J, Hey R, Zukin J. Comprehensive sonar imaging of the Easter microplate. Nature. 1989;341:701–5.
Fujio S, Imasato N. Diagnostic calculation for circulation and water mass movement in the deep Pacific. J Geophys Res (C Oceans). 1991;96(C1):759–74.
Lupton J. Hydrothermal helium plumes in the Pacific Ocean. J Geophys Res (C Oceans). 1998;103(C8):15853–68.
Won Y-J, Young CR, Lutz RA, Vrijenhoek RC. Dispersal barriers and isolation among deep-sea mussel populations (Mytilidae: Bathymodiolus) from eastern Pacific hydrothermal vents. Mol Ecol. 2003;12(1):169–84.
Hey RN, Massoth GJ, Vrijenhoek RC, Rona PA, Lupton J, Butterfield DA. Hydrothermal vent geology and biology at Earth’s fastest spreading rates. Mar Geophys Res. 2006;27(2):137–53.
Vrijenhoek RC. Gene flow and genetic diversity in naturally fragmented metapopulations of deep-sea hydrothermal vent animals. J Hered. 1997;88(4):285–93.
Ochman H, Elwyn S, Moran NA. Calibrating bacterial evolution. Proc Natl Acad Sci U S A. 1999;96(22):12638–43.
Ochman H, Wilson AC. Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J Mol Evol. 1987;26(1–2):74–86.
Brynnel EU, Kurland CG, Moran NA, Andersson S. Evolutionary rates for tuf genes in endosymbionts of aphids. Mol Biol Evol. 1998;15(5):574–82.
Von Damm K. Chemistry of hydrothermal vent fluids from 9–10°N, East Pacific Rise:“time zero,” the immediate posteruptive period. J Geophys Res Solid Earth. 2000;105(B5):11203–22.
Bowers TS, Campbell AC, Measures CI, Spivack AJ, Khadem M, Edmond JM. Chemical controls on the composition of vent fluids at 13–11°N and 21°N, East Pacific Rise. J Geophys Res Solid Earth. 1988;93(B5):4522–36.
Charlou JL, Fouquet Y, Donval JP, Auzende JM, Jean-Baptiste P, Stievenard M, Michel S. Mineral and gas chemistry of hydrothermal fluids on an ultrafast spreading ridge: East Pacific Rise, 17 to 19°S (Naudur cruise, 1993) phase separation processes controlled by volcanic and tectonic activity. J Geophys Res Solid Earth. 1996;101(B7):15899–919.
Duperron S, Guezi H, Gaudron S, Pop Ristova P, Wenzhöfer F, Boetius A. Relative abundances of methane-and sulphur-oxidising symbionts in the gills of a cold seep mussel and link to their potential energy sources. Geobiology. 2011;9(6):481–91.
Fisher C, Childress J, ARP A, Brooks J, Distel D, Favuzzi J, Macko S, Newton A, Powell M, Somero G, et al. Physiology, morphology, and biochemical composition of Riftia pachyptila at rose garden in 1985. Deep Sea Res Part A Oceanogr Res Pap. 1988;35(10–11):1745–58.
Dubilier N, Mulders C, Ferdelman T, de Beer D, Pernthaler A, Klein M, Wagner M, Erseus C, Thiermann F, Krieger J, et al. Endosymbiotic sulphate-reducing and sulphide-oxidizing bacteria in an oligochaete worm. Nature. 2001;411(6835):298–302.
Gayà-Vidal M, Albà M. Uncovering adaptive evolution in the human lineage. BMC Genomics. 2014;15(1):599–12.
Tiffin P, Ross-Ibarra J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol Evol. 2014;29(12):673–80.
Baas Becking LGM: Geobiologie of inleiding tot demilieukunde. The Hague, the Netherlands: W.P. Van Stoc-kum & Zoon (in Dutch); 1934.
De Wit R, Bouvier T. 'Everything is everywhere, but, the environment selects'; what did baas Becking and Beijerinck really say? Environ Microbiol. 2006;8(4):755–8.
Campbell BJ, Polson SW, Allen LZ, Williamson SJ, Lee CK, Wommack KE, Cary SC. Diffuse flow environments within basalt-and sediment-based hydrothermal vent ecosystems harbor specialized microbial communities. Front Microbiol. 2013;4:182.
Geographical structure of endosymbiotic bacteria hosted by Bathymodiolus mussels at eastern Pacific hydrothermal vents.doi: doi:10.5061/dryad.d3r64.
Acknowledgments
We thank the HOV Alvin pilots and crewmembers of the R/V Atlantis (Woods Hole Oceanographic Institution) for their expertise and able assistance at sea.
Funding
This work received funding from the following institutions: the Korea Polar Research Institute (KOPRI) (grants number PP13040 and PE15050) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (grant number NRF-2015R1A4A1041997) to YJW; the United States National Science Foundation (OCE8917311, OCE9217026, OCE-9529819, OCE9910799, OCE-0241613) to RCV.
Availability of data and materials
The data set supporting the results of this article is available in the Dryad repository, http://dx.doi.org/10.5061/dryad.d3r64 [83], and GenBank, KX987235-KX987241.
Authors’ contributions
Conceived and designed the study: PTH, EP, RCV, YJW. Performed the field work: RCV, YJW. Data generation of DNA sequences: EP, SGH. Analyzed the data: PTH, SJJ, KK, SGH, EHK. Wrote the paper: PTH, SGH, RCV, YJW. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
Permission for sampling the species in the US and Korea was not necessary. Animals were frozen and/or preserved in ethanol.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Primer sequences for 16S and six protein-coding genes of Bathymodiolus mussels’ symbionts. Table S2. Barcodes used for the amplification of (A) symbiont 16S rRNA encoding gene from 45 individual host mussels and (B) symbiont protein-coding genes from 72 individuals of host mussel. Table S3. Bacterial symbiont community in the gill tissues of host mussels, B. thermophilus and B. antarcticus, based on high-throughput data of 16S ribosomal RNA encoding gene from 45 individual host mussels. Table S4. Frequency distribution of Bathymodiolus symbiotic bacteria 16S phylotypes at seven vent localities. Table S5. Genetic diversity indices of symbiont 16S rRNA encoding gene from 45 individuals of host Bathymodiolus mussels in the EPR and PAR. Table S6. Summary table of synonymous and nonsynonymous substitutions of six protein-coding genes between two geographic groups, EPR + GAR and PAR. Table S7. Genetic diversity indices of symbiont protein-coding genes from 72 individuals of host Bathymodiolus mussels in the EPR, GAR, and PAR (based on 1% error correction criterion for the 454 pyrosequencing raw data). (DOCX 25 kb)
Additional file 2: Figure S1.
Alignment of seven examined genes and Bathymodiolus thiotrophs incomplete genome. Dark-green larger rectangle represents incomplete genome of thiotrophic endosymbionts obtained from Bathymodiolus host individual at latitude of 9°N, East Pacific Rise. Small light-green rectangles represent seven target genes used in the present study. Arrows represent direction of genes. Numbers indicate position of genes in the incomplete genome. Figure S2. Schematic overview of our protocol for the correction of 454-pyrosequencing data of protein-coding genes of symbiotic bacteria. Figure S3. Hierarchical fixation indices of F SC, F ST, and F CT calculated from each protein-coding gene across various error-correction criteria (1to 10%) applied to the raw high-throughput sequence reads. The setting of geographical groups into two regions (EPR + GAR and PAR) is shown in Figs. 1 and 2. Colors represent different protein-coding genes. As shown with the aid of lines, almost all the F statistics are stable irrespective of the different error-correction criteria. Figure S4. Mussel symbiont protein-coding gene trees from two representative sequences obtained from the EPR + GAR and PAR in this study and from other symbiont specific sequences [41]. Black squares (■) are the representative sequences of symbionts from this study. Colored boxes identify symbionts based upon ocean residence: West Pacific (orange), East Pacific (green), and Indian (blue). (DOCX 636 kb)
Additional file 3:
Developed genotyping script for correcting errors in protein-coding sequences. (DOCX 18 kb)
Additional file 4: Table S3.
Bacterial symbiont community in the gill tissues of host mussels, B. thermophilus and B. antarcticus, based on NGS data of 16S ribosomal RNA gene from 45 individual host mussels. (XLSX 16 kb)
Additional file 5: Table S5.
Genetic diversity indices of symbiont 16S rRNA gene from 45 individuals of host Bathymodiolus mussels in the EPR and PAR. (XLSX 13 kb)
Additional file 6: Table S7.
Genetic diversity indices of symbiont protein-coding genes from 72 individuals of host Bathymodiolus mussels in the EPR, GAR, and PAR (based on 1% error correction criterion for the 454 pryosequencing raw data). (XLSX 48 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ho, PT., Park, E., Hong, S.G. et al. Geographical structure of endosymbiotic bacteria hosted by Bathymodiolus mussels at eastern Pacific hydrothermal vents. BMC Evol Biol 17, 121 (2017). https://doi.org/10.1186/s12862-017-0966-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-017-0966-3