
Abstract
Background
Drosophila is an important model for studying the evolution of animal immunity, due to the powerful genetic tools developed for D. melanogaster. However, Drosophila is an incredibly speciose lineage with a wide range of ecologies, natural histories, and diverse natural enemies. Surprisingly little functional work has been done on immune systems of species other than D. melanogaster. In this study, we examine the evolution of immune genes in the speciose subgenus Drosophila, which diverged from the subgenus Sophophora (that includes D. melanogaster) approximately 25–40 Mya. We focus on D. neotestacea, a woodland species used to study interactions between insects and parasitic nematodes, and combine recent transcriptomic data with infection experiments to elucidate aspects of host immunity.
Results
We found that the vast majority of genes involved in the D. melanogaster immune response are conserved in D. neotestacea, with a few interesting exceptions, particularly in antimicrobial peptides (AMPs); until recently, AMPs were not thought to evolve rapidly in Drosophila. Unexpectedly, we found a distinct diptericin in subgenus Drosophila flies that appears to have evolved under diversifying (positive) selection. We also describe the presence of the AMP drosocin, which was previously thought to be restricted to the subgenus Sophophora, in the subgenus Drosophila. We challenged two subgenus Drosophila species, D. neotestacea and D. virilis with bacterial and fungal pathogens and quantified AMP expression.
Conclusions
While diptericin in D. virilis was induced by exposure to gram-negative bacteria, it was not induced in D. neotestacea, showing that conservation of immune genes does not necessarily imply conservation of the realized immune response. Our study lends support to the idea that invertebrate AMPs evolve rapidly, and that Drosophila harbor a diverse repertoire of AMPs with potentially important functional consequences.
Similar content being viewed by others
Background
The ability to defend oneself from parasites and pathogens (natural enemies) is essential for life, and animals have conserved sophisticated mechanisms of defence referred to as the innate immune system. The innate immune response requires recognition, signaling, and activation of defensive mechanisms. This defence response culminates in the synthesis and secretion of immune effectors, such as antimicrobial peptides (AMPs) -- host-encoded antibiotics that directly combat invading microorganisms [1]. For natural enemies, it is essential to overcome such host defences for success, thus setting the stage for antagonistic co-evolution. These evolutionary arms races have led to immune system genes typically evolving far more rapidly than other genes in the genome [2–5].
The genetically tractable model Drosophila melanogaster has been a workhorse of innate immunity, leading to the characterization of both the insect, and indeed animal, innate immune response [1]. Drosophila has also been of great importance to our understanding of the variability to which conserved genes may be expressed amongst closely related species, and how gene expression differences can result from interactions between genetics and environmental factors [6–8]. Upon the landmark sequencing of 12 Drosophila genomes in 2007, Drosophila researchers gained the ability to study the evolution of immune systems amongst closely related species [5, 9, 10]. An interesting pattern emerged, in that Drosophila immune signaling molecules were found to evolve rapidly, while immune effectors such as AMPs, did not [11, 12]. This pattern of AMP evolution was unexpected, given the importance of AMPs in the realized host response, and evidence for positive selection in AMPs of vertebrates [13–16] and social insects [17, 18].
Drosophila is an incredibly speciose lineage, however, with a wide range of ecologies, life histories, and specialized natural enemies (Fig. 1). Yet there have been almost no functional studies on Drosophila immune genes in species other than D. melanogaster (but see [19–21]). Along with D. melanogaster, most of the original 12 sequenced genomes are found in the subgenus Sophophora [22], with only three species from the diverse and speciose subgenus Drosophila (Drosophila grimshawi, D. virilis, and D. mojavensis), and no work on immune evolution has been done in any of the 300+ members of the immigrans-tripunctata radiation in this subgenus. Sequence data has recently become available for three species of this radiation: the genomes of Drosophila albomicans and D. guttifera [21, 23], and the transcriptome of Drosophila neotestacea [24] providing the opportunity to investigate the immune capacities of this relatively unexplored lineage of the subgenus Drosophila.

Phylogenetic relationships of the main lineages in Drosophila. Phylogeny constructed using maximum likelihood with concatenated Adh, amd, engrailed and glass protein sequences. Support values represent 100 bootstraps. Included species encompass much of the diversity of Drosophila, and form the basis of comparative work in this study. These sequences were generated from FlyBase curated protein translations and recently available sequence data from D. albomicans, D. guttifera, D. neotestacea, S. lebanonensis, and P. variegata. The subgenera Sophophora and Drosophila are estimated to have diverged 25-40 Ma (Obbard et al., 2012)
We start to explore Drosophila immune diversity by characterizing the immune repertoire of D. neotestacea, a mushroom-breeding species in the immigrans-tripunctata radiation, whose interactions with its natural enemies, particularly parasitic nematodes, are well-studied [25]. Drosophila neotestacea has also recently garnered attention for harboring a Spiroplasma bacterial symbiont that protects against nematodes and parasitic wasps [26, 27]. In general, we found that genes involved in the immune response of D. melanogaster were highly conserved in D. neotestacea, but found surprising evolutionary patterns for AMPs. We investigated two of these AMPs in more detail, the D. neotestacea orthologues of diptericin and drosocin. Using phylogenetic analysis, we describe the evolutionary history of the Drosophila diptericin gene family and the conservation of the Drosophila AMP drosocin in subgenus Drosophila flies. We found that the diptericin gene family rapidly diverged in the ancestors of the genus Drosophila, leading to not two, but three distinct Drosophila diptericins. We confirmed that these AMPs are induced by bacterial challenge in D. virilis, but were surprised to find that diptericin in D. neotestacea was not induced by infection. Along with other recent studies [21, 28, 29], this work suggests that invertebrate AMPs are more dynamic than previously thought. Our results further highlight that conservation of immune genes, even in closely related species, does not necessarily imply conservation of the realized immune response.
Methods
The immune repertoire of D. neotestacea
Using a recently sequenced transcriptome, we characterized the immune repertoire of D. neotestacea. This transcriptome was generated in order to understand how the bacterial symbiont Spiroplasma protects D. neotestacea against parasitic nematode infection. In brief, symbiont-positive and negative flies were either infected or uninfected with nematodes, resulting in four treatments; eggs, larvae, pupae and adult flies were included [24]. This transcriptome is expected to include a broad range of immune-related genes, as it includes diverse infections including parasitic nematodes and trypanosomatid gut parasites; nematode-exposed flies would have also been exposed to microorganisms entering the haemolymph via punctures in the larval cuticle following nematode attack.
Annotating the immune repertoire of D. neotestacea
We searched the D. neotestacea transcriptome using BLAST for immune genes that have been characterized in D. melanogaster. To generate a list of genes of interest, we conducted an extensive literature review to determine described constituents of the major D. melanogaster immune pathways: the Toll, Imd, JNK, JAK-STAT, and the melanization response pathways. We extracted D. melanogaster nucleotide sequences from FlyBase (vFB2015_04), and used BLASTn and tBLASTn to start to identify potential orthologous transcripts in the D. neotestacea transcriptome. When no significant hits (E < 0.1) were returned, we extracted corresponding orthologues from FlyBase for D. virilis, and/or D. mojavensis, and/or D. grimshawi, or from the D. albomicans [23], or D. guttifera genome [30], and again used BLASTn and tBLASTn. Sources used to generate a list of immune genes were: Lagueux et al. [31], De Gregorio et al. [32], Lemaitre and Hoffman [1], Starz-Gaiano et al. [33], Valanne et al. [34], Zaidman-Remy et al. [35], Hughes [36], Marchal et al. [37], An et al. [38], Binggeli and Lemaitre [39], Amoyel et al. [40], Salazar-Jaramillo et al. [20], and Yamamoto-Hino et al. [41].
When a significant BLAST hit was returned, the nucleotide sequence of the D. neotestacea transcript was then aligned with sequenced Drosophila orthologues as annotated by FlyBase for initial exploration. Transcript(s) were also codon-aligned with orthologues from D. melanogaster and D. virilis to confirm amino acid sequence similarity. In instances where amino acid sequence poorly resembled both D. melanogaster and D. virilis orthologues, or when multiple transcripts closely resembled these sequenced orthologues, additional genes from diverse Drosophila were extracted from FlyBase to provide outgroups for comparison. These genes were aligned using MUSCLE [42], followed by phylogenetic analysis using the Neighbour-joining method (1000 bootstraps); all analyses were performed in Geneious 7. When a D. neotestacea putative orthologue clustered with those from subgenus Drosophila flies, we considered this gene to be the true orthologue of the corresponding D. melanogaster gene.
Checking for presence of missing genes in the D. neotestacea genome
Drosophila melanogaster immune genes may be absent from the D. neotestacea transcriptome for a number of reasons; for instance, immune genes may be restricted to D. melanogaster and relatives (e.g. subgenus Sophophora, melanogaster subgroup). Alternatively, immune genes may be absent in the transcriptome despite their conservation in relatives of D. neotestacea. In this instance, there are two possibilities for this lack of expression. First, these immune genes may be absent from the D. neotestacea genome. Second, these immune genes might be present in the D. neotestacea genome, but were not expressed in the transcriptome.
Four genes were absent from the D. neotestacea transcriptome that were expected to be present. We followed up on these absent genes by designing PCR primers using sequenced Drosophila genomes, and tested these primers on a diversity of Drosophila including: D. neotestacea, D. falleni, and D. subobscura. Once the D. guttifera genome became available, we instead used the D. albomicans and D. guttifera genomes to determine if these genes were present or absent in the ancestor of D. neotestacea.
Three of the four genes apparently absent from the D. neotestacea transcriptome were short AMP genes. Due to the length of these AMP genes, BLAST was often unable to recover orthologues when searching the D. albomicans and/or D. guttifera genomes. To overcome this challenge, the synteny of the gene of interest in D. melanogaster was determined, and we then used longer genes that flanked the gene of interest as queries for BLAST searches. If an orthologue of a gene flanking the gene of interest was found in either the D. albomicans or D. guttifera genome, a manual search for the gene of interest was then conducted by identifying potential ORFs or conserved domains in the appropriate upstream or downstream gene region.
PCR primers, protocols, cloning, and sequencing
Primers used to successfully amplify immune genes absent from the D. neotestacea transcriptome can be found in Additional file 1: Table S1. Polymerase chain reactions were 12.5 μL in volume (1.25 μL 10× PCR mastermix, .2 mM dNTPs, 1.5 mM MgCl2, 0.625 μL of 0.25 μM forward and reverse primers, and 0.31 units of taq polymerase (Applied Biological Materials) with 0.5 μL of DNA template). All PCR products were Sanger sequenced to confirm that we were amplifying the correct sequence, and in the case of D. neotestacea genes of interest, to confirm transcriptome sequence. Sanger sequencing of PCR products was carried out by Macrogen USA. Sequences have been deposited in GenBank, under the following accession numbers: KX469340-KX469349.
Searching for novel immune genes in D. neotestacea
As the overwhelming majority of immune study in Drosophila has been done using D. melanogaster, it is possible that D. neotestacea transcribes as-yet uncharacterized immune genes that are restricted to D. neotestacea and related lineages (e.g. subgenus Drosophila). To examine this possibility, we looked for transcripts with homology to manually curated immune gene families from ImmunoDB [43]. Immune gene families were aligned with MUSCLE or MAFFT [44], after which profile HMMs were generated using hmmbuild in HMMer 3.1 (http://hmmer.org). We also included an alignment of Nimrod-like proteins extracted from a BLAST search using D. melanogaster nimrod on GenBank to generate a Nimrod-like domain profile, which is absent from ImmunoDB. We then searched all potential ORFs from the D. neotestacea transcriptome against these 39 profile HMMs using hmmsearch. Resulting significant matches were filtered for those that did not have an identified Drosophila orthologue from annotation by Hamilton et al. [24]. Finally, as the D. neotestacea transcriptome contains transcripts from Drosophila, nematodes, and trypanosomatids, we filtered these remaining ORFs for likely Drosophila transcripts, as annotated by Hamilton et al. [24]. The resulting list therefore contained likely Drosophila genes that lacked an orthologue in annotated Drosophila genomes on FlyBase.
Phylogenetic analysis of diptericin genes
We extracted annotated diptericins from FlyBase, and used BLAST to search GenBank, and recently sequenced drosophilid and dipteran genomes [45] for diptericin genes from a diversity of flies. The well-conserved glycine-rich domains (G domains) of these diptericins were then codon-aligned using MUSCLE. We used PhyML to construct a maximum likelihood phylogeny for these diptericin sequences with an AIC-selected best model of nucleotide substitution determined by Datamonkey.org model selection [46]. Diptericins from Mayetiola destructor, D. ananassae (Dana\GF11125) and D. simulans (Dsim\GD11418) were excluded from this phylogeny due to very long branches.
Synteny of diptericins in sequenced Drosophila genomes
We found three clades of Drosophila diptericins (hereon referred to as either Diptericin (Dpt) A, B, or C). To determine evolutionary relationships of Drosophila diptericins, we inspected the diptericin gene regions of drosophilid flies using FlyBase and sequenced drosophilid genomes. We extracted the diptericin-containing scaffold and manually searched for conserved diptericin motifs in this gene region to identify diptericin duplications if present. In its current genomic scaffold assembly, the signal peptide and P domain of the D. guttifera DptC gene was unavailable, and thus the N-terminus of this diptericin is not included in this analysis. Also, the intergenic region between the two D. guttifera diptericins was not fully sequenced, and thus the reported length for this intergenic region represents currently available sequence.
We aligned diptericin gene regions of D. melanogaster and D. virilis to related flies to generate an alignment encompassing divergent diptericins in diverse drosophilids. For some species, additional diptericin duplications were present, and we used flanking genes to determine the ancestral gene copy for alignment purposes. The P. variegata genome encodes two DptB orthologues not found on the same genomic scaffolds.
Positive selection on Drosophila diptericins
Intrigued by the degree of amino acid sequence similarity amongst Drosophila diptericins, we investigated rates of synonymous and non-synonymous change (dN/dS) in the diptericin G domain. We used Branch-site REL (BSR) [47] implemented in Datamonkey.org to identify lineages with elevated dN/dS in the diptericin G domain. To rule out the possibility that our results were sensitive to the presence of certain divergent diptericins, we repeated the analysis while removing divergent sequences.
Characterizing drosocin in the subgenus Drosophila
We recovered a drosocin-like gene (hereafter referred to as “drosocin”) with multiple tandem drosocin-domain repeats in the D. neotestacea transcriptome. We extracted similar drosocin gene sequences from sequenced genomes combining BLAST and manual gene region curation. Many, but not all, drosocin ORFs contained multiple tandem repeats, and so we aligned unique repeats of this drosocin gene with drosocins found in subgenus Sophophora flies using MUSCLE.
Fly cultures used in infection experiments
For infection experiments, we used a strain of D. neotestacea originally collected in W. Hartford, Connecticut, in 2006. The D. virilis strain used in this study was donated by Brent Sinclair (Western University, Canada), and the D. melanogaster strain (Oregon-R) used in this study was donated by Bruno Lemaitre (EPFL, Switzerland). All strains used were Wolbachia and Spiroplasma negative. All species were maintained at 21 °C with a 12-h light:dark cycle on Instant Drosophila Medium (Carolina Biological Supply). Approximately 10 females were allowed to lay on 1/2 tsp. Instant Drosophila Medium (1 tsp. for D. virilis) with 1:1 water; D. neotestacea vials were supplemented with ~0.5 g Agaricus bisporus. Newly emerged males were then collected daily and kept in isolation from females for 3–4 days on ~1/2 tsp Instant Drosophila medium with 1:1 water. All adults used in infection assays were 3–4 day old virgin males.
Immune challenge with Gram-negative bacteria (IMD pathway challenge)
Gram-negative bacteria induce the Imd immune pathway in Drosophila [1]. For our Imd pathway challenge, we used a pathogenic Serratia strain closely related to the soil bacterium Serratia marcescens, and isolated from mycophagous Drosophila cultures (Additional file 2: Figure S4). Bacteria were grown overnight at 37 °C and diluted in Luria-Bertani broth prior to wounding experiments.
Flies were lightly anaesthetized on CO2 and wounded in the left side of the thorax above the wing with a 0.6 μm tip tungsten needle. For septic woundings, this needle was dipped in OD600 = 0.15 ± 0.05 Serratia in Luria-Bertani broth. Flies were then left to recover in a clean polystyrene vial for 30 min prior to transfer to a vial containing ~1/2 tsp Instant Drosophila medium with 1:1 water. Six hours post-wounding, flies were flash frozen in liquid nitrogen and kept at −80 °C until RNA was extracted.
This experiment was performed three times for D. neotestacea and D. virilis, and once for D. melanogaster. Additionally, for one replicate experiment using D. virilis and D. neotestacea we also examined flies that were anaesthetized on CO2 and not wounded to provide a reference treatment for differences in AMP expression incurred by sterile wounding alone.
Immune challenge with fungi (Toll pathway challenge)
Pathogenic fungi induce the Toll signaling pathway in Drosophila [1]. We used the entomopathogenic fungus Beauveria bassiana (strain UAMH 1514) for our Toll pathway challenges. Beauveria cultures were provided by Will Hintz and Jon Leblanc (University of Victoria, Canada), and were grown on Potato-dextrose agar at 27 °C for one week until fungus was sporulating prior to exposures.
Flies were lightly anaesthetized on CO2 and transferred to either a sterile Potato-dextrose agar petri dish, or one containing sporulating Beauveria culture. Dishes were then shaken by hand for 30 s to cover the flies in fungal spores; we confirmed flies had been exposed to fungal spores using a dissecting microscope shortly after shaking. Flies were left to recover in a clean polystyrene vial for 30 min prior to transfer to a vial containing ~1/2 tsp Instant Drosophila medium with 1:1 water. Twenty-four hours post-exposure, flies were flash frozen in liquid nitrogen and kept at −80 °C until RNA was extracted.
This experiment was performed three times for D. virilis, and twice for D. melanogaster and D. neotestacea.
RNA extraction and cDNA synthesis
We extracted RNA from six to eight flies per treatment using Trizol-LS (Invitrogen) with the manufacturer’s protocol. Individual flies were added to microfuge tubes containing 300 μL Trizol and 5–15 0.1 mm silica/zirconia beads, and bead-beat for 3 s (BioSpec MiniBeadbeater 16). Following extraction, pellets were re-suspended in 20 μL RNAse-free water for five minutes at room temperature.
RNA purity was measured using 1 μL RNA on a Nanodrop 2000 Spectrophotometer (Thermo Scientific). The remaining 19 μL from each sample were then DNAse treated (Thermo Scientific DNAse I) according to the manufacturer’s protocol, with the DNAse heat inactivated. Extraction quality was assessed by agarose gel electrophoresis.
DNAse-treated RNA was reverse-transcribed using Applied Biological Materials 5X All-In-One RT MasterMix. Reverse transcription reactions were 20 μL containing 4 μL RT MasterMix and 16 μL of RNA in RNAse-free H2O with 300–1000 ng total RNA.
qPCR for gene expression and data analysis
Levels of expression for genes of interest were quantified using the qPCR primers listed in Additional file 1: Table S1. Primers were designed using Primer3, and primer efficiency was verified using a 5 × 5 fold dilution series; primer efficiencies are reported in Additional file 1: Table S1. All qPCR reactions used the following thermal cycling conditions: 95 °C for 10 min, then 35 cycles of 95 °C for 15 s followed by 60 °C for 45 s, with the product verified by melt curve analysis, as well as Sanger sequencing (Macrogen USA) once for each primer set. We used Applied Biological Materials, EvaGreen 2X qPCR MasterMix according to manufacturers protocol, with a BioRad CFX96 qPCR thermal cycler.
For all immune challenges, we assayed the expression of each fly’s respective diptericin orthologue. In Serratia challenges, we also assayed attacin B (AttB) and drosocin. In Beauveria challenges, we also assayed a bomanin (Bom) gene (CG5791 in D. melanogaster and its respective orthologues in D. virilis and D. neotestacea), drosomycin in D. melanogaster, and drosocin in D. neotestacea and D. virilis.
For all qPCR reactions, target genes were run alongside a normalizing control gene (RpL28, RpL32, and RpL11 for D. neotestacea, D. melanogaster, and D. virilis respectively). Each reaction was run in triplicate, and replicates were considered consistent if the threshold cycle (CT) of each replicate was contained within a 0.5 CT boundary.
Gene expression analysis was performed using the 2ΔΔCT method [48], and we report these data as boxplots using the ΔCT values (ΔCT = CT target gene – CT reference gene). Two-sample Welch’s T-tests of ΔCT values were used to determine differences in expression profile in R 3.1 statistical software.
Results
The immune repertoire of D. neotestacea
We found that 105 out of 108 genes expected to be involved in Drosophila immune pathways were present in the D. neotestacea transcriptome (Fig. 2). We did not recover the AMP metchnikowin (Mtk), nor could we amplify it from genomic DNA. We found Mtk in the genomes of the subgenus Drosophila flies D. mojavensis, D. virilis, and D. albomicans, but not D. guttifera; we were unable to determine if it is truly absent in D. guttifera, or if this absence is instead an artefact of the current genomic assembly. We did not recover an orthologue of PGRP-SC1 in the D. neotestacea transcriptome, but found that D. neotestacea harbours two copies of PGRP-SC2; PGRP-SC1 and SC2 have been shown to have mutually exclusive activities in D. melanogaster (Additional file 3: Figure S1) [49, 50]. Finally, diptericin B was not found in the D. neotestacea transcriptome, but we subsequently found an interesting pattern of diptericin evolution (see below).

The immune repertoire of D. neotestacea. This diagram is colour-coded to indicate: i) genes predicted to be present, and that were recovered in the transcriptome (light blue), ii) genes predicted to be absent in the transcriptome (because they are restricted to the subgenus Sophophora), and that were absent (dark blue), and iii) genes predicted to be present in the transcriptome, but were absent (red). The vast majority of immune genes matched predicted patterns of conservation in D. neotestacea, with the exception of the effector genes DptB and Mtk, and the Imd pathway inhibitor PGRP-SC1
Drosocin in the subgenus Drosophila
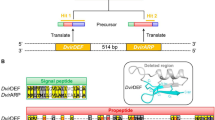
We searched for potentially novel immune genes in D. neotestacea with HMMer 3.1 using immune gene profiles. This search method recovered one immune gene of interest in the D. neotestacea transcriptome: a D. neotestacea drosocin; drosocin was previously thought to be absent in the subgenus Drosophila [19]. We further found orthologues of this D. neotestacea drosocin in other subgenus Drosophila flies. The signal peptide of this drosocin gene almost-perfectly matches that of drosocin genes in Sophophora species, however the D. neotestacea transcript contains multiple tandem repeats of drosocin protein domains (Fig. 3); this pattern of drosocin domain tandem repeats was recovered in some, but not all, of the other sequenced subgenus Drosophila genomes. Interspersed between each drosocin domain repeat are furin-like cleavage sites (e.g. RVVR), suggesting that the translated protein is likely cleaved into multiple mature drosocin peptides (Fig. 3a). Finally, while drosocin is found just upstream of the attacin gene region on chromosome 2R in D. melanogaster, subgenus Drosophila drosocin occurs in the gene region of the Drosophila down syndrome cell adhesion molecule (DSCAM1) and gustatory receptor 43a (Gr43a), ~7.36 million base pairs displaced from the attacin gene region, but still on chromosome 2R (Fig. 3b). We later confirmed that this subgenus Drosophila drosocin responds to immune challenge (see below).

Drosocin in the subgenus Drosophila. a The subgenus Drosophila drosocin has a strongly conserved ERPPY motif at the proline-rich N-terminus, followed by the drosocin domain PRPT, which includes a critical threonine residue. This domain is followed by furin-like cleavage sites (annotated as “Furin”). The presence of both furin-like cleavage sites and the key threonine residue indicate that this transcript is likely processed to produce multiple copies of a mature drosocin peptide glycosylated at its PRPT threonine. This alignment presents the signal peptide and first drosocin repeat in each species, and does not include tandem drosocin repeats, which vary in number and sequence depending on species. b Drosocin in the subgenus Drosophila is found within the gene region of DSCAM1 and Gr43a, ~7.36 million base pairs displaced from the drosocin gene region in the subgenus Sophophora. The D. virilis Gr43A gene region is included here
Diptericin in D. neotestacea and other Diptera
We did not recover a diptericin B orthologue in the D. neotestacea transcriptome, but found DptB sequences in both the D. albomicans and D. guttifera genomes; however the DptB molecule in D. guttifera has been pseudogenized by mid-exon frame shifts in both the diptericin P domain and G domain. We did however find a divergent diptericin previously annotated as diptericin B by Hamilton et al. [24] that, upon further inspection, was not highly similar to either of the diptericin genes (DptA and DptB) in D. melanogaster.
To determine the identity of this divergent D. neotestacea diptericin, we inspected diptericins from diverse Diptera, and using phylogenetic analysis, found that the D. neotestacea diptericin belongs to a clade of diptericins restricted to the subgenus Drosophila we term diptericin C (DptC) for clarity of discussion (Fig. 4a). Intriguingly, DptC genes clustered on a long branch separate from other Drosophila diptericins; to determine their evolutionary history we investigated the genomic positions of DptC genes in sequenced subgenus Drosophila flies.

The subgenus Drosophila encodes a highly divergent diptericin. a Maximum likelihood tree generated using a codon alignment of the well-conserved diptericin G domain from assembled diptericins of diverse brachyceran flies. Support values represent consensus from 100 bootstraps. Four distinct diptericin clades emerge, including three in the genus Drosophila: DptA (subgenus Sophophora), DptB (genus Drosophila), and a diptericin restricted to the subgenus Drosophila that we term DptC. b Synteny of diptericins in the genomes of sequenced drosophilids. DptA and DptC both occur upstream of DptB. We include D. neotestacea despite lacking a sequenced genome to indicate the lack of DptB recovered from the transcriptome. Diptericin B in D. guttifera is pseudogenized, and the intergenic region between DptC and DptB is listed as “>3000 bp” due to its current assembly. In S. lebanonensis, the DptA/DptC syntenic orthologue is present, but does not bear great similarity to any diptericin clade. c Summary phylogeny of Branch-site REL (BSR) analyses using only drosophilid diptericin G domains. Likelihood-ratio tests for branches with dN/dS > 1 consistently identified the branch leading to DptC as having evolved under diversifying (positive) selection (p < .05). Additional file 4: Figure S2 provides an example BSR analysis. d Amino acid alignment of the diptericin G domain from Drosophila diptericins. Numerous fixed differences are unique to each clade, particularly in the Gly22-Asp45 region. Greater conservation is observed in the Asn46-on region. The polymorphism at residue 71 described by Unckless et al. [21, 29] is indicated by a ★, and displays conserved differences amongst diptericin lineages in sequenced genomes
We found that DptC genes are encoded as only one exon, and are syntenic with the one-exon DptA genes of subgenus Sophophora flies, upstream of the two-exon DptB in the diptericin gene region (Fig. 4b). Moreover, we recovered this one-exon diptericin in the outgroup drosophilid Scaptodrosophila lebanonensis (Drosophilinae, Drosophilidae), but not in Phortica variegata (Steganinae, Drosophilidae), or in Ephydra gracilis (Ephydridae) (Fig. 4b). We recovered intact DptB genes in all sequenced drosophilids barring D. neotestacea (absent from transcriptome) and D. guttifera (pseudogenized).
The extreme divergence of these syntenic orthologues prompted us to search for signatures of positive selection in Drosophila diptericins (i.e. DptA, DptB, and DptC). Using Branch-site REL, we found that the branch leading to the DptC clade diverged under diversifying selection (likelihood ratio test (LRT); p < 0.05) (Fig. 4c). This result was robust to removal of the more divergent diptericins from the analysis. Additionally, we recovered some support for the hypothesis that DptA also diverged from DptB (LRT = 12.25; p = .017) through diversifying selection in the ancestor of the subgenus Sophophora (Additional file 4: Figure S2).
Comparing DptA, DptB, and DptC protein sequences, we found that the diptericin G domain has undergone considerable modification unique to but conserved within each diptericin clade (Fig. 4d). The Gly22-Asp45 region of the diptericin G domain was previously hypothesized by Cudic et al. [51] to be the region responsible for diptericin’s antibacterial activity. We found that 15 of the codons in this 23-residue region show lineage-restricted conserved differences, while the Asn46 to C-terminal region of the G domain shows greater conservation amongst Drosophila diptericin clades. Interestingly, in D. melanogaster and D. simulans, Unckless et al. [21] found that balancing selection is maintaining a polymorphism at residue 69 of the diptericin G domain (corresponding to residue 71 from Cudic et al. [51]), and that whether serine or an arginine was found at this site strongly affected resistance to pathogenic bacteria. We found this residue to be different between, but conserved within, lineages of DptB and DptC, yet polymorphic in DptA (Fig. 4d). We also found that diptericins in certain Drosophila species lacked a positively charged G domain (Additional file 5: Table S2); antimicrobial peptides are thought to require a positive net charge for bacterial killing [1].
AMP gene expression in subgenus Drosophila flies
We sought to determine if DptC and drosocin in subgenus Drosophila flies responded to immune challenge by Gram-negative bacteria and fungi. We found that drosocin was induced by Gram-negative bacterial challenge in both D. virilis and D. neotestacea (see below). We also found that while DptC was induced by Gram-negative challenge in D. virilis, surprisingly, DptC was not at all induced in D. neotestacea (see below). These two AMPs were not strongly induced by fungal challenge (see below).
Immune challenge with Gram-negative bacteria (Imd pathway challenge)
As expected, the Imd pathway-regulated genes AttB, drosocin, and DptA were induced by Serratia challenge in D. melanogaster (t(7.46) = 16.65, p < 0.0001, t(7.23) = 10.53, p < 0.0001, t(7.17) = 15.11, p < 0.0001, respectively) (Fig. 5a). This pattern of induction was also found in D. virilis for AttB, drosocin, and DptC (t(20.15) = 4.74, p < 0.0005, t(21.45) = 5.67, p < 0.0001, t(20.31) = 5.07, p < 0.0001, respectively) (Fig. 5b). However in D. neotestacea, while AttB and drosocin were both induced by Serratia infection (t(34.64) = 5.37, p < 0.0001; t(36.13) = 5.71, p < 0.0001, respectively), DptC (t(28.79) = 0.82, p = 0.42) was not (Fig. 5c). We also found that while AttB experiences an increase in expression in both D. virilis and D. neotestacea upon sterile wounding, we did not observe this level of induction in either D. virilis or D. neotestacea for drosocin and DptC (Additional file 6: Figure S3).

AMP gene expression following Serratia challenge. AMP expression was measured six hours after either sterile wounding (Sterile) or Serratia bacterial challenge (Septic) in a D. melanogaster, b D. virilis, and c D. neotestacea. N represents total sample size. Attacin B and drosocin were strongly induced in all three species (p < 0.001). However while DptA and DptC were strongly induced in D. melanogaster (p < 0.0001), and D. virilis (p < 0.0001) respectively, DptC in D. neotestacea was not (p = 0.42)
Immune challenge with fungi (Toll pathway challenge)
Due to the absence of many canonical Toll-regulated genes in the D. neotestacea transcriptome, we used a member of the recently described bomanin (Bom) gene family [52] to serve as a read-out of Toll pathway expression, the D. melanogaster Bom CG5791; Bom CG5791 was induced by septic injury and fungal infection in De Gregorio et al. [32]. In D. melanogaster, Beauveria infection strongly induced the Toll-regulated AMP drosomycin and also Bom CG5791 (t(19.05) = 5.59, p < .0001, t(17.85) = 8.45, p < .0001, respectively), but DptA expression was unaffected (t(24.86) = –0.01, p = 0.99) (Fig. 6a). This pattern of induction confirmed that CG5791 behaved as would be expected of a Toll-regulated AMP. We used this bomanin in D. virilis (GJ23146) and D. neotestacea (TSA Accession: GDUH01009588) as a read-out to confirm expression of the Toll pathway in our Beauveria infections. We found that Beauveria infection induced CG5791 in both D. virilis and D. neotestacea (t(23.18) = 3.55, p < .005, (t(27.42) = 3.24, p < .005, respectively) (Fig. 6c), though the change in expression (~1.1 ΔCT) was not as large as in D. melanogaster (2.7 ΔCT). Neither DptC nor drosocin were induced in D. neotestacea upon fungal exposure (t(29.94) = 0.50, p = 0.62, (t(26.96) = –0.42, p = 0.68, respectively) (Fig. 6). DptC was not induced in D. virilis (t(32.09) = 1.41, p = 0.17), though drosocin appeared to be induced in a few individuals (Fig. 6b); two D. virilis individuals had elevated bomanin, DptC, and drosocin, expression in the “Exposed” treatment.

AMP gene expression following Beauveria challenge. AMP expression was measured 24 h after either fungus-free controls or Beauveria challenge in a D. melanogaster, b D. virilis, and c D. neotestacea. N represents total sample size. Both Bom CG5791 and drosomycin were strongly induced in D. melanogaster (p < 0.0001). The orthologues of Bom CG5791 were induced in both D. virilis and D. neotestacea (p < .005), though to a lesser extent than in D. melanogaster. Drosocin and DptC in D. virilis and D. neotestacea were not strongly induced by Beauveria challenge; drosocin differential expression in D. virilis was marginally significant (t(28.11) = 2.18, p = .038). Diptericin A in D. melanogaster was not induced by Beauveria challenge (p = 0.99)
Discussion
Using a recently sequenced transcriptome as a starting point, we characterized the immune repertoire of D. neotestacea, a mushroom-breeding species in the subgenus Drosophila, which is estimated to have diverged from D. melanogaster and the subgenus Sophophora approximately 25–40 Ma ago [53]. The vast majority of immune genes were conserved and expressed in this transcriptome, with some interesting exceptions, particularly among AMPs. This finding opens a window into the diversity of the realized Drosophila immune response. The diversity of AMPs conserved in the subgenus Drosophila was unexpected and parallels renewed interest in Drosophila and arthropod AMPs [12, 21, 29]. Previous explorations of Drosophila immune evolution did not recover signals of selection in AMPs, but rather signaling pathway intermediates [4, 5, 9, 11]. As such, the predominant view of insect immune evolution holds that insect AMPs do not evolve rapidly, in contrast with many studies documenting balancing selection on AMPs in vertebrates (e.g. [13–16]). This view of AMP evolution in Drosophila may have resulted from two factors in particular. First, AMPs are often exceedingly short and therefore challenging to study using standard methods to examine homology, divergence, and signals of natural selection. Second, AMPs have been characterized in relatively few arthropod lineages.
The divergent evolution of diptericin, including finding a lineage (DptC) that is as deeply branching and diverse in Drosophila as DptA and DptB, is surprising. Diptericin has been the canonical readout for the Imd pathway in flies (induced by Gram-negative bacteria), and diptericins are especially well characterized in D. melanogaster [54, 55], although their structure and mode of action are yet to be fully described [1]. It would be very interesting to determine what are the functional consequences of diptericin variation, and whether the numerous conserved differences distinct to each diptericin lineage underlie unappreciated diversity in immune capacities of these genes, possibly representing adaptation to ecologically relevant natural enemies. Indeed, an interesting recent study found that variation at a single residue in DptA had striking consequences on the ability of D. simulans and D. melanogaster to resist infection by Providencia bacteria [21]. We found this residue to be highly variable across Drosophila (Fig. 4d).
Although DptC behaved as expected in response to microbial challenge in D. virilis, its lack of induction upon Serratia challenge in D. neotestacea is surprising and warrants further study. Traditionally, conservation of immune genes has been interpreted as representing a conservation of immune function. Yet D. neotestacea employs neither DptC, nor as far as we can tell, any other diptericin in its response to Serratia challenge. However DptC in D. neotestacea can be induced, as two adult females had elevated levels of DptC in Hamilton et al. [24], and they suspected these elevated DptC levels to have resulted from a cryptic bacterial infection, although we note that we only challenged adult males. Alternatively, tissue-specific AMP expression could account for the lack of DptC induction in D. neotestacea [56]. If DptC were involved in the local immune responses of surface epithelia such as in tracheae or the gut, septic wounding of the thorax may not induce DptC. Regardless, the lack of diptericin employed in response to Serratia infection implies that the D. neotestacea AMP arsenal combats certain bacteria without using any diptericins. As attacins and diptericins have common ancestry [54], it may be useful to consider the potentially redundant roles these AMPs play in the Drosophila immune response; attacin was highly expressed following D. neotestacea exposure to Serratia. It would also be interesting to challenge D. neotestacea with other gram-negative bacteria to see if DptC fails to be induced in general.
We also recovered and provide the first description, to our knowledge, of the AMP drosocin in the subgenus Drosophila. We found that in many species in this subgenus, drosocin contains multiple tandem repeats of the domains ERPPY and PRPT, which are likely proteolytically cleaved to produce multiple drosocin molecules at furin-like cleavage sites (e.g. RVVR) found between each repeat (Fig. 3). There are well-documented trade-offs with respect to mounting a host defence to infectious microbes [57–59], leading to the hypothesis that AMP expression should be optimized to expend only the minimum amount of energy required for an effective host defence [52]. The tandem-repeat drosocin genes of closely related subgenus Drosophila flies may allow researchers to test this hypothesis if flies optimize levels of drosocin expression and mature peptides produced. Additionally, there are many sequence differences amongst drosocins in the subgenus (Fig. 3), which may imply balancing selection [29]; we did not perform selection analyses for drosocin as the tandem-repeat structure of subgenus Drosophila drosocins make alignments somewhat subjective.
Our comparative approach allowed us to better characterize the conservation of metchnikowin (Mtk), a canonical read-out of the Toll pathway in D. melanogaster. Metchnikowin orthologues are annotated in FlyBase (vFB2015_04) in most species in the subgenus Sophophora (except obscura group species) as well as in D. grimshawi. Using manual curation, followed by BLAST, we recovered Mtk in the obscura group species D. pseudoobscura, D. persimilis, and Drosophila miranda, the subgenus Drosophila flies D. virilis, D. mojavensis, and D. albomicans, as well as D. busckii, S. lebanonensis, and P. variegata. We were not able to recover Mtk from D. neotestacea and D. guttifera. However, given our recovery of diptericin, drosocin, and Mtk from subgenus Drosophila flies, it seems that conservation of D. melanogaster AMPs is more widespread than previously described ([19]; FlyBase vFB2015_04).
Conclusions
This study lends further support to the idea that invertebrate AMPs evolve rapidly, and that Drosophila species harbor a diverse repertoire of AMPs with potentially important functional consequences. As such, investigating AMP polymorphisms promises to be an exciting field of research in coming years, both to understand factors contributing to susceptibility to infection [29], and perhaps even to provide templates for the discovery and development of novel antibiotics [60].
Abbreviations
- Adh:
-
Alcohol dehydrogenase
- AIC:
-
Akaike information criterion
- Amd:
-
Alpha methyldopa-resistant
- AMP:
-
Antimicrobial peptide
- AttB:
-
Attacin B
- Bom:
-
Bomanin
- BSR:
-
Branch-site REL
- Dpt:
-
Diptericin
- DSCAM1:
-
Drosophila down syndrome cell adhesion molecule
- Gr43a:
-
Gustatory receptor 43a
- HMM:
-
Hidden Markov model
- IM:
-
Immune-induced molecule
- Imd:
-
Immune-deficiency
- JAK-STAT:
-
Janus kinase- Signal-transducer and activator of transcription protein
- JNK:
-
Jun kinase
- LRT:
-
Likelihood ratio tests
- ML:
-
Maximum likelihood
- Mtk:
-
Metchnikowin
- PGRP:
-
Peptidoglycan recognition protein
- REL:
-
Random-effects likelihood
- RpL:
-
Ribosomal protein L
References
Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Ann Rev Imm. 2007;25:697–743.
Begun DJ, Whitley P. Adaptive evolution of relish, a Drosophila NF-kappaB/IkappaB protein. Genetics. 2000;154:1231–8.
Jiggins FW, Kim KW. The evolution of antifungal peptides in Drosophila. Genetics. 2005;171:1847–59.
Quesada H, Ramos-Onsins SE, Aguadé M. Birth-and-death evolution of the Cecropin multigene family in Drosophila. J Mol Evol. 2005;60:1–11.
Sackton TB, Lazzaro BP, Schlenke TA, Evans JD, Hultmark D, Clark AG. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 2007;39:1461–8.
Rifkin SA, Kevin PW, Junhyong K. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet. 2003;33:138–44. doi:10.1038/ng1086.
Sambandan D, Carbone MA, Anholt RRH, Mackay TFC. Phenotypic Plasticity and Genotype by Environment Interaction for Olfactory Behavior in Drosophila melanogaster. Genetics. 2008;179(2):1079–88. doi:10.1534/genetics.108.086769.
Hodgins-Davis A, Townsend JP. Evolving Gene Expression: From G to E to G X E. Trends Ecol Evol. 2009;24(12):649–58.
Jiggins FM, Kim KW. The evolution of antifungal peptides in Drosophila. Genetics. 2005;171(4):1847–59. doi:10.1534/genetics.105.045435.
Obbard DJ, Welch JJ, Kim KW, Jiggins FM. Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet. 2009;5:e1000698.
Lazzaro BP. Natural selection on the Drosophila antimicrobial immune system. Curr Opin Microbiol. 2008;11:284–9.
Rolff J, Schmid-Hempel P. Perspectives on the evolutionary ecology of arthropod antimicrobial peptides. Phil Trans R Soc B. 2016. doi:10.1098/rstb.2015.0297.
Hollox EJ, Armour JA. Directional and balancing selection in human beta-defensins. BMC Evol Biol. 2008. doi:10.1186/1471-2148-8-113.
Tennessen JA, Blouin MS. Balancing selection at a frog antimicrobial peptide locus: fluctuating immune effector alleles? Mol Biol Evol. 2008. doi:10.1093/molbev/msn208.
Hellgren O, Sheldon BC. Locus-specific protocol for nine different innate immune genes (antimicrobial peptides: beta-defensins) across passerine bird species reveals within-species coding variation and a case of trans-species polymorphisms. Mol Ecol Res. 2011. doi:10.1111/j.1755-0998.2011.02995.x.
Halldórsdóttir K, Árnason E. Trans-species polymorphism at antimicrobial innate immunity cathelicidin genes of Atlantic cod and related species. PeerJ. 2015. doi:10.7717/peerj.976.
Viljakainen L, Pamilo P. Selection on an antimicrobial peptide defensin in ants. J Mol Evol. 2008. doi:10.1007/s00239-008-9173-6.
Bulmer MS, Lay F, Hamilton C. Adaptive evolution in subterranean termite antifungal peptides. Insect Mol Biol. 2010. doi:10.1111/j.1365-2583.2010.01023.x.
Sackton TB, Clark AG. Comparative profiling of the transcriptional response to infection in two species of Drosophila by short-read cDNA sequencing. BMC Genomics. 2009;10:259.
Salazar-Jaramillo L, Paspati A, van de Zande L, Vermeulen CJ, Schwander T, Wertheim B. Evolution of a cellular immune response in Drosophila: A phenotypic and genomic comparative analysis. Gen Biol Evol. 2014. doi:10.1093/gbe/evu012.
Unckless RL, Howick VM, Lazzaro BP. Convergent balancing selection on an antimicrobial peptide in drosophila. Curr Biol. 2016. doi:10.1016/j.cub.2015.11.063.
Clark AG, Eisen MB, Smith DR, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–18.
Zhou Q, Zhu H, Huang Q, Zhao L, Zhang G, Roy SW, Vicoso B, Xuan Z, Ruan J, Zhang Y, Zhao R, Ye C, Zhang X, Wang J, Wang W, Bachtrog D. Deciphering neo-sex and B chromosome evolution by the draft genome of Drosophila albomicans. BMC Genomics. 2012. doi:10.1186/1471-2164-13-109.
Hamilton PT, Leong JS, Koop BF, Perlman SJ. Transcriptional responses in a Drosophila defensive symbiosis. Mol Ecol. 2014. doi:10.1111/mec.12603.
Jaenike J, Perlman SJ. Ecology and Evolution of Host-Parasite Associations: Mycophagous Drosophila and Their Parasitic Nematodes. Am Nat. 2002;160:S24–39.
Jaenike J, Unckless R, Cockburn SN, Boelio LM, Perlman SJ. Adaptation via Symbiosis: Recent Spread of a Drosophila Defensive Symbiont. Science. 2010;329:212–5.
Haselkorn TS, Jaenike J. Macroevolutionary persistence of heritable endosymbionts: acquisition, retention and expression of adaptive phenotypes in Spiroplasma. Mol Ecol. 2015;24:3752–65.
Gosset CC, Do Nascimento J, Augé M-T, Bierne N. Evidence for adaptation from standing genetic variation on an antimicrobial peptide gene in the mussel Mytilus edulis. Mol Ecol. 2014;23:3000–12.
Unckless RL, Lazzaro BP. The potential for adaptive maintenance of diversity in insect antimicrobial peptides. Phil Trans R Soc B. 2016;371:1695.
Koshikawa S, Giorgianni MW, Vaccaro K, Kassner VA, Yoder JH, Werner T, Carroll SB. Gain of cis-regulatory activities underlies novel domains of wingless gene expression in Drosophila. PNAS. 2015. doi:10.1073/pnas.1509022112.
Lagueux M, Perrodou E, Levashina EA, Capovilla M, Hoffmann JA. Constitutive expression of a complement-like protein in toll and JAK gain-of- function mutants of Drosophila. PNAS. 2000. doi:10.1073/pnas.97.21.11427.
De Gregorio E, Spellman PT, Rubin GM, Lemaitre B. Genome-Wide Analysis of the Drosophila Immune Response by Using Oligonucleotide Microarrays. PNAS. 2001;98:12590–5.
Starz-Gaiano M, Melani M, Meinhardt H, Montell D. Interpretation of the UPD/JAK/STAT morphogen gradient in Drosophila follicle cells. Cell Cycle. 2009;8:2917.
Valanne S, Wang J, Rämet M. The Drosophila Toll signaling pathway. J Imm. 2011;186:649.
Zaidman-Rémy A, Poidevin M, Hervé M, et al. Drosophila immunity: analysis of PGRP-SB1 expression, enzymatic activity and function. PLoS One. 2011;6:e17231.
Hughes AL. Evolution of the βGRP/GNBP/β-1,3-glucanase family of insects. Immunogenetics. 2012;64(7):549–58.
Marchal C, Vinatier G, Sanial M, Plessis A, Pret A, Limbourg-Bouchon B, Théodore L, Netter S. The HIV-1 vpu protein induces apoptosis in drosophila via activation of JNK signaling. PLoS One. 2012;7(3):e34310.
An C, Zhang M, Chu Y, Zhao Z. Serine protease MP2 activates prophenoloxidase in the melanization immune response of Drosophila melanogaster. PLoS One. 2013;8(11):e79533.
Binggeli O, Lemaitre B. Role and regulation of the melanization reaction in Drosophila immune response. Thèse EPFL. 2013;5840. doi:10.5075/epfl-thesis-5840.
Amoyel M, Anderson AM, Bach EA. JAK/STAT pathway dysregulation in tumors: A Drosophila perspective. Sem Cell Dev Biol. 2014;28C:96–103.
Yamamoto-Hino M, Muraoka M, Kondo S, Ueda R, Okano H, Goto S. Dynamic regulation of innate immune responses in Drosophila by senju- mediated glycosylation. PNAS. 2015. doi:10.1073/pnas.1424514112.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Waterhouse RM, et al. Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes. Science. 2007;316(5832):1738-1743. PMID:17588928.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Vicoso B, Bachtrog D. Numerous transitions of sex chromosomes in Diptera. PLoS Biol. 2015. doi:10.1371/journal.pbio.1002078.
Delport W, Poon AF, Frost SDW, Kosakovsky-Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;PMID:20671151.
Kosakovsky-Pond SL, Murrell B, Fourment M, Frost SDW, Delport W, Scheffler K. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol. 2011. doi:10.1093/molbev/msr125.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2011;25(4):402.
Garver LS, Wu LP, Wu J. The peptidoglycan recognition protein PGRP-SC1a is essential for toll signaling and phagocytosis of staphylococcus aureus in drosophila. PNAS. 2006. doi:10.1073/pnas.0506182103.
Guo L, Karpac J, Tran SL, Jasper H. PGRP-SC2 promotes gut immune homeostasis to limit commensal dysbiosis and extend lifespan. Cell. 2014;156:109–22.
Cudic M, Bulet P, Hoffmann R, Craik DJ, Otvos JL. Chemical synthesis, antibacterial activity and conformation of diptericin, an 82-mer peptide originally isolated from insects. Eur J Bioc. 1999;266(2):549.
Clemmons AW, Lindsay SA, Wasserman SA. An effector peptide family required for Drosophila toll-mediated immunity. PLoS Pathogens 2015;11(4): doi:10.1371/journal.ppat.1004876
Obbard DJ, Maclennan J, Kim K, Rambaut A, O’Grady PM, Jiggins FM. Estimating divergence dates and substitution rates in the Drosophila phylogeny. Mol Biol Evol. 2012;29:3459.
Hedengren M, Borge K, Hultmark D. Expression and evolution of the Drosophila Attacin/Diptericin gene family. Bioc Biophys Res Comm. 2000. doi:10.1006/bbrc.2000.3988.
Lee J, Lee J, Lee JH, Cho KS, Yoo J, Chung J. Diptericin-like protein: an immune response gene regulated by the anti-bacterial gene induction pathway in Drosophila. Gene. 2001;271:233–8.
Tzou P, Ohresser S, Ferrandon D, Capovilla M, Reichhart JM, Lemaitre B, Hoffmann JA, Imler JL. Tissue-Specific Inducible Expression of Antimicrobial Peptide Genes in Drosophila Surface Epithelia. Immunity. 2000;13:737–48.
Schmid-Hempel P. Variation in immune defence as a question of evolutionary ecology. Proc R Soc Biol Sci. 2003;270:357–66.
DiAngelo JR, Bland ML, Bambina S, Cherry S, Birnbaum MJ, Maniatis T. The Immune Response Attenuates Growth and Nutrient Storage in Drosophila by Reducing Insulin Signaling. PNAS. 2009;106:20853–8.
Buchon N, Silverman N, Cherry S. Immunity in Drosophila melanogaster—from microbial recognition to whole-organism physiology. Nat Rev Immunol. 2014. doi:10.1038/nri3763. PMID:25421701.
Mylonakis E, Podsiadlowski L, Muhammed M, Vilcinskas A. Diversity, evolution and medical applications of insect antimicrobial peptides. Phil Trans R Soc B. 2016;371:1695.
Acknowledgements
This work was supported by a Natural Sciences and Engineering Research Council of Canada Discovery Grant to SP.
Availability of data and materials
The data sets supporting the results of this article are included as supplementary data files as a .zip packaged file in this manuscript.
Authors’ contributions
MAH and SJP conceived and designed the study. MAH performed the laboratory work, and collected the data. MAH and PTH sorted and analysed the data, including bioinformatics analyses. All authors contributed to writing the manuscript and approved the final version of it.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethical approvals
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
PCR primers used in expression analyses and amplification of immune gene DNA from diverse Drosophila. (XLSX 39 kb)
Additional file 2: Figure S4.
Our Serratia strain is an oral pathogen of D. neotestacea related to Serratia marcescens. A) Flies were fed on mushroom agar with either 100uL of Luria-Bertani Broth (Control) or OD600 = 0.1 Serratia solution (Exposed) for 6 h, prior to transfer to sterile mushroom agar vials. Flies were turned over into new agar vials every 4 days, and mortality was recorded daily. Crosses indicate flies that were lost unrelated to treatment. Flies exposed to Serratia suffered significantly shorter lifespans compared to control treatments (n = 212; LR test: χ 2 = 11.8, p = 5.90e–4; GW test: χ 2 = 13.3, p = 2.66e–4). B) Maximum likelihood tree (100 bootstraps) of the isolated Serratia sp. 16S gene highlighted in red, with Rahnella sp. included as an outgroup. (PDF 390 kb)
Additional file 3: Figure S1.
Phylogenetic analysis of PGRP-SC1 and SC2 amino acid sequences using maximum likelihood. Support values indicate 100 bootstraps. Drosophila neotestacea has two PGRP-SC genes. The D. albomicans PGRP-SC1 signal peptide sequence is unresolved, and the current scaffold assembly in the D. guttifera genome does not contain the anterior region of its PGRP-SC1 orthologue. (PDF 45 kb)
Additional file 4: Figure S2.
Example branch-site REL analysis of Drosophila diptericins. Branch colour indicates the strength of selection, with red corresponding to dN/dS > 5, grey to dN/dS = 1, and blue to dN/dS = 0. The width of the colour on each branch represents the proportion of sites in the corresponding class. Bolded branches indicate branches that evolved under positive selection. In this BSR analysis, there is strong evidence that the root branch of the DptC clade diverged through positive selection (p < 0.001); there is also some evidence for positive selection at the divergence of DptA from DptB (p = .017). The codon alignment used in this analysis does not include D. ananassae diptericins, as D. ananassae DptA clustered with D. willistoni DptA on extremely long branches. This D. ananassae diptericin amino acid sequence can be seen in Fig. 4d, and consequences on net charge of D. ananassae DptA are shown in Additional file 5: Table S2. (PDF 47 kb)
Additional file 5: Table S2.
Net charges of DptA or DptC domains from Drosophila diptericins. Net charges were calculated using Protein Calculator v3.4 (http://protcalc.sourceforge.net/), and are given for each domain. DptC shows extensive differences at the amino acid sequence level. These differences have resulted in more extreme charges on each domain in the DptC molecule. DptC molecules generally have more negative P domains (except for D. albomicans), and more positive G domains, resulting in relatively similar net charges to DptA (except for D. albomicans). (XLSX 35 kb)
Additional file 6: Figure S3.
AMP gene expression following no-wound controls in (A) D. virilis and (B) D. neotestacea. Treatments involved a sterile wound (Sterile), Serratia bacterial challenge (Septic), or a no-wound control (No Wound). Comparing sterile wound treatments to no-wound controls, in D. virilis, AttB was induced 2.9-fold (t(6.79) = –2.22, p = .063), while in D. neotestacea AttB was induced by 4.2-fold (t(13.25) = –4.99, p < 0.0005). Drosocin and diptericin in both D. virilis and D. neotestacea were not strongly induced by sterile wounding (p > 0.1). The D. neotestacea DptC was not upregulated by Serratia challenge, even relative to unwounded control flies. Drosocin is not appreciably induced by sterile wounding in either species, despite drosocin being induced by sterile wounding in D. melanogaster (Lemaitre et al., 1997); this difference in expression may be due to drosocin’s shift in genomic position between these two lineages (Fig. 3b). (PDF 439 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hanson, M.A., Hamilton, P.T. & Perlman, S.J. Immune genes and divergent antimicrobial peptides in flies of the subgenus Drosophila. BMC Evol Biol 16, 228 (2016). https://doi.org/10.1186/s12862-016-0805-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-016-0805-y