
Abstract
Background
Recent improvements in sequencing technologies enabled detailed profiling of genomic features. These technologies mostly rely on short reads which are merged and compared to reference genome for variant identification. These operations should be done with computers due to the size and complexity of the data. The need for analysis software resulted in many programs for mapping, variant calling and annotation steps. Currently, most programs are either expensive enterprise software with proprietary code which makes access and verification very difficult or open-access programs that are mostly based on command-line operations without user interfaces and extensive documentation. Moreover, a high level of disagreement is observed among popular mapping and variant calling algorithms in multiple studies, which makes relying on a single algorithm unreliable. User-friendly open-source software tools that offer comparative analysis are an important need considering the growth of sequencing technologies.
Results
Here, we propose Comparative Sequencing Analysis Platform (COSAP), an open-source platform that provides popular sequencing algorithms for SNV, indel, structural variant calling, copy number variation, microsatellite instability and fusion analysis and their annotations. COSAP is packed with a fully functional user-friendly web interface and a backend server which allows full independent deployment for both individual and institutional scales. COSAP is developed as a workflow management system and designed to enhance cooperation among scientists with different backgrounds. It is publicly available at https://cosap.bio and https://github.com/MBaysanLab/cosap/. The source code of the frontend and backend services can be found at https://github.com/MBaysanLab/cosap-webapi/ and https://github.com/MBaysanLab/cosap_frontend/ respectively. All services are packed as Docker containers as well. Pipelines that combine algorithms can be customized and new algorithms can be added with minimal coding through modular structure.
Conclusions
COSAP simplifies and speeds up the process of DNA sequencing analyses providing commonly used algorithms for SNV, indel, structural variant calling, copy number variation, microsatellite instability and fusion analysis as well as their annotations. COSAP is packed with a fully functional user-friendly web interface and a backend server which allows full independent deployment for both individual and institutional scales. Standardized implementations of popular algorithms in a modular platform make comparisons much easier to assess the impact of alternative pipelines which is crucial in establishing reproducibility of sequencing analyses.
Similar content being viewed by others
Explore related subjects
Find the latest articles, discoveries, and news in related topics.Background
Sequencing technologies become more accessible as they generate more data in less time for diminishing costs [1]. However, the computational requirements of processing NGS data are much higher than what most clinical facilities and biomedical laboratories have. The level of programming skills required for using mapping, preprocessing and variant calling algorithms efficiently is fairly high [2]. Furthermore, concordance among sequencing algorithms is limited especially for cancer sequencing [2]. There are many sequencing algorithms that can perform well in different settings and choosing the best combination of sequencing algorithms for a dataset is very difficult. It’s been shown that combining multiple algorithms improves performance and ensemble methods are developed along this aim [3, 4]. Despite this improvement, relying on a single combination that would work well for all possible scenarios is impossible considering the heterogeneity of applications in research and clinic.
There have been many previous successful efforts to provide researchers with flexible open source sequencing analysis pipelines and platforms. Galaxy [5] and Terra [6], which are the most commonly used platforms, harbors many of the commonly used algorithms for the analysis of genomics, metagenomics, transcriptomics as well as other omics data. Both platforms allow users to create and share workflows on the workflow hub. All that flexibility brings a steeper learning curve for beginners. Even though it is possible to deploy them locally, they are essentially used as cloud services. This brings a barrier for many users as local local regulations can be restrictive to share data with third parties over the internet. With the advancement of hardware acceleration technology, especially GPU’s, the demand for local handling of the sequencing data is increasing. These platforms are yet to respond to that demand. Moreover, provided variant annotations are very limited despite the platform being under development for years. Sarek [7] is another NGS analysis workflow which is built on Nextflow [8] workflow language. It provides limited number algorithms for the detection and analysis of germline and somatic mutations. Also it does not have a user interface and backend to manage user files and runs. DNAScan [9] and Sequana [10] are other available predefined NGS analysis workflows. They have very limited interfaces just to upload data files and lack a broad range of algorithms for comparative analysis of variants. The Table 1 shows a summary of feature comparison of tools.
In order to address the above-discussed limitations of the sequence analysis tools, in this paper, we propose Comparative Sequencing Analysis Platform (COSAP) as an alternative analysis platform. COSAP offers multiple options for each step of the analysis pipeline and allows users to compare the effect of these choices by producing multiple VCF files each representing a particular combination. COSAP simplifies planning and running sequencing pipelines both for skilled developers and non-technical users. COSAP can run locally or be deployed on a server to be accessed by many users from their local devices via a web interface. Users can create desired pipelines from pre-installed algorithms for mapping/aligning, pre-processing, variant calling and annotation. COSAP first creates a template file that contains all of the information that is needed to execute every step. Execution can be done in two ways. First, most users can utilize COSAP’s user-friendly web interface to upload their files and run analysis by selecting dropdown menus, monitor the progress of running analyses as well as visualize the results. Second, advanced users can take this a step further and use COSAP’s underlying Python API. This API allows the construction of more complex pipelines. New pipeline steps can be introduced by coding them into COSAP’s codebase with a streamlined process due to COSAP’s intuitive software design. Since constructing and running pipelines are decoupled, template files can be created from the API and then run from UI and vice versa.
Available tools
Fastq preprocessing and short read mapping
Preprocessing and quality checking of raw reads is the first step in most NGS analysis. Fastp [11] which is an all-in-one tool for fastq file quality control and filtering is used for these tasks in COSAP. For the short read mapping, BWA [12] and Bowtie2 [13] are the most commonly used tools and are available in COSAP. The newer and faster version of BWA is also included [14].
Aligned read preprocessing and filtering
There are several BAM preprocessing steps before variant calling recommended in the GATK Best Practices guidelines [15]. COSAP utilizes GATK4 [16] and Samtools [17] to handle these steps. Sort, index and mpileup commands from Samtools and, all tools from GATK are currently available.
SNV, Indel and structural variant discovery
Variant calling is one of the most extensively studied areas in the NGS research and a whole raft of variant callers are developed and utilized in the literature. Currently, COSAP supports 11 variant callers and 1 deep learning based variant refinement tool [18]. These callers are HaplotypeCaller [19], Varscan2 [20], Strelka2 [21], and DeepVariant [22] for germline samples. The included somatic variant callers are Mutect2 [19], Varscan2 [20], Varnet [23], MuSe [24], VarDict [25], Octopus [26], SomaticSniper [27]. The structural variants are called with Manta [28].
Variant annotation
The variant annotation is an essential step to extract meaningful information from the variant sets. Variants are annotated by using many tools depending on the sample type, variant type and the need of the researcher. COSAP supports Ensembl VEP [29], SnpEFF [30], Annovar [31] for functional annotations of SNV’s. The AnnotSV [32] is used to annotate structural variants and ClassifyCNV [33] is the tool of choice for annotating copy number variations. Germline and somatic variants are automatically classified according to ACMG/AMP [34, 35]guidelines by InterVAR [36] and CancerVAR [37] accordingly. GenomeNexus [38] which is a tool to annotate variants from multiple sources is also available with COSAP.
Implementation
There is a plethora of NGS algorithms available for preprocessing, mapping, alignment, variant calling and annotation. Unfortunately, many of these algorithms are not well-documented and new methods or algorithms are developed that fulfills various needs regularly. Therefore we developed a fully customizable and open source platform and integrated the popular algorithms into this platform. This level of customizability is achieved by several design decisions.
COSAP has been built around the principle of modularity of pipeline steps. Every step is abstracted into a Python class where the dependencies and algorithms involved in that step are all included. These Python classes work in sync with whichever algorithm or algorithms they need to call and only exit once the processes are either finalized or an exception is encountered. The encapsulation of all pipeline steps with identical method signatures means that can move blocks around and build complex networks of algorithms without knowing the internal workings, like building Lego blocks.
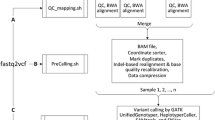
COSAP comes with the most popular algorithms used around the built-in NGS pipelines. This way for most use cases no new pipeline needs to be defined. These predefined steps are mainly targeted at somatic and germline pipelines. COSAP also includes predefined libraries for exome and genome panels. Custom libraries can be added in any location as long as their path is known to COSAP. The available tools, their outputs and general workflow of the COSAP pipeline is shown in Fig. 1.

Predefined pipeline steps have multiple algorithm/tool choices. The input and output of each step must be a list of files. The file names are for humans to understand, and steps know which file to read from a config file
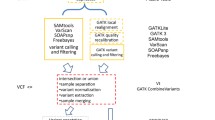
We also implemented fully functional web and backend applications as part of COSAP. The web application communicates with the backend via REST API. COSAP Docker containers are bound to the same storage with the backend and can work as celery workers which consume messages from the backend application. There may be multiple workers on the network which brings scalability to the platform. The message queuing architecture is shown in Fig. 2.

COSAP Docker container as a celery worker which consumes pipeline messages from the backend application
Software design
COSAP is a Python based modular tool that is designed to create NGS pipelines. Each pipeline step (aligning, variant calling, annotation, etc.) is written in its own classes following an abstract format. This abstraction allows each step to be created and run separately from each other. Custom steps can be written following the same abstract pattern. This means pipelines with any length and tool can be created like a graph. COSAP analysis consists of two major parts.
The first part contains the API that allows the user to define input files and pipeline steps, and then form one or more pipelines using these steps. The output of this creation is a pipeline state file in JSON format. This file contains all the information required to execute each step. It includes all input and inflight file names, location of algorithms and reference files, parameters for algorithms, timestamps and version information. The API has classes for complicated parameters to be passed along to algorithms and some predefined classes that can create most commonly used pipelines with a single or few lines of code.
The second part handles the running of the pipelines. This accepts a pipeline state file which is created by the first step automatically or manually edited by hand. There are two essential ways that a pipeline state file can be run. The legacy method runs each pipeline step defined in the state file in a sequence. The second method uses Snakemake [39] to optimize the resource usage (CPU and memory) by running steps in parallel if their requirements are satisfied. These requirements are essentially sufficient resources being available for the step to be run and files needed by the step are finalized or ready. Explained in detail in the performance section.
The decoupling of these two parts allows them to be assigned to different systems. The creation of pipeline states is not a resource heavy operation and can be handled by a smaller server whereas the second part (execution) can be split into clusters.
Results
Performance
Executing NGS pipelines may require large amounts of processing time and optimizing pipelines to leverage the full potential of modern hardware is extensively studied in the literature. Currently, hardware based accelerations such as NVIDIA Clara Parabrics [40] and Illumina Dragen [41] offer the best performance in terms of speed. These claim up to 80 × speed gain over the baseline GATK [16] tools. They also don't make a concession of variant calling accuracy as DRAGEN short-read call set was the top performer in PrecisionFDAv2 [42] as well as other studies [43] and Parabricks performed comparably in the benchmark study of Franke et al. [44]. The main of this approach is the requirement of specialized hardware such as FPGA and GPU which are inaccessible to many users. Software optimizations mostly aim for algorithmic improvements and better utilization of the hardware by adapting modern dataflow architectures and hyperthreading and may reach up to 16 × speed gain [45,46,47]. The problem with that approach is the modifications on the tools’ original code bases in order to make them compatible with other frameworks. These modifications are both error prone and forces users to stick to a specific version. In a previous study, Ahmad et al. [47] suggested that parallelizing GATK over RamDisk performed slightly worse than modified tools achieving ~ 3.5 × speed. Because of these reasons, we decided to utilize shared memory of Linux systems for in memory parallelization.
One of the most time consuming operations is the serialization and deserialization process, especially writing and reading files from the disks between pipeline steps. COSAP can be configured to use previously mentioned shared memory for these intermediate files. This means pipeline steps still need to perform serialization but the in memory read write speeds are incomparably higher than of reading from a disk. This kind of approach has two caveats. First, the memory requirement for naive algorithms is already high. When this is used along with pipeline step parallelization, the memory requirement will be a lot higher. The second issue is in case of a system failure, the memory (hot storage) may lose the data and pipeline steps would have to be run again. COSAP circumvents the second issue by writing the file into disk in a parallel thread. This momentarily slows down the computational capability of the system as the default compression and serialization algorithms used, usually heavily relies on CPU. However, compared to the alternative this hold up is negligible.
The scatter–gather method utilized in the COSAP achieves significant performance gains even when run on slow disk (~ 800 MB/s) (Fig. 3). The speed increases by a factor of 2 when the COSAP DNA pipeline runs on a NVMe drive (~ 2.8 GB/s). Ramdisk performs the best in all parallelized tools with up to 8 × speed gain compared to default implementation of the tools. COSAP currently accommodates parallelized versions of Mutect2, HaplotypeCaller from GATK toolset as well as Varscan2 [20] and SomaticSniper [27]. Other available variant callers have built-in multithreading support. The performance benchmarks are performed on an Intel Xeon E5-2680 v4 chip with 128 GB of memory using WES data from Sequencing Quality Control 2 [48] datasets with accession numbers of SRR7890850 and SRR7890851.

Performance of parallelized versions of the tools on different disk speed settings in comparison with the baselines
Python API
Python is a widely used programming language that can tackle both high and low level problems at the cost of performance in most cases. Since many of the applications and algorithms popular in NGS pipeline are coded in higher performing languages, Python language can be used to wrap around these libraries and only used for an adapter layer for enabling research. COSAP’s Python API allows Keras-like networks of NGS pipeline steps to be created. Custom or predefined pipeline step classes can be connected to create these networks.
Comparing pipelines and benchmarking
COSAP’s comparison and benchmark module help users to analyze their pipelines deeper and finetune accordingly. Users may have different motivations to make comparison and benchmarking analyses such as ensuring the instrument is working as expected or targeted methods capture all of the variants in clinically relevant regions, as Olson et al. argues [49]. They also present an “Overview first, zoom and filter, details on demand” framework for variant visualizations. COSAP’s comparison module follows a similar path where it visualizes the overall pipeline intersection and similarity which gives general understanding. It also gives users an option to draw double and triple venn diagrams of the pipelines or tools of their choice. Figure 4 shows an example comparison of variant callers on previously described seqc2 WES data.

Upset plot depicts the intersection between variant sets of several variant callers, and variant allele frequency distribution of each intersection
Depending on availability of the baseline variant set, COSAP can calculate precision and recall values to assess performance of each pipeline Fig. 5d. This option allows users to pick the best performing combination depending on their sensitivity and specificity needs. When a genome stratification bed file is supplied, the comparison and benchmarking module creates two of each graph enabling the user to see the effect of filtering on intersection and precision/recall values.

a Double venn diagram of chosen variant callers. b Triple venn diagram of chosen variant callers. c Jaccard similarities of each variant caller. d Precision and recall plot when ground truth set is available
Lastly, the comparison module can create bed files for any intersection including TP, TN, FP and FN sets and load them into IGV along with vcf tracks for detailed inspection of variants which might be needed especially for indels and structural variants (Additional file 1: Fig. 1).
User interface
The user interface of COSAP is implemented as a web application using ReactJS and MaterialUI design components and hosted at cosap.bio/portal. Although it is suggested, users can discover the COSAP web application and utilize it without creating an account. It should be noted that the main purpose of cosap.bio is to demonstrate strengths of the application and when the demand is high there could be long waiting times for jobs to start. The interface provides an easy way of creating projects with the available tools and presents workflow results in an understandable way. With the help of backend services which are powered by Django-Python, users can inspect the results broadly and deeply, modify their results and save them for future analyses. The user interface is also packed with igvJS [50] to help users to visually inspect variants. The main page which displays available services and latest actions is depicted in Fig. 4. On this page, the user can select the analysis type and navigate through other pages.
After selecting the analysis type, the user is directed to the project creation page where the name of the project, required files and desired algorithms are submitted. An example somatic project creation page is shown in Fig. 5. This page slightly changes depending on the required files and algorithms by analysis types.
On the project listing page which is shown in Fig. 6, users can track status of projects and see their details. Navigation to the project creation page is also possible.

Main page of the web application where users choose the analysis they want to perform and see their recent activity
Once the project run is finished, the user can navigate to the project results page on Fig. 7. On this page, a summary of results which includes quality control statistics, number of variants and MSI score is shown on top of the page. All the SNVs, INDELs, SVs, CNVs are listed on the page which allows users to filter the variants based on all available annotations. An example filter is shown in Fig. 8 (Figs. 9 and 10).

Project creation interface to create projects with input files and desired algorithms

Interface to track status of projects and manage them

Results page where basic stats of the run is displayed alongside with the detailed variant descriptions and classifications

Variant filtering example
Conclusions
COSAP enables technical and non-technical scientists to sequence and analyze DNA by predefined pipelines and with an easy to use UI. Among many other pipeline and sequencing platforms COSAP stands out with availability of comparative analyses, ease of use via the web UI, Python API and, reproducibility via Docker containers and pipeline config files. These design choices and good software practices remove obstacles that come with a plethora of algorithms that are loosely meant to fit together. Automatic generation of command line arguments completely eliminates human errors such as overwriting existing results or running the same experiment twice.
One of COSAP's strengths lies in its software design. COSAP can be conveniently extended with future algorithms which means it will be very difficult for COSAP to be outdated. DNA sequencing requires a lot of computational power which is expensive and unaffordable for most research institutes. We have designed COSAP to be deployable to every system possible with Docker containers. The container can be configured to be run on a laptop or on a cluster of computers with a large pool of resources. Multi-container setup controlled by Celery allows multiple users to run sequences of jobs or a huge number of jobs to be queued and run automatically over an extended period of time. This can all be managed from a simple web app with very low resource requirements.
As the area of genomics is in its infancy, there are a lot of algorithms with eccentric configurations. COSAP handling the execution of all algorithms means each algorithm's parameters, logging and error handling needs to be done for each algorithm. These algorithms have a huge range of methods on these problems varying from logging every debug, info, warning and error to giving successful run results for failed runs. Some algorithms can even cause segmentation faults. As in its current state COSAP can’t handle these types of problems. Another technical difficulty is to configure a cluster of computers to run COSAP containers. There are many solutions for distributed systems and supporting all of them is an impossible task. Therefore, distributed systems and multi-container setups require technical know-how.
In the future, addressing the problems mentioned above is a priority. Even though logging and exception handling is a ceaseless hassle, it can be improved to make debugging more comfortable. Some examples regarding cloud setup aimed at helping configuring multi-container setups are also in the works. Along with examples of infrastructure-as-a-code will be provided in order to make cloud deployment as painless and quick as possible.
Availability and requirements
Project name: COSAP.
Project home page: https://github.com/MBaysanLab/cosap
Operating system(s): Linux or Other via Docker.
Programming language: Python, JavaScript.
Other requirements: GATK Grch38 bundle and any other database access.
License: MIT.
Any restrictions to use by non-academics: None.
Availability of data and materials
COSAP repository access: https://github.com/MBaysanLab/cosap, https://github.com/MBaysanLab/cosap-webapi, https://github.com/MBaysanLab/cosap_frontend
Dataset access: WES data from Sequencing Quality Control 2 datasets with accession numbers of SRR7890850 and SRR7890851. The paper of the dataset is https://doi.org/10.1038/s41587-021-00993-6.
Abbreviations
- COSAP:
-
Comparative Sequencing Analysis Platform
- NGS:
-
Next generation sequencing
- SNV:
-
Single nucleotide variant
- SV:
-
Structural variant
- CNV:
-
Copy number variation
References
Reuter JA, Spacek DV, Snyder MP. High-throughput sequencing technologies. Mol Cell. 2015;58(4):586–97.
Cortés-Ciriano I, Gulhan DC, Lee JJ, Melloni GE, Park PJ. Computational analysis of cancer genome sequencing data. Nat Rev Genet. 2022;23(5):298–314.
Anzar I, Sverchkova A, Stratford R, Clancy T. NeoMutate: an ensemble machine learning framework for the prediction of somatic mutations in cancer. BMC Med Genomics. 2019;12:1–4.
Kisakol B, Sarihan Ş, Ergün MA, Baysan M. Detailed evaluation of cancer sequencing pipelines in different microenvironments and heterogeneity levels. Turk J Biol. 2021;45(2):114–26.
Afgan E, Baker D, Batut B, Van Den Beek M, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46(W1):W537–44.
Terra.bio. https://terra.bio/ Accessed 10 Nov 2023.
Garcia M, Juhos S, Larsson M, Olason PI, Martin M, Eisfeldt J, DiLorenzo S, Sandgren J, De Ståhl TD, Wirta V, Sarek NM, A portable workflow for whole-genome sequencing analysis of germline and somatic variants. BioRxiv 2018; 316976.
Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35(4):316–9.
Iacoangeli A, Al Khleifat A, Sproviero W, Shatunov A, Jones AR, Morgan SL, Pittman A, Dobson RJ, Newhouse SJ, Al-Chalabi A. DNAscan: personal computer compatible NGS analysis, annotation and visualization. BMC Bioinform. 2019;20(1):1.
Cokelaer T, Desvillechabrol D, Legendre R, Cardon M. “Sequana”: a set of snakemake NGS pipelines. J Open Source Softw. 2017;2(16):352.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997. 2013.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Vasimuddin M, Misra S, Li H, Aluru S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. In: 2019 IEEE international parallel and distributed processing symposium (IPDPS). IEEE; 2019. pp. 314–324.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Del Angel G, Rivas MA, Hanna M, McKenna A. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Sahraeian SM, Liu R, Lau B, Podesta K, Mohiyuddin M, Lam HY. Deep convolutional neural networks for accurate somatic mutation detection. Nat Commun. 2019;10(1):1041.
Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, Shakir K. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv. 2017; 201178.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–76.
Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Kim Y, Beyter D, Krusche P, Saunders CT. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15(8):591–4.
Poplin R, Chang PC, Alexander D, Schwartz S, Colthurst T, Ku A, Newburger D, Dijamco J, Nguyen N, Afshar PT, Gross SS. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018;36(10):983–7.
Krishnamachari K, Lu D, Swift-Scott A, Yeraliyev A, Lee K, Huang W, Leng SN, Skanderup AJ. Accurate somatic variant detection using weakly supervised deep learning. Nat Commun. 2022;13(1):4248.
Fan Y, Xi L, Hughes DS, Zhang J, Zhang J, Futreal PA, Wheeler DA, Wang W. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016;17(1):1–1.
Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, Dry JR. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44(11):e108.
Cooke DP, Wedge DC, Lunter G. A unified haplotype-based method for accurate and comprehensive variant calling. Nat Biotechnol. 2021;39(7):885–92.
Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28(3):311–7.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–2.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17(1):1–4.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H, Muller J. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics. 2018;34(20):3572–4.
Gurbich TA, Ilinsky VV. ClassifyCNV: a tool for clinical annotation of copy-number variants. Sci Rep. 2020;10(1):20375.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ, Younes A, Nikiforova MN. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19(1):4–23.
Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100(2):267–80.
Li Q, Ren Z, Cao K, Li MM, Wang K, Zhou Y. CancerVar: An artificial intelligence–empowered platform for clinical interpretation of somatic mutations in cancer. Sci Adv. 2022;8(18):eabj1624.
de Bruijn I, Li X, Sumer SO, Gross B, Sheridan R, Ochoa A, Wilson M, Wang A, Zhang H, Lisman A, Abeshouse A. Genome nexus: a comprehensive resource for the annotation and interpretation of genomic variants in cancer. JCO Clin Cancer Inform. 2022;6:e2100144.
Köster J, Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28(19):2520–2.
Nvidia Genome Sequencing Analysis [Internet]. NVIDIA. [cited 2023Apr13]. Available from: https://www.nvidia.com/en-us/clara/genomics/
Goyal A, Kwon HJ, Lee K, Garg R, Yun SY, Kim YH, Lee S, Lee MS. Ultra-fast next generation human genome sequencing data processing using DRAGEN bio-IT processor for precision medicine. Open J Genet. 2017;7(1):9–19.
Olson ND, Wagner J, McDaniel J, Stephens SH, Westreich ST, Prasanna AG, Johanson E, Boja E, Maier EJ, Serang O, Jáspez D. PrecisionFDA truth challenge V2: calling variants from short and long reads in difficult-to-map regions. Cell Genomics. 2022;2(5):100129.
Zhao S, Agafonov O, Azab A, Stokowy T, Hovig E. Accuracy and efficiency of germline variant calling pipelines for human genome data. Sci Rep. 2020;10(1):1–2.
Franke KR, Crowgey EL. Accelerating next generation sequencing data analysis: an evaluation of optimized best practices for genome analysis toolkit algorithms. Genomics & informatics. 2020;18(1):e10.
Wang S, Yang W, Zhang X, Yu R. Performance evaluation of IMP: a rapid secondary analysis pipeline for NGS data. In: 2018 IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE; 2018. pp. 1170–1176.
Herzeel C, Costanza P, Decap D, Fostier J, Wuyts R, Verachtert W. Multithreaded variant calling in elPrep 5. PLOS ONE. 2021;16(2):e0244471.
Ahmad T, Ahmed N, Al-Ars Z, Hofstee HP. Optimizing performance of GATK workflows using apache arrow in-memory data framework. BMC Genomics. 2020;21(10):1–4.
Fang LT, Zhu B, Zhao Y, Chen W, Yang Z, Kerrigan L, Langenbach K, de Mars M, Lu C, Idler K, Jacob H. Establishing community reference samples, data and call sets for benchmarking cancer mutation detection using whole-genome sequencing. Nat Biotechnol. 2021;39(9):1151–60.
Olson ND, et al. Variant calling and benchmarking in an era of complete human genome sequences. Nat Rev Genet. 2023;24:464–83.
Robinson JT, Thorvaldsdóttir H, Turner D, Mesirov JP. igv. js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics. 2023;39(1):btac830.
Acknowledgements
This paper would not have been completed without the endless support of Batuhan Kisakol and Sahin Sarihan.
Funding
The project is partially funded by the Health Institutes of Turkey. (Project No: 3899). The computing resources utilized in this work were made available through the National Center for High Performance Computing of Turkey (UHeM), supported by Grant No. 1011152021.
Author information
Authors and Affiliations
Contributions
MAE and OC are the primary coauthors. They have been mutually involved in researching, programming and documenting. BB has been involved in the back-end of the user interface. AAE contributed to the pipeline comparison module. MB is the professor conceptualizing, leading and observing the research.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The additional file shows an example IGV view with comparison tracks are loaded along with vcf files.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Ergun, M.A., Cinal, O., Bakışlı, B. et al. COSAP: Comparative Sequencing Analysis Platform. BMC Bioinformatics 25, 130 (2024). https://doi.org/10.1186/s12859-024-05756-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-024-05756-z