
Abstract
Background
The majority of the subspecies of Daucus carota have not yet been discriminated clearly by various molecular or morphological methods and hence their phylogeny and classification remains unresolved. Recent studies using 94 nuclear orthologs and morphological characters, and studies employing other molecular approaches were unable to distinguish clearly many of the subspecies. Fertile intercrosses among traditionally recognized subspecies are well documented. We here explore the utility of single nucleotide polymorphisms (SNPs) generated by genotyping-by-sequencing (GBS) to serve as an effective molecular method to discriminate the subspecies of the D. carota complex.
Results
We used GBS to obtain SNPs covering all nine Daucus carota chromosomes from 162 accessions of Daucus and two related genera. To study Daucus phylogeny, we scored a total of 10,814 or 38,920 SNPs with a maximum of 10 or 30% missing data, respectively. To investigate the subspecies of D. carota, we employed two data sets including 150 accessions: (i) rate of missing data 10% with a total of 18,565 SNPs, and (ii) rate of missing data 30%, totaling 43,713 SNPs. Consistent with prior results, the topology of both data sets separated species with 2n = 18 chromosome from all other species. Our results place all cultivated carrots (D. carota subsp. sativus) in a single clade. The wild members of D. carota from central Asia were on a clade with eastern members of subsp. sativus. The other subspecies of D. carota were in four clades associated with geographic groups: (1) the Balkan Peninsula and the Middle East, (2) North America and Europe, (3) North Africa exclusive of Morocco, and (4) the Iberian Peninsula and Morocco. Daucus carota subsp. maximus was discriminated, but neither it, nor subsp. gummifer (defined in a broad sense) are monophyletic.
Conclusions
Our study suggests that (1) the morphotypes identified as D. carota subspecies gummifer (as currently broadly circumscribed), all confined to areas near the Atlantic Ocean and the western Mediterranean Sea, have separate origins from sympatric members of other subspecies of D. carota, (2) D. carota subsp. maximus, on two clades with some accessions of subsp. carota, can be distinguished from each other but only with poor morphological support, (3) D. carota subsp. capillifolius, well distinguished morphologically, is an apospecies relative to North African populations of D. carota subsp. carota, (4) the eastern cultivated carrots have origins closer to wild carrots from central Asia than to western cultivated carrots, and (5) large SNP data sets are suitable for species-level phylogenetic studies in Daucus.
Similar content being viewed by others
Background
The center of diversity of the genus Daucus is in the Mediterranean region [1]. Daucus species also occur elsewhere, with one species (D. glochidiatus [Labill.] Fischer & al.) in Australia, four species in the American continent (D. carota L., D. montanus Humb. & Bonpl. ex Schütt., D. montevidensis Link ex Sprengel, D. pusillus Michx.); D. carota occurs in many continents worldwide. Wild and cultivated carrots (D. carota L. sensu lato) belong to the D. carota complex. Its constituent taxa all possess 2n = 18 chromosomes, have weak biological barriers to interbreeding, and some of these taxa are difficult to define taxonomically [2, 3], making the D. carota complex the most problematic species group in the Apiaceae family [4].
The classification of the members of the D. carota complex has attracted the interest of various researchers [3]. Germplasm curators have relied on local floras for identifying Daucus such as those from Algeria [5], the Azores [6], Europe [7], the Iberian Peninsula and Balearic Islands [8], Libya [9], Morocco [10], Palestine [11], Portugal [12], Syria [13], Tunisia [14, 15], and Turkey and the East Aegean Islands [16]. Currently, there is no consensus about the number of subspecies of D. carota. More than 60 species have been proposed for the phenotypic variants observed within the D. carota complex [3]. For instance, 11 wild subspecies were recognized by Heywood [2, 17], five by Arenas and García-Martin [18], and five in the latest comprehensive morphoanatomical classification of Daucus by Sáenz [1] (subsp. carota, subsp. gummifer, subsp. hispanicus, subsp. maritimus, and subsp. maximus). Pujadas Salvà [19] proposed nine subspecies for the Iberian Peninsula plus Balearic Islands (subsp. carota, subsp. cantabricus, subsp. commutatus, subsp. gummifer, subsp. halophilus, subsp. hispanicus, subsp. majoricus, subsp. maximus, and subsp. sativus).
Several molecular approaches have been used to examine the diversity and genetic relationships of D. carota. St. Pierre et al. [20] used isozymes to study 168 accessions of the D. carota complex from 32 countries and could not separate them into distinct groups. Random amplified polymorphic DNA (RAPD) and amplified fragment length polymorphisms (AFLP) were employed by Nakajima et al. [21] and showed that all accessions of D. carota group into a major clade. Vivek and Simon [22, 23] used restriction fragment length polymorphisms (RFLPs) of nuclear, plastid, and mitochondrial DNA and interpreted their results to be generally concordant with the classification proposed by Sáenz [1]. However, only one additional subspecies was studied (subsp. drepanensis). Using AFLPs, Shim and Jørgensen [24] showed wild and cultivated carrot clustered separately. Similar results were obtained by Bradeen et al. [25] with AFLPs and inter-simple sequence repeats (ISSR) and they concluded wild carrots had no substructure. Rong et al. [26] obtained a Daucus phylogeny using SNPs and found the subspecies of D. carota to be intermixed. Later, Lee and Park [27] mentioned D. sahariensis, D. syrticus and D. gracilis are probably the closest relatives to D. carota. In an attempt to characterize the populations of D. carota present in São Miguel Island (Azores, Portugal), Matias Vaz [28] used one nuclear ortholog, nuclear ribosomal DNA ITS, and morphological descriptors, and concluded that the classification of D. carota remained problematic. Other morphological studies [3, 29–32] did not distinguish the subspecies of D. carota. However, Iorizzo et al. [33] developed 3,326 single nucleotide polymorphisms (SNPs) to study the genetic structure and domestication of carrot. Using seven wild subspecies of D. carota (other than subsp. carota) as outgroups, they found a clear separation between wild (subsp. carota) and cultivated (subsp. sativus) accessions of Daucus.
Other studies distinguished Daucus species outside of the complex. Using an integrated approach consisting of morphology, together with the ribosomal internal transcribed spacers (ITS), the plastid trnQ-rps16 intergenic spacer and plastid rps16 intron sequences, the existence of a neglected species from North Africa, D. mauritii (Sennen ex Maire) Sennen, was confirmed [34]. More recently, Spooner et al. [35] demonstrated the utility of eight nuclear orthologs to infer the phylogeny of Daucus. This study revealed all the subspecies of D. carota, D. capillifolius (that they named as D. carota subsp. capillifolius (Gilli) Arbizu) and D. syrticus are closely related, but was ineffective at separating the subspecies. Arbizu et al. [36] used 94 nuclear orthologs obtained by next-generation sequencing technology to examine multiple accessions per species of Daucus, and could distinguish the species well, including the 2n = 18 species D. syrticus, but not the subspecies of D. carota. Similarly, a recent investigation [37] used ITS sequences and three plastid DNA sequences to study the phylogeny of subtribe Daucinae and showed accessions of the D. carota complex did not group the subspecies together.
The last decade has seen tremendous advances in genome-scale data collection and analysis, allowing researchers from various disciplines to address new questions. A major innovation for the plant systematics community is high-throughput DNA sequencing [38] to infer phylogenetic relationships among recently diverged species or populations [39, 40]. Challenges for taxonomic resolution at low taxonomic levels mainly arise due to biological events such as gene flow by hybridization and introgression [41], gene duplication [42], horizontal gene transfer [43], and incomplete lineage sorting [44]. To address the challenge of resolving the phylogenetic relationships among very closely related species, large genome-scale data sets can be used [45]. Reduced-representation methods provide powerful and cost-effective tools, producing abundant large-scale genomic data [46] and have been used in many phylogenetic studies [45, 47–51].
Genotyping-by-sequencing (GBS) is one such genome-wide reduced representation method that generates sequence variants by utilizing next-generation sequencing technology, producing a powerful and cost-effective genotyping procedure [52]. A large number of variants are generated by GBS [53] and have been applied to a wide range of crops such as barley [54], lentils [51], maize [55], potato [56], reed canarygrass [57], rice [58], soybean [59], switchgrass [60], and wheat [61]. GBS has been used to infer the classification of wild species like chickpea [62], lentils [51], sedges [45], and tomatoes [63].
Daucus would benefit from a modern taxonomic monograph, but currently there is no consensus about the number of species in the genus or the number of subspecies within the D. carota complex. We here explore the use of GBS to discover SNPs distributed along all nine chromosomes, and evaluate their phylogenetic utility within Daucus, through the examination of 164 accessions, and analyses using concatenated data and a coalescent model. We then integrate our GBS results with prior morphological data for a coordinated approach to distinguish subspecies within the D. carota complex.
Methods
Plant materials
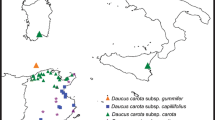
We examined 162 accessions of Daucus, and two accessions of related outgroup genera, Orlaya and Rouya (164 accessions in total) (Additional file 1: Table S1). A previous study [36] confirmed that the phylogeny of Daucus is divided into two main clades, A (including A′) and B. Sub-clade A′ contains members of the Daucus carota complex. In the present study, the Daucus carota complex is represented by 144 accessions. All accessions were obtained from the United States National Plant Germplasm System, maintained at the North Central Regional Plant Introduction Station (NCRPIS) in Ames, Iowa. As indicated above, subspecies boundaries in D. carota are controversial and no two authors agree on how many there are. This issue was investigated by Arbizu et al. [29] and Spooner et al. [31] who recognized three wild subspecies (subsp. capillifolius, subsp. carota, subsp. gummifer) and one cultivated subspecies (subsp. sativus). We chose all accessions available as germplasm (collected in 35 countries), and used these names. Based on the present results (below, partly supporting D. carota subsp. maximus, although on a clade with some accessions of subsp. carota), we reevaluated the characters differentiating subsp. carota and subsp. maximus and reanalyzed the morphological data of Spooner et al. [31]. Further details of the accessions examined in this study are available at the Germplasm Resources Information Network - GRIN (https://npgsweb.ars-grin.gov/gringlobal/search.aspx).
Genotyping-by-sequencing data set
All 164 accessions were planted in a greenhouse at the University of Wisconsin-Madison and genomic DNA was extracted using the CTAB method [64] from young leaves. Genomic DNA quality was initially assessed by visualizing 900 ng of each sample on a 1% agarose gel and twenty random samples were digested with EcoRI following the manufacturer′s protocol (Promega, Madison, WI). In addition, DNA quality was verified by measuring the optical density at a wavelength of 260 and 280 nm with a ND-1000 Nanodrop Spectrophotometer (NanoDrop Technologies, Wilmington, DE). Samples were sent to the UW-Madison Biotechnology Center for DNA sequencing. First, quantity of the genomic DNA was evaluated with Quant-iT™ PicoGreen® dsDNA kit (Life Technologies, Grand Island, NY). Then, the protocol described by Elshire et al. [52] was followed. In short, samples of genomic DNA were digested using a methylation-sensitive restriction enzyme, ApeKI (New England Biolabs, Ipswich, MA) with recognition site GCWGC, where W is A or T. Barcoded adapters amenable to Illumina sequencing were then added by ligation with T4 ligase (New England Biolabs, Ipswich, MA). Adapter-ligated samples were pooled and amplified to provide library quantities amenable for sequencing, and adapter dimers were removed by SPRI bead purification. Quality and quantity of the finished libraries were assessed using the Agilent Bioanalyzer High Sensitivity Chip (Agilent Technologies, Inc., Santa Clara, CA) and Qubit® dsDNA HS Assay Kit (Life Technologies, Grand Island, NY), respectively. Libraries were standardized to 2 μM. Cluster generation was performed using HiSeq SR Cluster Kit v3 cBot kits (Illumina Inc, San Diego, CA, USA). Finally, 100 bp single-end reads were sequenced using the HiSeq SBS Kit v3 (50 Cycle) (Illumina Inc.) on a HiSeq2000 sequencer. Images were analyzed using the standard Illumina Pipeline, version 1.8.2. A second set of libraries was sequenced using the same protocol described above. Here we included samples of the first libraries that produced a low number of reads, and additional accessions to increase the representation of the D. carota complex. To quantify the number of reads per library, we processed the raw GBS data (FASTQ format) using the program process_radtags.pl, which is part of the STACKS pipeline [65].
Data were then analyzed using the bioinformatics pipeline TASSEL-GBS version 4.3.13 [66, 67]. Briefly, GBS sequence tags were identified in all FASTQ files by indicating the restriction enzyme (ApeKI) and the DNA barcodes for each sample used for library preparation. Only reads having an intact barcode sequence were kept. Then, unique tags from each sequence were merged producing a master tag count file in FASTQ text format (argument –t was used), which was used as an input to the program Burrows-Wheeler Aligner (BWA) version 0.7.12 [68], to align all the tags against the carrot reference genome LNRQ01000000.1 [69]. The alignment file in SAM format was converted into a binary tagsOnPhysicalMap (.topm) file. We then used the SeqToTBTHDF5 plug-in to obtain a TagsByTaxa (TBT) file in HDF5 format containing the number of times each GBS tag was observed in each sample. The TBT file was transposed into a tag-optimized orientation, and together with the .topm file, they were used as input for the SNP calling plug-in. For each carrot chromosome, one HapMap genotype file was generated (nine in total), followed by the search of duplicate SNPs and their corresponding merging in each HapMap file (options -misMat = 0.1 and -callHets were used). GBSHapMapFilters plug-in was then used to discard samples with a SNP call rate and minimum R-square value for LD filter less than 0.1 (arguments -mnTCov = 0.1 and -mnR2 = 0.1 were called). Further data curation was conducted using VCFtools version 0.1.14 [70] with the following criteria of retention: (i) minimum minor allele frequency of 0.1, (ii) maximum minor allele frequency of one, (iii) number of alleles less than or equal to two, and (iv) maximum missing data of 0.1 or 0.3. To impute missing genotypes, we used the program Beagle version 4.0 [71] with arguments burnin-its = 10, phase-its = 10, impute-its = 10. Beagle software uses a Hidden Markov model for inferring haplotype phase of the samples and then filling in missing genotypes.
Phylogenetic analyses
All SNPs were concatenated into a single alignment. Maximum likelihood and Bayesian analyses were conducted via the CIPRES [72] portal at the San Diego Supercomputer Center (http://www.phylo.org) with the GTR + G nucleotide substitution model using RAxML version 8.2.4 [73] and MrBayes version 3.2.6 [74], respectively. Selection of the best-fit evolutionary model for our GBS data set was attempted using jModelTest version 2.1.4 [75] on CIPRES. However, this gave an error message indicating Phyml command line cannot run. Phyml might be crashing due to the elevated number of samples (164 and 150). The most common model of evolution for DNA analysis is general time-reversible (GTR) [76] and that is the main reason only GTR-based models are implemented in RAxML [77]. In addition, the RAxML manual [77] strongly suggests avoiding Pinv + Gamma model. Hence, we continued our phylogenetic analyses with GTR + G. We obtained the best-scoring ML tree from 100 independent ML tree searches, and then 1,000 nonparametric bootstrap inferences were performed with the same program. All ML analyses were performed using four data sets: (1) 164 accessions with 10% missing imputed genotypes, (2) 164 accessions with 30% missing imputed genotypes, (3) 150 accessions with 10% missing imputed genotypes, and (4) 150 accessions with 30% missing imputed genotypes. The data set with 164 accessions represents five sub-species of Daucus carota, eight Daucus species and two related genera (Orlaya and Rouya). On the other hand, the 150 accessions are comprised of five sub-species of Daucus carota and one species, D. syrticus, as outgroup. We first used all 164 samples to reconstruct the phylogeny of Daucus to confirm the sister group of the D. carota complex. To do so, we rooted our tree on Orlaya daucoides based on Arbizu et al. [36]. Because of this wider analysis, subsequent ML analyses of the D. carota complex were executed using D. syrticus as outgroup (150 accessions in total).
Bayesian analysis was carried out using only the data set of the D. carota complex (150 accessions) containing 10% missing imputed genotypes. We conducted two independent four-chain 50 million generation runs per input file, sampled every 1,000 generations. Tracer v1.6 (http://tree.bio.ed.ac.uk/software/tracer/) was used to analyze the convergence to the stationary distribution and the effective sample size (ESS) of each parameter of each input file. We discarded the first 25% of generations as burn-in.
A lineage tree was constructed considering only members of the D. carota complex, including D. syrticus, using the two data sets containing imputed genotypes of maximum missing rate of 0.1 or 0.3. The method we used was implemented in the software SVDquartets [78], which assumes each SNP has its own genealogical history and relationships among quartets of taxa are inferred under the coalescent model. In addition, we inferred a species level phylogeny by renaming the 150 accessions employed for the lineage tree construction with new proposed subspecies names determined in this study (Additional file 1: Table S1). The name for the plants initially identified as putative hybrids of subsp. carota and subsp. capillifolius is subsp. carota, as proposed by Spooner et al. [31]. To conduct these analyses, we evaluated all possible quartets including 100 nonparametric bootstrap replicates. SVDquartets was executed in the program PAUP* version 4.0a147 [79] and the resulting trees were viewed in FigTree version 1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/).
Population structure
The composition of population structure in the 150 accessions belonging to the D. carota complex, including D. syrticus, was determined using all original filtered SNPs in the two data sets comprising 10 or 30% missing imputed data. First, our VCF data sets were converted into PLINK PED [80] format using the --plink option in software VCFtools. Then, the PGDSpider program [81] was used to convert PLINK PED format into STRUCTURE format. We employed the Bayesian clustering program STRUCTURE version 2.3.4 [82] with populations (K value) ranging from 1 to 14, replicated ten times, with a burn−in length of 20,000 and 50,000 Monte Carlo iterations. An admixture model with no previous population information was considered; all other parameters were set to default values. STRUCTURE results were then processed in the software STRUCTURE HARVESTER [83], detecting the most likely number of clusters by using the rate of change in the log probability of our data between successive values of K, also known as Delta K [84]. To minimize variance across all the ten iterations of the selected K values, we used the program CLUMPP version 1.1.2 [85] utilizing the LargeKGreedy algorithm with 1,000 permutations. Population structure was then visualized using DISTRUCT software version 1.1 [86].
Morphological analysis
We analyzed the data set of morphological characters previously obtained by Spooner et al. [31] for the D. carota complex. Accessions identified as D. carota subsp. sativus (cultivated carrot) were not included in the present analysis; only wild Daucus carota accessions. Samples of wild carrots were classified into five groups (D. carota subsp. capillifolius, subsp. carota, subsp. gummifer, subsp. maximus, and hybrids between subsp. carota with subsp. capillifolius) based on our molecular phylogenetic analyses, population structure results, and the previous study conducted by Spooner et al. [31], with most accessions examined in common among studies.
A data set consisting of 127 accessions of the D. carota complex (minus subsp. sativus) and 23 continuous characters (stem, leaf, flower and mericarp) was examined with a stepwise discriminant analysis (linear, common variance) in JMP® version 11.2 (SAS Institute Inc.) using a backward selection method. A model with significant variables in identifying accession structure was obtained by removing characters one at a time until the model F-test p value ≤ 0.05. We then performed a canonical variate analysis (CVA). A descriptive statistical analysis was conducted to verify the mean, median, standard deviation and range of values. Utilizing R software version 3.2.0 [87], box plots were constructed as a graphic tool to visualize comparisons across accessions of wild carrots.
Results
The GBS analysis pipeline implemented in TASSEL-GBS version 4.3.13 [66, 67] identified a total of 16,291,308 and 14,548,150 unique 100-bp sequence tags for the following two data sets, respectively: (1) 162 accessions of Daucus and two accessions of related genera (Orlaya and Rouya), and (2) 144 accessions of the D. carota complex and six accessions of D. syrticus. Moreover, 889,445 and 789,311 SNPs were obtained, respectively for each data set. Then, after filtering out SNPs using different criteria (see Methods), our final data sets with a maximum missing data of 10% consisted of 10,814 or 18,565 SNPs, respectively (Additional file 2: Table S2).
Phylogeny inference
The maximum likelihood (ML) phylogenetic reconstruction of Daucus with 10 or 30% missing imputed genotypes allowed us to determine the outgroup of the D. carota complex to be D. syrticus (Additional file 3: Figure S1 and Additional file 4: Figure S2), fully in agreement with prior molecular studies in Daucus [27, 35, 36, 88–90]. Our ML trees, using varying numbers of SNPs, are highly resolved with two main clades, A and B, both with more than 97% bootstrap support (Additional file 3: Figure S1 and Additional file 4: Figure S2). Members of the D. carota complex together with D. syrticus possess 2n = 18 chromosomes, while the remaining taxa in clades A and B possess 2n = 20 and 22 chromosomes. Maximum likelihood trees show that species Rouya polygama (corrected from Margotia gummifera in Spooner et al. [35] and Arbizu et al. [36]) is placed within a monophyletic Daucus clade and is sister to a clade formed by the D. carota complex, and D. syrticus. In addition, D. aureus is sister to the clade mentioned above, and sister to them is D. crinitus. Members of the D. guttatus complex were placed within clade B. Daucus involucratus is sister to a clade formed by D. guttatus and D. littoralis (Additional file 3: Figure S1 and Additional file 4: Figure S2). Using 10,814 SNPs (10% missing imputed genotypes), D. setulosus is sister to D. pusillus. On the other hand, the ML tree using a data set containing 38,920 SNPs (30% missing imputed genotypes) places D. pusillus as sister to a clade formed by D. guttatus, D. littoralis, D. involucratus, and D. setulosus. For both data sets of missing imputed genotypes (10 or 30%), bootstrap values of most clades were higher than 90%. However, the data set containing 38,920 SNPs showed higher bootstrap support for the following three clades of D. carota: (i) Balkan Peninsula and Middle East, wild, (ii) North America and Europe, wild; and (iii) North Africa, wild (Additional file 3: Figure S1 and Additional file 4: Figure S2).
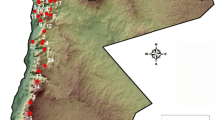
Employing data sets including only accessions of the D. carota complex, with D. syrticus as outgroup and using 18,565 and 43,713 SNPs with a maximum of 10% (Fig. 1) and 30% (Additional file 5: Figure S3) missing imputed genotypes, respectively, ML analyses revealed that within clade A′, members of the D. carota complex were placed in six clades (labeled clades 1–6 in Fig. 1 and Additional file 5: Figure S3) according to their cultivation type and geographic locality: (1) Western, cultivated, (2) central Asia, wild and Eastern, cultivated, (3) Balkan Peninsula and Middle East, wild, (4) North America and Europe, wild (5) North Africa, wild, and (6) Iberian Peninsula and Morocco, wild. The landrace accessions from the Middle East and Europe form a grade between clades 2 and 3.

Maximum likelihood reconstruction and structure of the genetic diversity of 144 accessions of the Daucus carota complex and outgroups using 18,565 SNPs (10% missing imputed genotypes) obtained by GBS. Each accession is represented by a horizontal bar, and each color corresponds to a population (nine in total). Numbers above the branches represent bootstrap values, with only values higher than 70% shown. Names given to clades refer to the geographic origin and improvement status of the accessions of the D. carota complex. The outgroup taxon is D. syrticus
We followed the same nomenclature used by Iorizzo et al. [33, 69] for western cultivated carrots (Europe and American continent) and eastern cultivated carrots (Middle East and Asia). All cultivated carrots (D. carota subsp. sativus) were monophyletic (clades 1,2 + grade; Fig. 1), but wild members of D. carota from central Asia were placed together with eastern members of subsp. sativus. Consequently, our study does not recover a subsp. sativus monophyletic clade as was reported in Iorizzo et al. [33], but it is concordant with the phylogenetic result of Iorizzo et al. [69]. Interestingly, four geographically coherent sub-clades (northern Asia, southern Asia, central Asia and Middle East) of clade 2 containing cultivated accessions were recovered (Fig. 1 and Additional file 5: Figure S3). However, two accessions, PI 430525 (Afghanistan) and PI 163234 (India), depicting extensive allelic admixture were not placed in those four sub-clades.
Maximum likelihood analyses using both data sets of 10 or 30% missing imputed genotypes recovered, with 100% bootstrap, two individual sub-clades of clade 3 containing accessions collected in Turkey and Syria (Fig. 1 and Additional file 5: Figure S3). Wild accessions of D. carota from France were also placed with 100% bootstrap support in a single sub-clade within the North America and Europe, wild clade (clade 4), depicting a separation between accessions of subspecies carota and gummifer (Fig. 1 and Additional file 5: Figure S3). All accessions of D. carota subsp. capillifolius collected in Tunisia and immediately adjacent western Libya are in the clade of subsp. carota from North Africa, wild.
Accessions PI 478873 and PI 478874 collected in Sardinia and Sicily (Italy), respectively, were on the North Africa, wild clade (clade 5). Both islands are located close to North Africa. Moreover, accession Ames 7674, donated from Tuscany (Italy) is not placed within the North America and Europe, wild clade (like in Additional file 3: Figure S1 and Additional file 4: Figure S2). Rather, it is sister to a clade formed by all members of the D. carota complex, except the clade placing those D. carota accessions from the Iberian Peninsula and Morocco, wild (Fig. 1 and Additional file 5: Figure S3).
All accessions from Portugal and Morocco that were initially named by GRIN as D. carota subsp. maximus and subsp. maritimus were on the Iberian Peninsula and Morocco, wild clade (clade 6); except PI 652225 which is in the Middle East and Europe, landrace grade. In addition, all subsp. gummifer from Portugal (except A26381) were on a sub-clade with 100 and 98% bootstrap support using 10 or 30% missing imputed genotypes, respectively (Fig. 1 and Additional file 5: Figure S3). Accessions from Portugal were placed, according to their geographic areas, in three sub-clades: northern Portugal, central Portugal and southern Portugal. There are, however, a few exceptions: (i) accession PI 652222, which was collected in Vila Real (north of Portugal) was not contained in the northern Portugal sub-clade but in the central Portugal sub-clade, and (ii) accession PI 295862 (collected in Spain) was placed in the central Portugal sub-clade for ML tree obtained using 10% missing imputed genotypes. The northern Portugal sub-clade has a bootstrap support of 77 and 99% for data set using 18,565 and 43,713 SNPs, respectively. (Fig. 1 and Additional file 5: Figure S3). Accessions collected in the north central region of Morocco were on a sub-clade with 100% bootstrap support for both data sets used in our ML analyses. Accession PI 280706 collected in Chile was on the Iberian Peninsula and Morocco, wild clade (Fig. 1 and Additional file 5: Figure S3). Bootstrap supports were higher than 90% for both data sets for the six clades mentioned above (Fig. 1 and Additional file 5: Figure S3).
As expected, with almost all posterior probability values of one, our Bayesian tree (Additional file 6: Figure S4) has the same overall topology as the ML tree (Fig. 1), including the cultivation status and geographic locality of the accessions.
The SVDquartets analyses using 18,565 and 43,713 SNPs with a maximum of 10 or 30% missing imputed genotypes, respectively, also recovered the same six clades of the D. carota complex (Additional file 7: Figure S5 and Additional file 8: Figure S6). There are, however, some differences with the trees obtained using a concatenated approach: (1) clades North Africa, wild and North America, wild are sisters, (2) the lineage tree using 43,713 SNPs (Additional file 8: Figure S6) shows wild carrots from central Asia and cultivated carrots (subsp. sativus) from eastern did not form a clade. Rather, with a low bootstrap value of support of 41%, the clade of western cultivated carrots was sister to the members of wild carrots from central Asia, (3) using data sets with 43,713 and 18,565 SNPs, wild carrots from central Asia form an individual clade with 97 and 54%, respectively. However, the data set using 43,713 SNPs did not include accession PI 274297 (collected in Pakistan), and (4) lineage trees using the two data sets misplaced accessions relative to the dominant tree topologies obtained by ML analyses. The most notable case is that the subsp. gummifer clade from France is sister to the clade containing other accessions of the D. carota complex. Both data sets show that bootstrap values are higher than 90% for most of the clades of the D. carota complex. In addition, accessions from Turkey were in a clade with 100 and 94% bootstrap support on lineage tree using 18,565 and 43,713 SNPs, respectively (Additional file 7: Figure S5 and Additional file 8: Figure S6). With a bootstrap support of 50%, the Syria sub-clade is recovered using 18,565 SNPs, but it is not fully recovered when 43,713 SNPs were used since accession PI 652338 is not contained in the Syria sub-clade. Rather, this accession was sister to the Turkey sub-clade (Additional file 8: Figure S6). Wild accessions of D. carota subsp. carota collected in France form a clade in both data sets. The three sub-clades containing accessions collected in Portugal named north, central and south were also obtained in the SVDquartets analyses, but accessions PI 26404 and PI 26405, both collected in southern Portugal, were not placed in the clade expected.
Utilizing a coalescent approach consisting of 18,565 SNPs, the species tree (Additional file 9: Figure S7) indicates that D. carota subsp. capillifolius was sister to a clade formed by subsp. carota and subsp. sativus; and sister to them, a clade formed by subsp. gummifer and subsp. maximus. The species tree using 43,713 SNPs (figure not shown in this study) demonstrated the same topology as mentioned above, but with a higher bootstrap value for the clade containing subsp. carota and sativus: 100 vs. 53%.
Population structure
The Evanno method [84] indicated the best K value (number of populations) is two for both data sets. However, the next largest peak is at K = 9 and K = 3 for data matrix of 18,565 SNPs and 43,713 SNPs, respectively. Further, the third highest peak for data set with 43,713 SNPs (30% missing imputed genotypes) was obtained at K = 9 (Additional file 10: Figure S8 and Additional file 11: Figure S9). Waples and Gaggiotti [91] and Frantz et al. [92] indicate that the Evanno method tends to underestimate the number of genetic clusters. Other studies [33, 93] similarly obtained a false highest peak at K = 2 with the Evanno method due to a strong rejection of the null hypothesis of no structure (K = 1). Hence, the second highest peak obtained with 43,713 SNPs (K = 3) is likely caused by a strong rejection of the hypothesis of two clusters only. On the contrary, the highest likelihood value was obtained at K = 9 for both data sets (Additional file 10: Figure S8 and Additional file 11: Figure S9), which is concordant with the ML analyses of the Daucus carota complex (Fig. 1 and Additional file 5: Figure S3); so further discussion follows K = 9. STRUCTURE analyses show abundant admixture (Fig. 1 and Additional file 5: Figure S3), except for all accessions placed in cluster Iberian Peninsula and Morocco wild, which exhibit very low admixture. The D. carota complex is clustered according to wild vs. cultivated and its geographic origin. In addition, eastern members of subsp. sativus (cultivated carrot) are clustered with wild carrots from central Asia, rather than with all cultivated carrots. Furthermore, wild carrots from Syria also form a single cluster. All accessions of D. syrticus also cluster together and show no intraspecific allelic admixture (Fig. 1 and Additional file 5: Figure S3).
Morphological analysis
Graphical analyses of the 23 character state distributions used in our morphological study are shown in Additional file 12: Figure S10. This demonstrates tremendous variation and overlap of character states across all taxa, similar to the results obtained by Spooner et al. [31] and Arbizu et al. [29]. Hence, only a multivariate analysis using a combination of morphological characters distinguishes members of the D. carota complex (except subsp. capillifolius). The following 11 out of the 23 continuous morphological characters were significant in the F test for this analysis of D. carota, P ≤ 0.05 in the stepwise discriminant analysis (from most to least significant): plant height, mericarp length, number of spines on the four secondary mericarp ribs, secondary umbel diameter, number of bract lobe pairs, stem diameter, peripheral petal length, petiole diameter, number of umbel rays, leaf-sheath width, and leaf length including petiole.
Canonical variate analysis shows that all taxa are separated into four groups (D. carota subsp. capillifolius, subsp. carota, subsp. gummifer, and subsp. maximus). Accessions of D. carota hybrid are intermingled within the subsp. carota and subsp. capillifolius group. In addition, there are one and three accessions of subsp. gummifer and subsp. carota, respectively mixed with the subsp. maximus group (Fig. 2). Character state distributions of six significant discriminators are presented in Fig. 3, and character state overlap within these discriminators was very common.

Canonical variate analysis of the Daucus carota complex (subsp. sativus not considered) using 23 continuous morphological characters from the stem, leaf, flower, and mericarp structures

Box plots of six significant morphological characters obtained by canonical variate analysis that distinguish subspecies of the Daucus carota complex (subsp. sativus not considered). The box plots display individual plant values for median, 25 and 75% percentile, range, and outliers
Key to Daucus carota subsp. capillifolius, subsp. carota, subsp. gummifer, subsp. maximus and subsp. sativus
Note: As discussed in the text, there is tremendous overlap in the ranges of these key morphological data. These character state ranges are from Spooner et al. [31] reanalyzed here, considering subsp. maximus as a valid subspecies (with the caveat that a few accessions of subsp. carota are on this clade 6), and using only the interquartile ranges of the data (Fig. 3 and Additional file 12: Figure S10).
-
1.
Mature root not woody and edible, and highly pigmented (rarely white), root branching generally absent.........................subsp. sativus.
-
1.
Mature root woody and inedible, white to white-yellow, roots commonly branched.
-
2.
Petal color yellow, number umbel rays generally 18–25, mericarp length 4.5–5.5 mm...............................subsp. capillifolius.
-
2.
Petal color white, number of umbel rays generally 40–70, mericarp length 2–4 mm.
-
3.
Plant height 25–60 cm, mature primary umbel shape generally flat, mainly growing in maritime environments, leaves shiny and thick with broad segments................................subsp. gummifer.
-
3.
Plant height 70–110 cm, mature primary umbel shape generally convex, mainly growing in continental environments, leaves dull and thin with narrow segments.
-
4.
Leaf-sheath width 7.5–10 mm, bract 30–45 mm x 30–45 mm, number of spines on the secondary mericarp ribs 11–15, length of secondary mericarp spines 1.0–1.5 mm ...........subsp. carota.
-
4.
Leaf-sheath width 5.5–7.0 mm, bract 20–35 mm x 20–35 mm, number of spines on the secondary mericarp ribs 8–10, length of secondary mericarp spines 1.5–2.0 mm.........subsp. maximus.
-
4.
-
3.
-
2.
Discussion
Genotyping-by-sequencing data sets
Genome-wide data sets provide an opportunity to resolve difficult phylogenetic problems on species groups [94] that have failed using gene sequence phylogenies [45, 95, 96]. We here investigated the utility of SNPs obtained through GBS to resolve the classification of the D. carota complex. As expected, GBS identified abundant genome-wide SNPs (Additional file 2: Table S2), with varying levels of missing genotype calls. Due to low coverage sequencing, missing data has been reported as a problem in GBS [51, 97]. The impact of missing data on phylogenetic analyses has been extensively analyzed. Some studies [98–100] concluded that as long as the number of characters is abundant, phylogenetic inference is not sensitive to missing data. Other studies [101–107] concluded that missing data might affect phylogenetic reconstruction. Missing data can be addressed with imputation [108], a statistical method that replaces missing data by estimated values [97]. Yang et al. [109], using simulated data, reported that the accuracy of Beagle imputation software is 0.99 for a missing rate of 10 or 30%. Therefore, to explore further the utility of missing imputed genotypes on phylogenetic analysis, we employed missing rates of 10 or 30%.
Phylogenomics of Daucus
The phylogenetic relationships of Daucus were previously reported by Spalik and Downie [88], Spalik et al. [89], Spooner et al. [35], Arbizu et al. [36, 90], Lee and Park [27], and Banasiak et al. [37]; all findings supported two main clades. Our two data sets with 10 or 30% missing imputed genotypes also reconstructed the two main clades of Daucus, A and B (Additional file 3: Figure S1 and Additional file 4: Figure S2). Significant differences were not observed between the two Daucus topologies using two data sets (10,914 SNPs vs. 38,920 SNPs), even though the data set with 10% missing imputed genotypes had 72% fewer SNPs. Arbizu et al. [36] also mentioned that a reduction in the amount of DNA sequence data did not influence the topology of Daucus. However, bootstrap values of some clades using 30% missing imputed genotypes (38,920 SNPs) were higher. Kimball and Braun [110] also indicated that they obtained a modestly greater bootstrap support by increasing their number of sites approximately 3-fold.
Similar to the consensus trees of Daucus obtained by Spooner et al. [35] and Arbizu et al. [36, 90], clade A in our ML tree consists of the D. carota complex, D. syrticus, D. aureus, D. crinitus and a related ingroup genus (Rouya). Concordant with these results, D. syrticus is the appropriate outgroup for the study of the D. carota complex. In addition, clade B consists of species of the D. guttatus complex and close relatives. Maximum likelihood analyses confirmed accessions of D. guttatus were recovered as a monophyletic clade as similarly reported in Arbizu et al. [90]. Originally, accession numbers A25731, A25732 and A25778 were identified by the GRIN database as D. carota. However, based on our ML results and morphological analysis of members of the D. guttatus complex [90, 111], these three accessions were re-identified as D. guttatus. The paraphyly of the genus Daucus (circumscribed containing Rouya polygama) was extensively documented using plastid or ribosomal DNA characters [35, 37]. Recent studies [37, 112], using nrDNA ITS sequences and plastid sequences also showed that Rouya polygama groups within the Daucus clade.
Classification of the D. carota complex
Daucus carota sensu lato is a taxonomically complicated group that has attracted the attention of many investigators. Previous molecular and morphological studies have failed to distinguish many of the subspecies of the D. carota complex. Our present GBS data using imputed data for missing rates of 10 or 30% recovered a clade containing the members of the D. carota complex and D. syrticus, all consisting of 2n = 18 chromosomes, matching the results of prior investigators. Furthermore, concordant with previous work conducted by Iorizzo et al. [33, 69], our Bayesian and ML trees partitioned accessions of D. carota according to their improvement status (cultivated, landrace, wild) and geographic origin. We here identified one grade and one extra clade, respectively: (1) Middle East and Europe, landrace, and (2) Iberian Peninsula and Morocco, wild (Fig. 1 and Additional file 5: Figure S3). Using 3,326 SNPs in expressed genes, Iorizzo et al. [33] indicated that eastern wild accessions of carrots (i.e., collected in the Middle East, central Asia and eastern Asia) are most closely related to cultivated carrots. Our present data, using additional samples from more diverse geographic origins and all genomic regions rather than only expressed genes, revealed that eastern members of subsp. sativus (cultivated carrot) are in a clade with wild carrots from central Asia, rather than with all cultivated carrots as shown in the ML tree of Iorizzo et al. [33]. Considerable allelic admixture shown by STRUCTURE results is in agreement with the cross-fertilization habit within carrots and the introgression between wild populations and cultivated carrots [113–115]. Even though China and Pakistan geographically do not belong to the central Asia region, our study placed two accessions collected in northern Pakistan (Parachinar) and one in northwestern China (Xinjiang) in the central Asia wild clade, yet these collections are very close to the central Asia region. Future work will benefit from extra samples from neighboring countries of the central Asia geographic region. Rong et al. [26] also used SNPs and samples of wild carrots from different geographic origins except central Asia, and their Bayesian tree showed that wild carrots from western Asia are the closest relatives to eastern cultivated carrots.
Extensive morphological studies conducted by Small [3], Reduron [116], Arbizu et al. [29] and Spooner et al. [31] identified two comprehensive morphological groups of wild D. carota (carota and gummifer). The latter authors proposed two morphologically supported taxa for the D. carota complex, D. carota subsp. carota and subsp. gummifer. However, these subspecies have not been recognized by a recent molecular study [36]. Similarly, the present study failed to identify two unique groups, even though we used genome-wide SNPs and 144 samples of D. carota.
Using fruit morphological descriptors, Mezghani et al. [30] separated subsp. maximus from the other subspecies of D. carota. An investigation [32] attempting to distinguish the subspecies of D. carota native to Portugal provided further evidence to separate subsp. maximus from other taxa by morphometric analysis of the fruits and chemical characterization of essential oils, concluding, as Sáenz de Rivas and Heywood [117], that subsp. maximus should be considered a species (i.e., D. maximus) rather than a subspecies of D. carota. Our discriminant analysis also separated subsp. maximus from the other subspecies of D. carota, but there are no individual characters that alone affect this separation and they extensively overlapped with other subspecies (Additional file 12: Figure S10). These facts, and the intercrossability of subsp. maximus with the other subspecies, and all cladistics analyses to date, lead us to retain ‘maximus′ as a provisional subspecies of D. carota, awaiting further studies using additional collections. Arbizu et al. [29] made the same case to recognize D. capillifolius as a subspecies of D. carota.
A single clade exclusively containing D. carota subsp. carota was not obtained. Rather, at 100% bootstrap value or posterior probability of 1, we observed a single clade containing cultivated carrot (subsp. sativus), and the wild subspecies capillifolius, carota (except those from northern and central Portugal), and gummifer from France. Similar to Iorizzo et al. [33], subsp. carota clade was subdivided according to their geographic origins as follows: (i) central Asia, wild, (ii) the Balkan Peninsula and the Middle East, wild (iii) North America and Europe, wild, (iv) North Africa, wild, and (v) Iberian Peninsula and Morocco, wild. Our ML and STRUCTURE results demonstrate that eastern cultivated carrots originated from populations of D. carota from central Asia.
A strict concept for monophyly failed for the subspecies of the D. carota complex. Daucus carota subsp. gummifer, used here is a broad sense to include other potential subspecies, as outlined in Spooner et al. [31] is a morphologically coherent set of morphotypes restricted to areas within a few km of the Atlantic Ocean and the Mediterranean Sea [31], and subsp. capillifolius is a morphologically distinctive taxon from Tunisia and Libya. Subspecies capillifolius occurs within the clade containing subsp. carota. Moreover, subsp. gummifer occurs in different clades containing geographically isolated populations of subsp. carota in Europe (France, Portugal, and Italy) and subsp. maximus in northern Africa (Morocco). GBS results, therefore, do not support the subspecies of D. carota to form monophyletic lineages, except subsp. capillifolius, which is an apospecies (sensu Olmstead [118]).
Accessions originally labeled as D. carota subsp. maritimus were placed in the Iberian Peninsula and Morocco, wild clade. Arbizu et al. [36], using 94 nuclear orthologs, also reported that accessions of D. carota from Portugal, Spain and Morocco were placed in an individual sub-clade with 100% bootstrap support. Rong et al. [26] marked one group of their Bayesian tree containing subspecies of D. carota as “Mediterranean, southern Europe”. Within this group, there is a sub-clade consisting of samples from Portugal and Spain labeled subsp. carota, subsp. maritimus and subsp. maximus. However, our results do not distinguish them from other subspecies (subsp. azoricus and subsp. hispanicus not examined here). Spooner et al. [31] considered subsp. maritimus and subsp. major in the D. carota subsp. carota group. Similarly, Bolòs and Vigo [119] and Pujadas Salvà [19] considered that subsp. maritimus corresponded to subsp. carota. Consequently, we suggest that morphotypes labeled as subsp. carota, subsp. major and subsp. maritimus from the Iberian Peninsula are subsp. carota. In addition, we consider that accessions from the Iberian Peninsula and Morocco originally labeled as subsp. maximus represent this subspecies since our morphological analysis distinguish them from subsp. carota. Within Portugal, it is very likely that subsp. carota predominates in the northern area of the country, while subsp. maximus is restricted to parts of central and southern Portugal and Morocco.
Using nuclear ribosomal DNA ITS, Lee and Park [27] grouped subsp. halophilus, subsp. azoricus, subsp. gadecaei, subsp. drepanensis and subsp. gummifer into a single clade. The last two subspecies correspond to subsp. gummifer [31] and in the present study accession Ames 31194, initially labeled as subsp. halophilus, is placed together with other accessions of subsp. gummifer. Matias Vaz [28] also reported that samples from São Miguel Island are more closely related to subsp. gummifer and subsp. halophilus. Therefore, we propose subsp. azoricus should be considered as D. carota subsp. gummifer. Another subspecies of D. carota, subsp. majoricus (A. Pujadas) is distributed in the Mallorca, Cabrera and surrounding islands [19], and based on the detailed morphological description provided by Martínez-Flores et al. [120], we consider it very likely this subspecies also corresponds to the subsp. gummifer.
Lineage trees, obtained using 10 or 30% missing imputed genotypes and a very fast quartet-based method under the coalescent model implemented in SVDquartets software [78], show differences in the topology compared to Bayesian and ML trees. Discordance between ML and quartet phylogenies of Carex sect. Racemosae also were reported by Massatti et al. [49]. A primary concordance tree indicated that the subspecies of D. carota have low concordance factors [36], suggesting high discordance between genes in wild carrots; probably caused by recombination, hybridization and introgression [41], and incomplete lineage sorting (ILS) [44]. Concatenation methods are often more accurate than coalescent-based methods if ILS is low [121–123]. Therefore, perhaps the main cause of discordance between genes of the subspecies of D. carota is not high ILS, but other factors. A species tree using HKY model of evolution reported by Arbizu et al. [36] shows that subsp. carota is sister subsp. gummifer, and sister to them is subsp. capillifolius. In the present study, we obtained a similar species tree (Additional file 9: Figure S7). The difference is that subsp. maximus was considered as a distinct subspecies in the present study.
Cultivated carrot (D. carota subsp. sativus) is the most important member in the Apiaceae family in terms of economy and nutrition [115, 124, 125], and is considered the second most popular vegetable in the world after potato [126]. Genetic diversity of wild carrots can be exploited to meet future challenges such as nutraceutical food for a growing population, cultivars adapted to climate change, among others. Wild Daucus species possessing 2n = 18 chromosomes represent the main gene pool for breeding work in carrots. Here we also propose that wild carrots (subsp. maximus) from the Iberian Peninsula and Morocco may be used as a new source of genes for the development of new carrot cultivars. Unique essential oils are present in D. carota subsp. maximus [32, 127] and are considered to confer insecticidal properties. Indeed, this compound may be useful in new carrot commercial hybrids for organic production system.
Conclusions
Single nucleotide polymorphisms (SNPs) provide robust markers for the study of population structure and phylogenetics of Daucus carota. Genotyping-by-sequencing identified numerous SNPs distributed along the nine chromosomes of D. carota. Our data sets with 10 or 30% missing data with imputed data additions tested the utility of genome-wide SNPs in clarifying the phylogenetics and population structure of D. carota and related species. The phylogenomics of Daucus was well resolved, showing higher bootstrap support for most clades using the GBS data set with a larger number of SNPs, 38,920 sites (30% missing imputed data). However, both data sets used to study the D. carota complex (18,565 SNPs vs. 43,713 SNPs) revealed equal topology and very similar bootstrap values for most clades. One new clade and one grade were detected (Iberian Peninsula and Morocco wild, and Middle East and Europe landrace). The Bayesian structure results are concordant with the maximum likelihood tree, except that an extra cluster consisting of wild carrots from Syria was detected (nine in total). Wild carrots from central Asia were more related to eastern cultivars further supporting the hypothesis that cultivated carrot originated in central Asia. Low allelic admixture was shown for wild carrots belonging to the Iberian Peninsula and Morocco clade. The morphologically distinct wild carrot subsp. capillifolius, from Tunisia and immediately adjacent Libya, was placed in a sub-clade of subsp. carota from North Africa and is supported as an apospecies. Daucus carota subsp. gummifer is a morphologically coherent set of morphotypes restricted to areas within a few km of the Atlantic Ocean and the Mediterranean Sea. It is here supported as a series of apospecies with separate and independent origins from subsp. carota and subsp. maximus in similar maritime habitats; the nomenclature of these awaits further study. Discriminant analysis distinguished 11 significant morphological descriptors that best distinguish the subspecies of the D. carota complex. Phylogenies obtained by Bayesian and ML method were very similar. However, these concatenation approaches showed disagreement with a coalescent-based method probably due to varying ILS levels in the genome of the subspecies of D. carota. Our study demonstrates the ability of GBS to distinguish population and subspecies structure in D. carota. Further studies using additional collections of D. carota from a wider geographic area and other potential subspecies (these field collections are in progress), and examination of relevant type material, are needed to provide a better understanding of taxonomic variation and nomenclature in the D. carota complex. This was the very process by which Arbizu et al. [90] and Martínez-Flores et al. [111] solved the species boundaries in another problematical group in Daucus, a procedure we are pursuing here.
References
Sáenz Laín C. Research on Daucus L. (Umbelliferae). An Jard Bot Mad. 1981;37:481–533.
Heywood VH. The Daucus carota-gingidium complex. Flora Europaea Notulae Systematicae. 1968;7:65–8.
Small E. A numerical taxonomic analysis of the Daucus carota complex. Can J Bot. 1978;56:248–76.
Thellung A: Daucus. In: Hegi G, editor. Illustrierte Flora von Mitteleuropa. München: JF Lehmanns Verlag; 1926. p. 1501–26.
Quézel P, Santa S: Daucus. In: Quézel P, Santa S, editors. Nouvelle Flore de L’Algérie et des Régions Désertiques Méridionales. Paris: Éditions du Centre National de la Recherche Scientifique; 1963. p. 659–63.
Schäfer H. Flora of the Azores. Weikersheim: Margraf Publishers; 2005.
Heywood VH. Daucus. Feddes Repert. 1968;79:1–68.
Pujadas Salvà AJ. Daucus. In: Nieto-Feliner G, Jury SL, Herrero A, editors. Flora Iberica: Plantas vasculares de la Península Ibérica e islas Baleares. Madrid: Real Jardín Botánico CSIC; 2003. p. 97–125.
Jafri SMH, El-Gadi A. Daucus. In: Jafri SMH, El-Gadi A, editors. Flora of Libya. Tripoli: Al-Faateh University, Faculty of Science, Department of Botany;1985. p. 130–44.
Jury JL. Daucus. In: Valdés B, Rejdali M, Achhal El Kadmiri A, Jury JL, Montserrat JM, editors. Catalogue des plantes vasculaires du Nord de Maroc, incluant des clés d’identification. Madrid: Consejo Superior de Investigaciones Científicas; 2002. p. 467–9.
Zohary M. Flora Palaestina. Jerusalem: The Israel Academy of Sciences and Humanities; 1972.
Franco JA. Nova Flora de Portugal (Continente e Açores). Lisbon: Sociedade Astória; 1971.
Mouterde P. Nouvelle Flore de Liban et de la Syrie. Beirut: Dar El-Machreq; 1966.
Pottier-Alapetite G. Daucus. In: Pottier-Alapetite G, editor. Flore de la Tunisie: Angiospermes-Dicotylédones (Apetales – Dialypetales). Tunis: Ministère de l’Enseignement Supérieur et de la Recherche Scientifique et le Ministère de l’Agriculture; 1979. p. 615–21.
Le Floc’h É, Boulos L, Véla E. Catalogue synonymique commenté de la flore de Tunisie. Tunis: République Tunisienne, Ministère de l’Environnement Et Du Développment Durable Banque Nationale de Gènes; 2010.
Cullen J. Daucus. In: Davis PH, editor. Flora of Turkey and the East Aegean Islands. Edinburgh: Edinburgh University Press; 1972. p. 531–6.
Heywood VH. Daucus. In: Tutin TG, Heywood VH, Burges NA, Valentine DH, Walters SM, Webb DA, editors. Flora Europaea. Cambridge: Cambridge University Press; 1968. p. 373–5.
Arenas JA, García-Martin F. Atlas carpológico y corológico de la subfamilia Apioideae Drude (Umbelliferae) en España peninsular y Baleares. Ruizia. 1993;12:222–34.
Pujadas Salvà AJ. El complejo de Daucus carota L. (Apiaceae) en la flora Ibérica. An Jard Bot Mad. 2002;59:368–75.
St. Pierre MD, Bayer RJ, Weis IM. An isozyme-based assessment of the genetic variability within the Daucus carota complex (Apiaceae: Caucalideae). Can J Bot. 1990;68:2449–57.
Nakajima Y, Oeda K, Yamamoto T. Characterization of genetic diversity of nuclear and mitochondrial genomes in Daucus varieties by RAPD and AFLP. Plant Cell Rep. 1998;17:848–53.
Vivek BS, Simon PW. Genetic relationships and diversity in carrot and other Daucus taxa based on nuclear restriction fragment length polymorphisms. J Amer Soc Hort Sci. 1998;123:1053–7.
Vivek BS, Simon PW. Phylogeny and relationships in Daucus based on restriction fragment length polymorphisms (RFLPs) of the chloroplast and mitochondrial genomes. Euphytica. 1999;105:183–9.
Shim SI, Jørgensen RB. Genetic structure in cultivated and wild carrots (Daucus carota L.) revealed by AFLP analysis. Theor Appl Genet. 2000;101:227–33.
Bradeen JM, Bach IC, Briard M, le Clerc V, Grzebelus D, Senalik DA, Simon PW. Molecular diversity analysis of cultivated carrot (Daucus carota L.) and wild Daucus populations reveals a genetically nonstructured composition. J Amer Soc Hort Sci. 2002;127:383–91.
Rong J, Lammers Y, Strasburg JL, Schidlo NS, Ariyurek Y, de Jong TJ, Klinkhamer PGL, Smulders MJM, Vrieling K. New insights into domestication of carrot from root transcriptome analyses. BMC Genomics. 2014;15:895.
Lee BY, Park C. Molecular phylogeny of Daucus (Apiaceae): Evidence from nuclear ribosomal DNA ITS sequences. J Species Res. 2014;3:39–52.
Matias Vaz AM. Estudo morfológico e filogenético das subespécies Daucus carota ssp. azoricus e Daucus carota ssp. maritimus na ilha de S. Miguel. Ponta delgada: Universidade dos Açores, M.Sc. Thesis; 2014.
Arbizu C, Reitsma KR, Simon PW, Spooner DM. Morphometrics of Daucus (Apiaceae): A counterpart to a phylogenomic study. Am J Bot. 2014;101:2005–16.
Mezghani N, Zaouali I, Amri WB, Rouz S, Simon PW, Hannachi C, Ghrabi Z, Neffati M, Bouzbida B, Spooner DM. Fruit morphological descriptors as a tool for discrimination of Daucus L. germplasm. Genet Resour Crop Evol. 2014;61:499–510.
Spooner DM, Widrlechner MP, Reitsma KR, Palmquist DE, Rouz S, Ghrabi-Gammar Z, Neffati M, Bouzbida B, Ouabbou H, El Koudrin M, Simon PW. Reassessment of practical subspecies identifications of the USDA Daucus carota L. germplasm collection: Morphological data. Crop Sci. 2014;54:706–18.
Tavares AC, Loureiro J, Castro S, Coutinho AP, Paiva J, Cavaleiro C, Salgueiro L, Canhoto JM. Assessment of Daucus carota L. (Apiaceae) subspecies by chemotaxonomic and DNA content analyses. Biochem Syst Ecol. 2014;55:222–30.
Iorizzo M, Senalik DA, Ellison SL, Grzebelus D, Cavagnaro PF, Allender C, Brunet J, Spooner DM, Van Deynze A, Simon PW. Genetic structure and domestication of carrot (Daucus carota subsp. sativus) (Apiaceae). Am J Bot. 2013;100:930–8.
Martínez-Flores F, Juan A, Crespo MB. Molecular and morphological evidence supports the resurrection of a neglected species of Daucus (Apiaceae) from North Africa. Bot J Linn Soc. 2012;169:320–37.
Spooner D, Rojas P, Bonierbale M, Mueller LA, Srivastav M, Senalik D, Simon P. Molecular phylogeny of Daucus (Apiaceae). Syst Bot. 2013;38:850–7.
Arbizu C, Ruess H, Senalik D, Simon PW, Spooner DM. Phylogenomics of the carrot genus (Daucus, Apiaceae). Am J Bot. 2014;101:1666–85.
Banasiak L, Wojewódzka A, Baczyński J, Reduron J-P, Piwczyński M, Kurzyna-Młynik R, Gutaker R, Czarnocka-Cieciura A, Kosmala-Grzechnik S, Spalik K. Phylogeny of Apiaceae subtribe Daucinae and the taxonomic delineation of its genera. Taxon. 2016;65:563–85.
Lemmon EM, Lemmon AR. High-throughput genomic data in systematics and phylogenetics. Annu Rev Ecol Evol Syst. 2013;44:99–121.
Felsenstein J. Inferring Phylogenies. Massachusetts: Sunderland; 2004.
Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 2006;55:21–30.
Rieseberg LH, Baird SJE, Gardner KA. Hybridization, introgression, and linkage evolution. Plant Mol Biol. 2000;42:205–24.
Page RDM, Charleston MA. From gene to organismal phylogeny: Reconciled trees and the gene tree/species tree problem. Mol Phylogenet Evol. 1997;7:231–40.
Doolittle WF. Lateral genomics. Trends Cell Biol. 1999;9:M5–8.
Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol. 1988;5:568–83.
Escudero M, Eaton DAR, Hahn M, Hipp AL. Genotyping-by-sequencing as a tool to infer phylogeny and ancestral hybridization: A case study in Carex (Cyperaceae). Mol Phylogenet Evol. 2014;79:359–67.
Good JM. Reduced representation methods for subgenomic enrichment and next-generation sequencing. In: Orgogozo V, Rockman MV, editors. Molecular Methods for Evolutionary Genetics, Methods in Molecular Biology. New York: Springer; 2011. p. 85–103.
Emerson KJ, Merz CR, Catchen JM, Hohenlohe PA, Cresko WA, Bradshaw WE, Holzapfel CM. Resolving postglacial phylogeography using high-throughput sequencing. Proc Natl Acad Sci U S A. 2010;107:16196–200.
Hipp AL, Eaton DAR, Cavender-Bares J, Fitzek E, Nipper R, Manos PS. A framework phylogeny of the American oak clade based on sequenced RAD data. PLoS One. 2014;9:e93975.
Massatti R, Reznicek AA, Knowles LL. Utilizing RADseq data for phylogenetic analysis of challenging taxonomic group: A case study in Carex sect. Racemosae. Am J Bot. 2016;103:337–47.
Wang XQ, Zhao L, Eaton DAR, Li DZ, Guo ZH. Identification of SNP markers for inferring phylogeny in temperate bamboos (Poaceae: Bambusoideae) using RAD sequencing. Mol Ecol Resour. 2013;13:938–45.
Wong MML, Gujaria-Verma N, Ramsay L, Yuan HY, Caron C, Diapari M, Vandenberg A, Bett KE. Classification and characterization of species within the genus Lens using genotyping-by-sequencing (GBS). PLoS One. 2015;10:e122025.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:e19379.
Beissinger TM, Hirsch CN, Sekhon RS, Foerster JM, Johnson JM, Muttoni G, Vaillancourt B, Buell CR, Kaeppler SM, de Leon N. Marker density and read depth for genotyping populations using genotyping-by-sequencing. Genetics. 2013;193:1073–81.
Liu H, Bayer M, Druka A, Russell JR, Hackett CA, Poland J, Ramsay L, Hedley PE, Waugh R. An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genomics. 2014;15:104.
Lu F, Romay MC, Glaubitz JC, Bradbury PJ, Elshire RJ, Wang T, Li Y, Li Y, Semagn K, Zhang X, Hernandez AG, Mikel MA, Soifer I, Barad O, Buckler ES. High-resolution genetic mapping of maize pan-genome sequence anchors. Nat Commun. 2015;6:6914.
Uitdewilligen JGAML, Wolters AA, D’hoop BB, Borm TJA, Visser RGF, van Eck HJ. A next-generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS One. 2013;8:e62355.
Ramstein GP, Lipka AE, Lu F, Costich DE, Cherney JH, Buckler ES, Casler MD. Genome-wide association study based on multiple imputation with low-depth sequencing data: application to biofuel traits in reed canarygrass. G3:Genes|Genomes|Genet. 2015;5:891–909.
Spindel J, Wright M, Chen C, Cobb J, Gage J, Harrington S, Lorieux M, Ahmadi N, McCouch S. Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor Appl Genet. 2013;126:2699–716.
Jarquín D, Kocak K, Posadas L, Hyma K, Jedlicka J, Graef G, Lorenz A. Genotyping by sequencing for genomic prediction in a soybean population. BMC Genomics. 2014;15:740.
Lipka AE, Lu F, Cherney JH, Buckler ES, Casler MD, Costich DE. Accelerating the switchgrass (Panicum virgatum L.) breeding cycle using genomic selection approaches. PLoS One. 2014;9:e112227.
Poland J, Endelman J, Dawson J, Rutkoski J, Shuangye W, Manes Y, Dreisigacker S, Crossa J, Sánchez-Villeda H, Sorrels M, Jannink J-L. Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome. 2012;5:103–13.
Bajaj D, Das S, Badoni S, Kumar V, Singh M, Bansal KC, Tyagi AK, Parida SK. Genome-wide high-throughput SNP discovery and genotyping for understanding natural (functional) allelic diversity and domestication patterns in wild chickpea. Sci Rep. 2015;5:12468.
Labate JA, Robertson LD, Strickler SR, Mueller LA. Genetic structure of the four wild tomato species in the Solanum peruvianum s.l. species complex. Genome. 2014;57:169–80.
Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1990;12:13–5.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–40.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One. 2014;9:e90346.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Iorizzo M, Ellison S, Senalik D, Zeng P, Satapoomin P, Huang J, Bowman M, Iovene M, Sanseverino W, Cavagnaro P, Yildiz M, Macko-Podgórni A, Moranska E, Grzebelus E, Grzebelus D, Ashrafi H, Zheng Z, Cheng S, Spooner D, Van Deynze A, Simon P. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asteroid genome evolution. Nat Genet. 2016;48:657–66.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–23.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPREs science gateway for inference of large phylogenetic trees. New Orleans: In Proceedings of the Gateway Computing Environments Workshop (GCE); 2010. p. 1–8.
Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: Mode models, new heuristics and parallel computing. Nat Methods. 2012;9:772.
Summer JG, Jarvis PD, Fernández-Sánchez J, Kaine BT, Woodhams MD, Holland BR. Is the general time-reversible model bad for phylogenetics? Syst Biol. 2012;61:1069–74.
Stamatakis A. The RAxML v8.2.X manual. Heidelberg: Heidelberg Institute for Theoretical Studies; 2016.
Chifman J, Kubatko L. Quartet inference from SNP under the coalescent model. Bioinformatics. 2014;30:3317–24.
Swofford D. PAUP*: Phylogenetic analysis using parsimony (*and other methods). version 4.0a147. Sunderland: Sinauer Associates; 2002.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Lischer HEL, Excoffier L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 2012;28:298–9.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Earl DA, von Holdt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet Resour. 2012;4:359–61.
Evanno G, Regnaut S, Gouder J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–6.
Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–8.
R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2015.
Spalik K, Downie SR. Intercontinental disjunctions in Cryptotaenia (Apiaceae, Oenantheae): an appraisal using molecular data. J Biogeogr. 2007;34:2039–57.
Spalik K, Piwczyński M, Danderson CA, Kurzyna-Młynik R, Bone TS, Downie SR. Amphitropic amphiantarctic disjunctions in Apiaceae subfamily Apioideae. J Biogeogr. 2010;37:1977–94.
Arbizu CI, Simon PW, Martínez-Flores F, Ruess H, Crespo MB, Spooner DM. Integrated molecular and morphological studies of the Daucus guttatus complex (Apiaceae). Syst Bot. 2016;41:479–92.
Waples RS, Gaggiotti O. What is a population? an empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol. 2006;15:1419–39.
Frantz AC, Cellina S, Krier A, Schley L, Burke T. Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: clusters or isolation by distance? J Appl Ecol. 2009;46:493–505.
Vigouroux Y, Glaubitz JC, Matsuoka Y, Goodman MM, Sánchez GJ, Doebley J. Population structure and genetic diversity of new world maize races assessed by DNA microsatellites. Am J Bot. 2008;95:1240–53.
Rannala B, Yang Z. Phylogenetic inference using whole genomes. Annu Rev Hum Genet. 2008;9:217–31.
Qiu Y, Lee J, Bernasconi-Quadroni F, Soltis DE, Soltis PS, Zanis M, Zimmer EA, Chen Z, Savolainen V, Chase MW. The earliest angiosperms: evidence from mitochondrial, plastid and nuclear genomes. Nature. 1999;402:404–7.
Soltis PS, Soltis DE, Chase MW. Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature. 1999;402:402–4.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Rev Genet. 2011;12:499–510.
Philippe H, Snell EA, Bapteste E, Lopez P, Holland PWH, Casane D. Phylogenomics of eukaryotes: impact of missing data on large alignments. Mol Biol Evol. 2004;21:1740–52.
Wiens JJ, Morrill MC. Missing data in phylogenetic analysis: reconciling results from simulations and empirical data. Syst Biol. 2011;60:719–31.
Jiang W, Chen S-Y, Wang H, Li D, Wiens JJ. Should genes with missing data be excluded from phylogenetic analyses? Mol Phylogenet Evol. 2014;80:308–18.
Hartmann S, Vision TJ. Using ESTs for phylogenomics: can one accurately infer a phylogenetic tree from a gappy alignment? BMC Evol Biol. 2008;8:95.
Lemmon AR, Brown JM, Stanger-Hall K, Lemmon EM. The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst Biol. 2009;58:130–45.
Simmons MP. Misleading results of likelihood-based phylogenetic analyses in the presence of missing data. Cladistics. 2012;28:208–22.
Hovmöller R, Knowles LL, Kubatko LS. Effects of missing data on species tree estimation under the coalescent. Mol Phylogenet Evol. 2013;69:1057–62.
Kvist S, Siddall ME. Phylogenomics of Annelida revisited: a cladistics approach using genome-wide expressed sequence tag data mining and examining the effects of missing data. Cladistics. 2013;29:435–48.
Xi Z, Liu L, Davis CC. The impact of missing data on species tree estimation. Mol Biol Evol. 2015;33:838–60.
Zheng Y, Wiens JJ. Do missing data influence the accuracy of divergence-time estimation with BEAST? Mol Phylogenet Evol. 2015;85:41–9.
Poland JA, Rife TW. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome. 2012;5:92–102.
Yang Y, Wang Q, Chen Q, Liao R, Zhang X, Yang H, Zheng Y, Zhang Z, Pan Y. A new genotype imputation method with tolerance to high missing rate and rare variants. PLoS One. 2014;9:e101025.
Kimball RT, Braun EL. Does more sequence data improve estimates of galliform phylogeny? analyses of a rapid radiation using a complete data matrix. PeerJ. 2014;2:e361.
Martínez-Flores F, Arbizu CI, Reitsma K, Juan A, Simon PW, Spooner DM, Crespo MB. Lectotype designation for seven species names in the Daucus guttatus complex (Apiaceae) from the central and eastern Mediterranean basin. Syst Bot. 2016;41:464–78.
Lyskov D, Degtjareva G, Samigullin T, Pimenov M. Systematic placement of the Turkish endemic genus Ekimia (Apiaceae) based on morphological and molecular data. Turk J Bot. 2015;39:673–80.
Heywood VH. Relationships and evolution in the Daucus carota complex. Israel J Bot. 1983;32:51–65.
Wijnheijmer EHM, Brandenburg WA, Ter Borg SJ. Interactions between wild and cultivated carrots (Daucus carota L.) in the Netherlands. Euphytica. 1989;40:147–54.
Simon PW. Domestication, historical development, and modern breeding of carrot. Plant Breed Rev. 2000;19:157–90.
Reduron J-P. Ombellifères de France, vol. 2. Nercillac: Société botanique du Centre-Ouest; 2007. p. 960–1099.
Sáenz de Rivas C, Heywood VH. Estudio preliminar sobre los Daucus de la España Peninsular. Anal Inst Bot Cavanilles. 1974;31:97–118.
Olmstead RG. Species concepts and plesiomorphic species. Syst Bot. 1995;20:623–30.
Bolòs O, Vigo J. Flora dels Països Catalans. Barcelona: Editorial Barcino; 1990.
Martínez-Flores F, Crespo MB, Juan Gallardo A. Nuevos datos sobre el endemismo balear Daucus carota L. subsp. majoricus A. Pujadas (Apiaceae) en el archipiélago de Cabrera. Orsis. 2011;25:7–28.
Mirarab S, Bayzid MS, Boussau B, Warnow T. Statistical binning enables an accurate coalescent-based estimation of the avian tree. Science. 2014;346:1250463.
Mirarab S, Bayzid MS, Warnow T. Evaluating summary methods for multilocus species tree estimation in the presence of incomplete lineage sorting. Syst Biol. 2014;65:366–80.
Chou J, Gupta A, Yaduvanshi S, Davidson R, Nute M, Mirarab S, Warnow T. A comparative study of SVDquartets and other coalescent-based species tree estimation methods. BMC Genomics. 2015;16(Suppl10):S2.
Rubatzky VE, Quiroz CF, Simon PW. Carrots and related vegetable Umbelliferae. New York: CABI; 1999.
Vilela NJ. Cenoura: Um alimento nobre na mesa popular. Hort Bras. 2004;22:1–2.
Heywood VH. The socio-economic importance of the Apiales. J Fac Pharm Istambul. 2014;44:113–30.
Saad H-EA, El-Sharkawy SH, Halim AF. Essentials oils of Daucus carota ssp. maximus. Pharm Acta Helv. 1995;70:79–84.
Acknowledgments
The authors thank the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing facilities and services. We are grateful to Kathleen Reitsma and staff at the North Central Regional Plant Introduction Station in Ames, Iowa for providing germplasm. C. Arbizu thanks David Baum for discussion of the preliminary results of this study. This paper represents partial fulfillment of a Ph.D. degree for CA in Plant Breeding and Plant Genetics at the University of Wisconsin-Madison.
Funding
This work is supported by the USDA Agricultural Research Service. C. Arbizu is partly funded by the National Council of Science and Technology of Perú (Concytec).
Availability of data and material
The data sets supporting the conclusions of this study are available in the Dryad Digital Repository http://dx.doi.org/10.5061/dryad.319sr.
Authors’ contributions
C. Arbizu, P. Simon and D. Spooner conceived and designed the study. C. Arbizu and S. Ellison performed the experiments. C. Arbizu, S. Ellison and D. Senalik analyzed the data. D. Spooner and P. Simon supervised the work. C. Arbizu wrote the manuscript. All authors read, revised and approved the final manuscript.
Competing interests
The authors declare that they do not have competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
The 162 accessions of Daucus, and two accessions of related genera characterized in this study, improvement status, locality information and new identification. (PDF 308 kb)
Additional file 2: Table S2.
Summary of processing GBS data of Daucus. (PDF 98 kb)
Additional file 3: Figure S1.
Phylogenomics of Daucus from a maximum likelihood analysis using 164 accessions and 10,814 SNPs (10% missing imputed genotypes) obtained by GBS. Numbers above branches represent bootstrap values, with only values higher than 70% shown. Names given to clades refer to the geographic origin and improvement status of the accessions of the D. carota complex. Clades A and B corresponds to the two main groups of the Daucus phylogeny. (PDF 1.39 mb)
Additional file 4: Figure S2.
Phylogenomics of Daucus from a maximum likelihood analysis using 164 accessions and 38,920 SNPs (30% missing imputed genotypes) obtained by GBS. Numbers above branches represent bootstrap values, with only values higher than 70% shown. Names given to clades refer to the geographic origin and improvement status of the accessions of the D. carota complex. Clades A and B corresponds to the two main groups of the Daucus phylogeny. (PDF 1.34 mb)
Additional file 5: Figure S3.
Maximum likelihood reconstruction and structure of the genetic diversity of 144 accessions of the Daucus carota complex and outgroups using 43,713 SNPs (30% missing imputed genotypes) obtained by GBS. Each accession is represented by a horizontal bar, and each color corresponds to a population (nine in total). Numbers above branches represent bootstrap values, with only values higher than 70% shown. Names given to clades refer to the geographic origin and improvement status of the accessions of the D. carota complex. The outgroup taxon is D. syrticus. (PDF 1.46 mb)
Additional file 6: Figure S4.
Bayesian phylogenetic tree of 144 accessions of the Daucus carota complex and outgroups using 18,565 SNPs (10% missing imputed genotypes) obtained by GBS. Numbers above the branches represent posterior probabilities, with only values higher than 0.7 shown. Names given to clades refer to the geographic origin and improvement status of the accessions of the D. carota complex. The outgroup taxon is D. syrticus. (PDF 1.33 mb)
Additional file 7: Figure S5.
Relationships among 144 accessions of the Daucus carota complex and outgroups from an exhaustive quartet sampling inference using 18,565 SNPs (10% missing imputed genotypes) obtained by GBS. Numbers above the branches represent bootstrap values, with only values higher than 70% shown. Names given to clades refer to the geographic origin and improvement status of the accessions of the D. carota complex. ME & E refers to Middle East & Europe. Accessions designated by double stars are misplaced relative to the maximum likelihood topology of the Daucus carota complex using the same number of SNPs. The outgroup taxon is D. syrticus. (PDF 1.30 mb)
Additional file 8: Figure S6.
Relationships among 144 accessions of the Daucus carota complex and outgroups from an exhaustive quartet sampling inference using 43,713 SNPs (30% missing imputed genotypes) obtained by GBS. Numbers above branches represent bootstrap values, with only values higher than 70% shown. Names given to clades refer to the geographic origin and improvement status of the accessions of the D. carota complex. ME & E refers to Middle East & Europe. Accessions designated by double stars are misplaced relative to the maximum likelihood topology of the Daucus carota complex using the same number of SNPs. The outgroup taxon is D. syrticus. (PDF 142 kb)
Additional file 9: Figure S7.
Species tree of the Daucus carota complex based on a coalescent model using an exhaustive quartet sampling inference and 18,565 SNPs (10% missing imputed genotypes) obtained by GBS. Numbers above the branches represent bootstrap values. The outgroup taxon is D. syrticus. (PDF 104 kb)
Additional file 10: Figure S8.
Number of populations. A. Plot of Delta K (ΔK). B. Plot of the log likelihood; internal plot corresponds to the log likelihood (thousands) for K ranging from 1 to 9. All values were obtained from STRUCTURE HARVESTER analysis. Fourteen populations were considered in a data set of 18,565 SNPs (10% missing imputed genotypes) and 150 samples. (PDF 893 kb)
Additional file 11: Figure S9.
Number of populations. A. Plot of Delta K (ΔK). B. Plot of the log likelihood; internal plot corresponds to the log likelihood (thousands) for K ranging from 1 to 9. All values were obtained from STRUCTURE HARVESTER analysis. Fourteen populations were considered in a data set of 43,713 SNPs (30% missing imputed genotypes) and 150 samples. (PDF 167 kb)
Additional file 12: Figure S10.
Box plot analyses of the 23 morphological characters examined for members of Daucus carota complex (subsp. sativus not included) in this study. The box plot displays individual plant values for median, 25 and 75% percentile, range, and outliers. (PDF 17.1 mb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Arbizu, C.I., Ellison, S.L., Senalik, D. et al. Genotyping-by-sequencing provides the discriminating power to investigate the subspecies of Daucus carota (Apiaceae). BMC Evol Biol 16, 234 (2016). https://doi.org/10.1186/s12862-016-0806-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-016-0806-x