
Abstract
Background
Quaternary climatic changes led to variations in sea level and these variations played a significant role in the generation of marine terrace deposits in the South Atlantic Coastal Plain. The main consequence of the increase in sea level was local extinction or population displacement, such that coastal species would be found around the new coastline. Our main goal was to investigate the effects of sea level changes on the geographical structure and variability of genetic lineages from a Petunia species endemic to the South Atlantic Coastal Plain. We employed a phylogeographic approach based on plastid sequences obtained from individuals collected from the complete geographic distribution of Petunia integrifolia ssp. depauperata and its sister group. We used population genetics tests to evaluate the degree of genetic variation and structure among and within populations, and we used haplotype network analysis and Bayesian phylogenetic methods to estimate divergence times and population growth.
Results
We observed three major genetic lineages whose geographical distribution may be related to different transgression/regression events that occurred in this region during the Pleistocene. The divergence time between the monophyletic group P. integrifolia ssp. depauperata and its sister group (P. integrifolia ssp. integrifolia) was compatible with geological estimates of the availability of the coastal plain. Similarly, the origin of each genetic lineage is congruent with geological estimates of habitat availability.
Conclusions
Diversification of P. integrifolia ssp. depauperata possibly occurred as a consequence of the marine transgression/regression cycles during the Pleistocene. In periods of high sea level, plants were most likely restricted to a refuge area corresponding to fossil dunes and granitic hills, from which they colonized the coast once the sea level came down. The modern pattern of lineage geographical distribution and population variation was established by a range expansion with serial founder effects conditioned on soil availability.
Similar content being viewed by others


Background
The severe climatic oscillations that occurred during the Pleistocene produced major changes in species distribution and consequently in their genetic diversity. These changes strongly impacted the vegetation at different latitudes and longitudes [1,2]. Some species became extinct over much of their range, some dispersed to new locations, and others survived in refuges and then expanded again; all of these events could have occurred repeatedly during the period [3,4]. Fragmentation, contraction, and expansion of species’ distribution occurred during the Quaternary climate fluctuations and have been suggested as an explanation for the current patterns of genetic diversity found in different lineages [5-7].
The response of vegetation in the Southern Hemisphere to the glacial-interglacial oscillations has been substantially different from that of the Northern Hemisphere [8,9]. Studies from several taxa have reported deep phylogeographic structures in South America dating to the Neogene, possibly as a consequence of old marine transgressions and the uplift of the Andes (for review see [10]). Nevertheless, genetic consequences of recolonization after the last glacial maximum (LGM) ca. 20 000 years before the present have also been described in species from South America (e.g., [4,10-12]).
Quaternary climatic changes also led to variations in sea level, which was lower during glacial periods and higher during interglacial periods [13]. Periodical transgressions of 100 m above the present level have been reported for the Quaternary [14,15], and some studies [16,17] have indicated that sea level changes during the Quaternary played a significant role in the generation of marine terrace deposits in the South Atlantic Coastal Plain (SACP). Studies along the east-northeast Atlantic Coast in South America have shown that during high sea level, barrier island lagoon systems were the dominant mode of sedimentation (reviewed by [18]). In the South Atlantic, these systems resulted in a geological formation known as the SACP, which consists of four barrier lagoon depositional systems (Barrier I, II, III and IV) representing the sedimentary record of a marine transgression: three from the Pleistocene and one from the Holocene, dating to 400 000, 325 000, 125 000, and 7000 years before the present, respectively [19].
The main consequence of the increase in sea level was local extinction or population displacement, such that coastal species would be found around the new coastline. Thus, for coastal species, estuaries and slopes of the mountains may have acted as refuge areas during interglacial periods. Under the refuge hypothesis, one could expect to find evidence of high genetic diversity in areas of stability and lower diversity and the molecular signatures of recent range expansion in the species in unstable, recently recolonized regions [1,20]. The refuge theory has been widely tested for tropical Neotropical biomes [10], but its general relevance for non-forested biomes has been less explored [10,12].
If refuge areas were important in the SACP during the Pleistocene, it should be possible to find a correlation between genetic diversity and putative ancient refuge areas (see [9,20]). More specifically, one would expect to find areas of high genetic diversity and no signal of recent population growth in refuge areas, whereas recently colonized habitats would have lower genetic diversity and signals of population expansion. Moreover, the age of genetic lineages should be congruent with geographical distribution, and population expansion must postdate SACP origin.
Petunia integrifolia is a bee-pollinated species that presents purple flowers [21] and is part of the short tube group [22,23] and lowland clade of Petunia [24]. According to Stehmann and Bohs [25], this species comprises two subspecies (Figure 1): P. integrifolia ssp. integrifolia and P. integrifolia ssp. depauperata (Fries) Stehmann and Semir. The former is widespread in the Pampas region (Uruguay, part of Argentina, and southern part of Rio Grande do Sul, Brazil), while the latter is endemic to the SACP, from Florianópolis (Santa Catarina, Brazil) to Chuí (border between Brazil and Uruguay). Each subspecies represents a different evolutionary lineage based on sequences of the internal transcribed spacers of ribosomal nuclear DNA and plastid DNA [26].

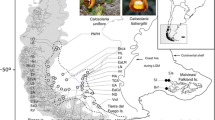
Plant material. Map of the sampling sites for the two Petunia integrifolia subspecies, where black circles (numbers 1 to 30) represent Petunia integrifolia ssp. depauperata samples and black squares correspond to Petunia integrifolia ssp. integrifolia. More information on collection sites is available in Additional file 2. Right side: representatives of two Petunia integrifolia subspecies, general view of individual and flower detail.
In this work, we used a phylogeographic approach to characterize the genetic diversity of a comprehensive sample of P. integrifolia ssp. depauperata, looking for geographical patterns of genetic variation and lineage distribution that could have emerged during the Pleistocene. Our main aim was to investigate the effects of sea level changes and the putative role of refuge areas for P. integrifolia ssp. depauperata using sequences from two plastid intergenic spacers. We chose these genetic markers because they have been successfully used in phylogeographic studies in Petunia [24,26-30] and in a closely related genus, Calibrachoa Cerv. [31].
Results and discussion
Sequence analysis
A general characterization of each marker is shown in Table 1. For P. integrifolia ssp. depauperata, the concatenated alignment resulted in 1120 bp, with 25 variables sites and 19 parsimoniously informative sites, resulting in 26 haplotypes. For P. integrifolia ssp. integrifolia, the concatenated alignment resulted in 1071 bp, with 15 variable sites and seven parsimoniously informative sites, resulting in 13 haplotypes. Variable and informative indels produced the discrepancies between the complete alignment and individual sequence length ranges. Despite the great morphological similarity and their taxonomic status, these two taxa did not share haplotypes, which is surprising because haplotype sharing is widely reported among Petunia species [24,28]. Another example of haplotype sharing is the three P. axillaris (Lam.) Britton, Sterns and Poggenb subspecies, which cannot be identified based on these plastid sequences alone [29].
As described in Table 2, the molecular diversity presented for P. integrifolia ssp. depauperata was h = 0.77 ± 0.01 and π (%) = 0.13 ± 0.09. All parameters were in accordance with other data obtained for other Petunia species in different surveys, despite the wide difference in geographic distribution range and sample sizes among species. In a previous study including P. integrifolia ssp. depauperata, Longo et al. [26] found only nine haplotypes and similar diversity indices using far fewer individuals. P. exserta Stehmann, a species with a restricted geographic distribution, was analyzed using the same genetic markers and a similar sample size and presented only six haplotypes but similar nucleotide diversity [28]. The three subspecies of P. axillaris, that together cover a geographic range larger than that of P. integrifolia ssp. depauperata, presented 35 haplotypes, but nucleotide and haplotype diversity values [29] were close to those reported in the present study.
Phylogenetic relationships and molecular dating
The 39 different haplotypes of P. integrifolia were used in the network and phylogenetic analyses (Additional file 1). The median-joining network divided the sample into four major haplogroups (Figure 2). The first three haplogroups were composed of haplotypes found in individuals of P. integrifolia ssp. depauperata (H1 to H26) and will be further discussed below. The last haplogroup contains all haplotypes of P. integrifolia ssp. integrifolia individuals (numbers 27 to 39). Haplotype H19 (identified by an asterisk in Figure 2) was an exception, grouping in the same haplogroup as P. integrifolia ssp. integrifolia haplotypes but appearing in two individuals from population 17 of P. integrifolia ssp. depauperata. We cannot discard the possibility of hybridization between these species, but natural hybridization is rare in Petunia and is limited to P. exserta and P. axillaris [28]. It is most likely that these individuals considered as P. integrifolia ssp. depauperata were misidentified during collection because the respective population is located at the limit of the taxa distribution. The haplogroup of P. integrifolia ssp. integrifolia was separated from P. integrifolia ssp. depauperata haplogroups by two mutational steps. This fact makes these subspecies suitable for gene studies related to adaptations to osmotic and saline stresses, which could be involved in the differentiation process in this taxa complex.

Evolutionary relationships among haplotypes of Petunia integrifolia subspecies. (A) Bayesian phylogenetic tree with posterior probabilities (PP > 0.9) shown above the branches and ages indicated below branches for selected nodes in thousands of years (Kya). Confidence intervals are presented in parenthesis; selected nodes marked with “a” indicate the higher probability of Most Recent Common Ancestor origin in Petunia integrifolia ssp. integrifolia group and “b” and “c” indicate Center and Southern Groups of Petunia integrifolia ssp. depauperata, respectively (see Table xx for probabilities); (B) Haplotype median-joining network. Sizes of the circles are proportional to the overall frequency of the haplotypes, and the color within each circle represents different genetic groups, according to the colors on the left and the Bayesian inference. Crossed lines represent inferred differences between haplotypes. The haplotype marked by asterisk (H19) belongs to Petunia integrifolia spp. depauperata population 17 (more details in the main text).
The three haplogroups of P. integrifolia ssp. depauperata had a striking geographical structure (Figure 2), with one haplogroup distributed in the central part of the species distribution (Center Group), one in the northern part (Northern Group), and the last distributed in the southern part (Southern Group). Haplotypes from the Center Group were observed in individuals from a region of a fossil dune environment and granitic hills that were not affected by changes in the sea level during the Pleistocene [31,32] and were reminiscent of the oldest sea level transgression/regression cycle (Barrier I; [19]). Haplotypes from the Northern Group were distributed in populations localized in regions associated with Barrier II [19], which is younger than Barrier I. Finally, haplotypes from the Southern Group occurred in a region corresponding to the more recent Barriers III and IV [19]. Notably, the Northern Group was separated by one mutational step from the central haplogroup, while the southern haplogroup was separated from the northern haplogroup by one mutational step. The northern and southern haplogroups exhibited a marked star-like shape, which might indicate a population expansion.
Different populations shared the four most frequent haplotypes (H1, H4, H5, and H10). Only six populations were monomorphic, and 18 haplotypes were exclusives (Additional file 2). In general, neighbor populations shared haplotypes. Sharing ancestral polymorphisms is common in Petunia species populations [24,28,29] and Calibrachoa [30,33], independent of the geographic distance between the populations. However, neighbor populations may also share haplotypes due to gene flow mediated by pollen and seeds between these populations because the plastid is maternally inherited (through seeds) in Petunia species [34]. The nearly continuous geographical distribution of P. integrifolia ssp. depauperata along the coast may facilitate gene flow either due to higher pollinator displacement or due to seed dispersal caused by the strong winds in the SACP [35,36].
The Bayesian phylogenetic tree displayed two main clades with maximum support values corresponding to P. integrifolia subspecies and a divergence time of approximately 635 thousand years ago (Kya; Figure 2 top). The P. integrifolia ssp. depauperata clade showed the same three haplogroups previously identified in the evolutionary network (Figure 2 bottom). The Center Group was sister to the group formed by individuals from the Northern and Southern Groups. The Northern and Southern Groups diverged from one another ca. 361 Kya, whereas both diverged from the Center Group ca. 488 Kya. These ages are broadly compatible with the estimates of ages for SACP origin and the associated depositional systems [19], which suggest that the diversification of these haplogroups was possible as soon as new habitat was available for plant colonization in 400 kya (Barrier I), 325 kya (Barrier II), 125 kya (Barrier III), and 7 kya (Barrier IV), respectively.
The striking geographic structure of these clades makes the phylogeographic reconstruction very clear for most nodes of the tree (Figure 2), the only exceptions for which the reconstruction is not obvious are the root of the tree and some of the nodes closer to the root (reconstructions marked by letters in Figure 2). Table 3 presents the posterior probabilities for these reconstructions. The scenario with highest posterior probability puts the most recent common ancestor (MRCA) of the whole clade in the region currently occupied by subsp. integrifolia, and subsequent migration of the lineage-giving rise to the depauperata subspecies to the central region. Our estimates are not clear, however, regarding the MCRA of the populations currently in the southern and northern regions, with high posterior probability of being in either region (Table 3). We explored Bayes factors to find significant migrations between geographic locations, however due to some topographic uncertainty at the most recent nodes of the tree and the few inferred migrations along the tree, most of these results were non-significant.
The phylogeographic model assumes constant migration rates throughout evolutionary history. While this is clearly not the case for our analyses because some of the regions were submerged and thus unavailable for plant migration, the results highlight our hypothesis of a refuge in the center of distribution and posterior migration to the north and to the south. Even without informing the model that the southern and northern portions of the SAPC were not available habitats early in their evolutionary history, we infer the origin of these populations of P. integrifolia ssp. depauperata to be in the western regions, from P. integrifolia ssp. integrifolia individuals, and migration to the coastal regions to only happen in more recent times (Figure 2).
Population structure and spatial genetic analysis
A two-level AMOVA (i.e., subpopulations vs. the total population) resulted in an estimate of 66% of the total genetic variance found among populations, while the remaining 34% of total genetic variation was found within populations (Table 4). The Mantel test indicated a non-significant association (P = 0.2) between genetic and geographic distances for populations, which may suggest that the genetic differences among populations reflect the underlying genealogical structure of the plastid spacers more strongly than gene flow between neighbor populations.
The Samova analysis suggested that three groups (K = 3) maximize the genetic structure in the among-group level (Φ CT = 0.642; P < 0.001) (Figure 3). For K ≥ 4, Samova found similar or lower Φ CT values. Populations 1-8 formed Samova’s Group 2 (solid line). Most of these populations only had haplotypes from the Center Group, but populations 7 and 8 also presented, in low frequency, haplotypes H5 and H14 from the Northern Group, respectively. Populations 9-10 and 12-22 formed Samova’s Group 1 (dashed line). Most of these populations only had haplotypes from the Southern Group, except for population 9 (two individuals presenting haplotype H4, from the Center Group), population 13 (one individual presenting haplotype H5, from the Northern Group), and population 17 (two individuals presenting haplotype H19, which is more related to P. integrifolia ssp. integrifolia). Finally, populations 11 and 23-30 formed Samova’s Group 3 (dotted line). Populations 23-30 only had haplotypes from the Northern Group. The presence of population 11 in SG3, however, is more difficult to explain. This assignment may have occurred because this population had a high frequency of haplotypes H5 and H16 from the Northern Group (but also haplotype H10, from the Southern Group), despite this population being geographically closer to SG1. However, H5 was already present in another population from SG2 (Pop 7), though with minor frequency. Thus, population 11 may actually be part of SG1, but due to stronger drift or founder effect, a higher frequency of Northern Group haplotypes assigns it to SG3. More genetic markers will be necessary to clarify this issue. Notably, the geographical limits of these groups do not match the known putative actual or historical barriers (rivers or other geographic discontinuities) proposed as drivers of population structure of Calibrachoa heterophylla (Sendtn.) Wijsman from the same geographic distribution [30].

Geographical groups identified by SAMOVA analyses. Each group is indicated by a border style. The colors displayed in the populations correspond to genetic groups found in the phylogeny and the median-joining haplotype network. The gray dots indicate Petunia integrifolia ssp. integrifolia populations. The black dots highlighted with larger fonts are the populations with haplotypes from different genetic groups. Lines represent SAMOVA groups according the legend.
Demographic analyses
Fu’s F S value was negative and significant (-16.5, P < 0.02), whereas Tajima’s D was also negative but marginally non-significant (-1.31, P = 0.05) for P. integrifolia ssp. depauperata. Tajima’s D and Fu’s F s are classical neutrality tests used to assess population demographic history. Both assume that populations have been in mutation–drift balance for a long period of time [37]. Negative values in both tests are indicative of a demographic expansion. Together with the star-like shape observed in the networks, particularly for the Northern and Southern Groups, this may suggest a population expansion associated with SACP colonization.
The Northern group presented the lower haplotype diversity (h = 0.27) and a significant Fu’s F S (-4.09, P < 0.02), while the Southern group displayed the lower nucleotide diversity (π = 0.03) and significant and negative values for both neutrality tests (Tajima’s D = -1.92, P < 0.05; Fu’s F S = -11.30, P < 0.02) (Table 2).
The higher genetic diversity and lack of indication of population growth are perfectly compatible with Center group representing a refuge for P. integrifolia ssp. depauperata. However, the smaller genetic diversity and signals of population expansion suggest that Southern and Northern groups represent distinct events of range expansion from the refuge as long as SACP became available for this taxon.
The Bayesian Skyline Plot for P. integrifolia ssp. depauperata may be indicative of smooth yet constant population growth from ~20 000 years ago (Figure 4). This date roughly corresponds to the LGM and is a time when the majority of land was already established in the SACP (the extreme south of this region is considered completely stable only from around 7 000 years ago). This result should be taken with caution given the large credible intervals associated with population size estimates. Interestingly, though, if these estimates are correct, then the signal of population growth exhibited in the neutrality tests may reflect a more recent expansion rather than the original expansion of Southern and Northern groups, which should be older. We also tried to replicate this analysis separately for each haplogroup, but the effective sample sizes were very low, most likely due the lack of enough genetic variation for this estimate.

Historical changes in the effective size. Bayesian skyline plot showing the effective population size fluctuation throughout time for Petunia integrifolia ssp. depauperata (solid line, median estimations; grey area, confidence interval).
There are several species endemic to the SACP, but few studies have used a phylogeographic perspective to put in evolutionary context the genetic variation in this region. Mäder et al. [30], studying C. heterophylla, found that this species migrated from the west to colonize the SACP, with paleo river channels associated with the main genealogical lineages differentiation at SACP. Lopes et al. [38] studied a subterranean rodent in the same geographic area and identified (modern) rivers acting as effective barriers to gene flow in this species. Strikingly, P. integrifolia ssp. depauperata seems to be less sensitive to rivers in the SACP, as its genetic groups occurred along a wide area. This may be related to a different seed dispersal efficiency compared with C. heterophylla, for which rivers were more efficient barriers [30].
Single-locus studies may be problematic because single markers may tell a different evolutionary history compared to the rest of the genome [39-41]. However, it has also been recently suggested that one or a few small fragments presenting highly informative content could be better in solving the evolutionary relationships among lineages than a large genome survey with incongruent results [42]. In this sense, cpDNA markers applied to Petunia and in the closely related genus Calibrachoa [33,43] have been able to clarify the evolutionary relationships among species and among lineages within species [24,27,29,44-46].
Considering the results presented here and the absence of many studies in the South Atlantic Coastal Region, especially in regards to plant species and the influence of changes in the sea level on genetic variability distribution, this is an important contribution to the understanding of the processes that drove evolution.
Conclusion
Climate changes that occurred during the Pleistocene influenced the sea level in the South Atlantic Coastal Plain and promoted genetic differentiation and speciation in the herbaceous annual plant Petunia integrifolia ssp. depauperata. During this period, plants were restricted to a refuge area, represented by the central part of its current distribution, corresponding to fossil dunes and granitic hills, from which plants colonized the coast as soon as marine regressions exposed suitable lands. This inference is supported by genetic diversity and population expansion statistics. This is the first time that a refuge is proposed on the south edge of the Atlantic Coast.
Methods
Plant material, DNA extraction, PCR amplification and sequencing
A total of 291 individuals of P. integrifolia ssp. depauperata were identified and sampled from 30 locations, with regular distance intervals, covering all of the known distribution of this taxon, including the adjacent northern and southern areas (Figure 1, locations numbered from 1 to 30, hereafter named as populations). In addition, 85 individuals of P. integrifolia ssp. integrifolia were used in comparative approaches and in dating estimates. We obtained the geographic coordinates by Global Positioning System (GPS), and vouchers were deposited in the BHCB herbarium, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil. Total DNA was extracted from young leaves carefully collected for genetic analysis. Silica gel dried leaves were frozen in liquid nitrogen and ground to a fine powder, and DNA was extracted with cetyltrimethyl ammonium bromide (CTAB) protocol as described by Roy et al. [47]. This work was conducted under permit MP 2.186/16 of the Brazilian Federal Government to access plant genetic information to develop evolutionary or taxonomic studies. No specific collection permits were required because neither taxa are endangered or protected and because no population occurs on protected areas.
Polymerase chain reaction (PCR) was employed to amplify the non-coding plastid trnH-psbA and trnS-trnG intergenic spacers, using the universal primers previously described ([48,49], respectively), and following amplification conditions described in Lorenz-Lemke et al. [27]. The PCR products were purified according to Dunn and Blatnner [50] and sequenced in a MegaBACE1000 (GE Healthcare Bio Sciences Corp., Piscataway, NY, USA) automatic sequencer according to the manufacturer’s instructions and the DYEnamicET Terminator Sequencing Premix Kit (GE Healthcare). Tables S1 and S2 in Additional information provide voucher information, GenBank accession numbers, and the general geographic information for each sample.
Sequence analysis and molecular diversity
For each marker, both forward and reverse strands were checked using Chromas (available at website: http://technelysium.com.au/) and aligned manually in GeneDoc [51]. Because the plastid segments are naturally linked and this genome is usually non-recombining, a common situation in plant phylogenies, the two markers were combined in a single concatenated set. All insertion/deletion events (indels) that involved poly A/T were eliminated from the analyses because their homologies cannot be adequately accessed [52]. Contiguous indels of more than one base pair (bp) were treated as one mutational event [53]. Descriptive statistics of genetic variability, such as haplotype (h) and nucleotide (π) diversities [54], were estimated in Arlequin 3.5 [55].
Phylogenetic relationships
The evolutionary relationships between haplotypes were estimated using the median-joining network method (e = 0; [56]) as implemented in the Network 4.6 software (available at website: http://www.fluxus-engineering.com/sharenet.htm).
The dated Bayesian phylogenetic tree of haplotypes was inferred using Beast 1.8.0 [57] with a Yule tree prior and the HKY substitution model with four gamma-distributed rate categories based on results from the Akaike Information Criterion in jModelTest 2 [58]. We also used a lognormal relaxed clock and went to the literature for an informed prior for the nucleotide substitution rate. We compiled rates calculated for plastid markers for shrubs or herbaceous plants with generation time of up to three years, features that are similar to Petunia. We found eight published rates that fit these criteria, varying between 1 x 10-9 and 8.24 x 10-9 substitutions per site per year (s/s/y) [59-63]. To take into account this rate heterogeneity, we used a gamma distribution prior with a shape parameter 1.6 and scale parameter 1.6 x 10-9 as prior. We assumed an offset value of 1 x 10-9 s/s/y, such that the median of the prior was 3.05 x 10-9 s/s/y, allowing rate values of 8.24 x 10-9 s/s/y to be reached with low probability (distribution graphical result is available in Additional file 3).
Discrete phylogeographical analysis
For the phylogeographic reconstruction, we modeled geography through the discrete phylogeographic diffusion model of [64] and used the Bayesian stochastic search variable selection (BSSVS) procedure in Beast. We classified sampling locations in four discrete states: southern coastal plain (Southern), northern coastal plain (Northern), Center, and “integrifolia” (corresponding to inland individuals of P. integrifolia ssp. integrifolia). We then estimated a reconstruction of the phylogeographic history of the clade. We also explored different choices of prior distributions for migration rates in this model, including a uniform prior and a distance-informed prior, and we found that additional information on geographic distances between locations had little impact on the estimated rates.
For each of the above analyses, two independent runs consisting of 1 x 108 Markov chain Monte Carlo (MCMC) iterations were performed, sampling every 1000 generations; 10% of iterations were removed as burn-in. Convergence was checked by visual inspection of the independent runs in Tracer 1.6 [65] so that all parameters had effective sample sizes (ESS) > 200. We obtained the maximum clade credibility (MCC) tree and the posterior probabilities (PP) for each node [66] using the TreeAnnotator from Beast package, and finally, we used Figtree 1.4.0 [67] to draw and edit the resulting phylogenetic trees.
Geographic structure
We also used Arlequin to perform an analysis of molecular variance (AMOVA; [68]) to quantify the degree of genetic structure among groups of populations, which was performed using Φ-statistics and 1000 permutations to test significance. The Mantel test was used to evaluate the correlation between genetic and geographic distances, using 1000 permutations in the program Alleles in Space 1.0 [69]. We used Samova 1.0 [70] to estimate the number of genetic homogeneous groups (K) for P. integrifolia ssp. depauperata through a spatial analysis of molecular variance. Following this, we used Arlequin to estimate AMOVA and genetic variability statistics for the groups defined by Samova, as described previously.
Demographic analysis
We also used Arlequin to perform the neutrality tests Tajima’s D [71] and Fu’s Fs [72]. Historical changes in the effective size of P. integrifolia ssp. depauperata were also inferred through the Bayesian Skyline Plot (BSP; [73]) method implemented in Beast, using the same prior and conditions described previously except that all individual sequences were included and the Bayesian Skyline tree prior was used. The BSP reconstruction was done using Tracer 1.6.
Availability of supporting data
Sequences are deposited in GenBank (accession numbers available in Additional file 1).
References
Bennett KD, Provan J. What do we mean by ‘refugia’? Quat Sci Rev. 2008;27:2449–55.
Stewart JR, Lister AM, Barnes I, Dalén L. Refugia revisited: individualistic responses of species in space and time. Proc Roy Soc Lond B. 2010;277:661–71.
Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Phil Trans Roy Soc B. 2004;359:183–95.
Hewitt GM. Quaternary phylogeography: the roots of hybrid zones. Genetica. 2011;139:617–38.
Hewitt GM. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc. 1996;58:247–76.
Hewitt GM. Post glacial recolonization of European biota. Biol J Linn Soc. 1999;68:87–112.
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF. Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol. 1998;7:453–64.
Markgraf V, McGlone M, Hope G. Neogene paleoenvironmental and paleoclimatic change in southern temperate ecosystems a southern perspective. Trends Ecol Evol. 1995;10:143–7.
Keppel G, van Niel KP, Wardell-Johnson GW, Yates CJ, Byrne M, Mucina L, et al. Refugia: identifying and understanding safe havens for biodiversity under climate change. Gl Ecol Biogeogr. 2012;21:393–404.
Turchetto-Zolet AC, Pinheiro F, Salgueiro F, Palma-Silva C. Phylogeographical patterns shed light on evolutionary process in South America. Mol Ecol. 2013;22:1193–213.
Hewitt GM. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–13.
Beheregaray LB. Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Mol Ecol. 2008;17:3754–74.
Lambeck K, Esat TM, Potter EK. Links between climate and sea levels for the past three million years. Nature. 2002;419:199–206.
Yokoyama Y, Lambeck K, Johnston P, Deckker P, Fifield K. Timing of the last glacial maximum from observed sea level minima. Nature. 2000;406:713–6.
Ponce JF, Rabassa J, Coronato A, Borromei AM. Paleogeographical evolution of the Atlantic coast of Pampa and Patagonia from the last glacial maximum to the Middle Holocene. Biol J Linn Soc. 2011;103:363–79.
Bigarella JJ, Andrade GO. Contributions to the study of the Brazilian Quaternary. Geol Soc Spec Publ. 1965;84:433–51.
Suguio K, Martin L, Bittencourt ACSP, Dominguez JML, Flexor JM, Azevedo AEG. Quaternary emergent and submergent coast: comparison of the Holocene sedimentation in Brazil and Southeastern United States. An Acad Bras Cienc. 1984;56:163–7.
Dominguez JML, Bittencourt ACSP, Martin L. Controls on Quaternary coastal evolution of the east northeastern coast of Brazil: roles of sea level history, trade winds and climate. Sed Geol. 1992;80:213–32.
Martin L, Suguio K, Flexor JM. Hauts niveaux marins pléistocènes du littoral Brésilien. Palaeogeogr Palaeoclim Palaeoecol. 1988;68:231–9.
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C. Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science. 2009;323:785–9.
Gübitz T, Hoballah ME, Dell’Olivo A, Kuhlemeier C. Petunia as a model system for the genetics and evolution of pollination syndromes. In: Gerats T, Strommer J, editors. Petunia evolutionary, developmental and physiological genetics. New York: Springer; 2009. p. 29–49.
Stehmann JR, Lorenz-Lemke AP, Freitas LB, Semir J. The genus Petunia. In: Gerats T, Strommer J, editors. Petunia evolutionary, developmental and physiological genetics. New York: Springer; 2009. p. 1–28.
Reck-Kortmann M, Silva-Arias GA, Segatto ALA, Mäder G, Bonatto SL, Freitas LB. Multilocus phylogeny reconstruction: new insights into the evolutionary history of the genus Petunia. Mol Phylogen Evol. 2014;81:19–28.
Lorenz-Lemke AP, Togni PD, Mäder G, Kriedt RA, Stehmann JR, Salzano FM, et al. Diversification of plant species in a subtropical region of eastern South American highlands: a phylogeographic perspective on native Petunia (Solanaceae). Mol Ecol. 2010;19:5240–51.
Stehmann JR, Bohs L. Nuevas combinaciones en Solanaceae. Darwiniana. 2007;45:240–1.
Longo D, Lorenz-Lemke AP, Mäder G, Bonatto SL, Freitas LB. Phylogeography of the Petunia integrifolia complex in southern Brazil. Bot J Linn Soc. 2014;174:199–213.
Lorenz-Lemke AP, Mäder G, Muschner VC, Stehmann JR, Bonatto SL, Salzano FM, et al. Diversity and natural hybridization in a highly endemic species of Petunia (Solanaceae): a molecular and ecological analysis. Mol Ecol. 2006;15:4487–97.
Segatto ALA, Cazé ALR, Turchetto C, Klahre U, Kuhlemeier C, Bonatto SL, et al. Nuclear and plastid markers reveal the persistence of genetic identity: A new perspective on the evolutionary history of Petunia exserta. Mol Phylogen Evol. 2014;70:504–12.
Turchetto C, Fagundes NJR, Segatto ALA, Kuhlemeier C, Solís-Neffa VG, Speranza PR, et al. Diversification in the South American Pampas: The genetic and morphological variation of the widespread Petunia axillaris complex (Solanaceae). Mol Ecol. 2014;23:374–89.
Mäder G, Fregonezi JN, Lorenz-Lemke AP, Bonatto SL, Freitas LB. Geological and climatic changes in quaternary shaped the evolutionary history of Calibrachoa heterophylla, an endemic South Atlantic species of petunia. BMC Evol Biol. 2013;13:178.
Villwock JA, Tomazelli LJ, Loss EL, Dehnhardt EA, Horn NO, Bachi FA, et al. Geology of the Rio Grande do Sul Coastal Province. Quat South Amer Antarct Penin. 1986;4:79–97.
Pessenda LCR, Gouveia SEM, Ribeiro AS, Oliveira PE, Aravena R. Late Pleistocene and Holocene vegetation changes in northeastern Brazil determined from carbon isotopes and charcoal records in soils. Palaeogeog Palaeoclim Palaeoecol. 2010;297:597–608.
Fregonezi JN, Turchetto C, Bonatto SL, Freitas LB. Biogeographical history and diversification of Petunia and Calibrachoa (Solanaceae) in the Neotropical Pampas grassland. Bot J Linn Soc. 2013;171:140–53.
Derepas A, Dulieu H. Inheritance of the capacity to transfer plastids by pollen parent in Petunia hybrida Hort. J Hered. 1992;83:6–10.
Garzoli S, Simionato C. Baroclinic instabilities and forced oscillations in the Brazil/Malvinas confluence front. Deep Sea Res. 1990;37:1053–74.
Goni GJ, Wainer I. Investigation of the Brazil Current front variability from altimer data. J Geophys Res. 2001;106:31117–28.
Nei M, Kumar S. Molecular evolution and phylogenetics. New York: Oxford University Press; 2000.
Lopes CM, Ximenes SSF, Gava A, Freitas TRO. The role of chromosomal rearrangements and geographical barriers in the divergence of lineages in a South American subterranean rodent (Rodentia: Ctenomyidae: Ctenomys minutus). Heredity. 2013;111:293–305.
Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 2006;55:21–30.
Townsend JP. Profiling phylogenetic informativeness. Syst Biol. 2007;56:222–31.
Townsend JP, Su Z, Tekle YI. Phylogenetic signal and noise: predicting the power to resolve a phylogeny. Syst Biol. 2012;61:835–49.
Horreo JL. ‘Representative Genes’, is it OK to use a small amount of data to obtain a phylogeny that is at least close to the true tree? J Evol Biol. 2012;25:2661–4.
Fregonezi JN, Freitas LB, Bonatto SL, Semir J, Stehmann JR. Infrageneric classification of Calibrachoa (Solanaceae) based on morphological and molecular evidence. Taxon. 2012;61:120–30.
Ando T, Kokubun H, Watanabe H, Tanaka N, Yukawa T, Hashimoto G, et al. Phylogenetic analysis of Petunia sensu Jussieu (Solanaceae) using chloroplast DNA RFLP. Ann Bot. 2005;96:289–97.
Kulcheski FR, Muschner VC, Lorenz-Lemke AP, Stehmann JR, Bonatto SL, Salzano FM, et al. Molecular phylogenetic analysis of Petunia Juss. (Solanaceae). Genetica. 2006;126:3–14.
Chen S, Matsubara K, Omori T, Kokubun H, Kodama H, Watanabe H, et al. Phylogenetic analysis of the genus Petunia (Solanaceae) based on the sequence of the HF1 gene. J Plant Res. 2007;120:385–97.
Roy A, Frascaria N, MacKay J, Bousquet J. Segregating random amplified polymorphic DNAs (RAPDs) in Betula alleghaniensis. Theor App Genet. 1992;85:173–80.
Hamilton MB. Four primers pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Mol Ecol. 1999;8:513–25.
Sang T, Crawford DJ, Stuessy TF. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am J Bot. 1997;84:1120–36.
Dunn IS, Blattner FR. Charons 36–40: multi enzyme, high capacity, recombination deficient replacement vectors with polylinkers and polystuffers. Nucl Acids Res. 1987;15:2677–98.
Nicholas KB, Nicholas Jr HB, Deerfield II DW. GeneDoc: analysis and visualization of genetic variation. Embnew News. 1997;4:14.
Aldrich J, Cherney BW, Merlin E, Christopherson L. The role of insertion/deletions in the evolution of the intergenic region between psbA and trnH in the chloroplast genome. Curr Genet. 1988;14:137–46.
Simmons MP, Ochoterena H. Gaps as characters in sequence based phylogenetic analyses. Syst Biol. 2000;49:369–81.
Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987.
Excoffier L, Lischer HEL. Arlequin suite ver. 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res. 2010;10:564–7.
Bandelt HJ, Forster P, Röhl A. Median joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 2005;16:37–48.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nature Meth. 2012;9:772.
Zurawski G, Clegg MT, Brown AHD. The nature of nucleotide sequence divergence between barley and maize chloroplast DNA. Genetics. 1984;106:735–49.
Wolfe KH, Li WH, Sharp PM. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci U S A. 1987;84:9054–8.
Richardson JE, Pennington RT, Pennington TD, Hollingsworth PM. Rapid diversification of a species rich genus of Neotropical rain forest trees. Science. 2001;293:2242–5.
Mummenhoff K, Linder P, Friesen N, Bowman JL, Lee JY, Franzke A. Molecular evidence for bicontinental hybridogenous genomic constitution in Lepidium sensu stricto (Brassicaceae) species from Australia and New Zealand. Am J Bot. 2004;91:254–61.
Yamane K, Yano K, Kawahara T. Pattern and rate of indel evolution inferred from whole chloroplast intergenic regions in sugarcane, maize and rice. DNA Res. 2006;13:197–204.
Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comp Biol. 2009;5, e1000520.
Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer v1.5. http://beast.bio.ed.ac.uk/Tracer.
Rannala B, Yang ZH. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J Mol Evol. 1996;43:304–11.
Rambaut A. FigTree v1.4: Tree Fig. drawing tool. http://tree.bio.ed.ac.uk/software/figtree/.
Excoffier L, Smouse PE, Quattro M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91.
Miller M. Alleles in space: computer software for the joint analysis of interindividual spatial and genetic information. J Hered. 2005;96:722–4.
Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11:2571–81.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Drummond AJ, Suchard MA. Bayesian random local clocks or one rate to rule them all. BMC Biol. 2010;8:114.
Acknowledgements
The authors thank A.P. Lorenz-Lemke, G. Mäder, and P.D. Togni for help with fieldwork and J.R. Stehmann for taxonomic assistance. This project was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and the programa de Pós-Graduação em Genética e Biologia Molecular da Universidade Federal do Rio Grande do Sul (PPGBM-UFRGS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LBF and AMCRF planned, designed, and led the project; AMCRF conducted the experiments; AMCRF, JNF, NJRF, GC, and SLB ran the analyses; LBF, AMCRF, and GC wrote most of the text; LBF and SLB provided reagents and equipment to develop the experiments. All authors contributed in the preparation of the study and have commented on and approved the final manuscript.
Authors’ information
Collectively, the group is interested in investigating evolutionary processes, plant speciation, conservation genetics, phylogeographic patterns and molecular systematics.
Additional files
Additional file 1: Table S1.
Haplotypes information.
Additional file 2: Table S2.
Sampling information.
Additional file 3: Figure S1.
Plastid evolutionary rates distribution.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Ramos-Fregonezi, A.M., Fregonezi, J.N., Cybis, G.B. et al. Were sea level changes during the Pleistocene in the South Atlantic Coastal Plain a driver of speciation in Petunia (Solanaceae)?. BMC Evol Biol 15, 92 (2015). https://doi.org/10.1186/s12862-015-0363-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-015-0363-8