
Abstract
Background
Next Generation Sequencing (NGS) is a commonly used technology for studying the genetic basis of biological processes and it underpins the aspirations of precision medicine. However, there are significant challenges when dealing with NGS data. Firstly, a huge number of bioinformatics tools for a wide range of uses exist, therefore it is challenging to design an analysis pipeline. Secondly, NGS analysis is computationally intensive, requiring expensive infrastructure, and many medical and research centres do not have adequate high performance computing facilities and cloud computing is not always an option due to privacy and ownership issues. Finally, the interpretation of the results is not trivial and most available pipelines lack the utilities to favour this crucial step.
Results
We have therefore developed a fast and efficient bioinformatics pipeline that allows for the analysis of DNA sequencing data, while requiring little computational effort and memory usage. DNAscan can analyse a whole exome sequencing sample in 1 h and a 40x whole genome sequencing sample in 13 h, on a midrange computer. The pipeline can look for single nucleotide variants, small indels, structural variants, repeat expansions and viral genetic material (or any other organism). Its results are annotated using a customisable variety of databases and are available for an on-the-fly visualisation with a local deployment of the gene.iobio platform. DNAscan is implemented in Python. Its code and documentation are available on GitHub: https://github.com/KHP-Informatics/DNAscan. Instructions for an easy and fast deployment with Docker and Singularity are also provided on GitHub.
Conclusions
DNAscan is an extremely fast and computationally efficient pipeline for analysis, visualization and interpretation of NGS data. It is designed to provide a powerful and easy-to-use tool for applications in biomedical research and diagnostic medicine, at minimal computational cost. Its comprehensive approach will maximise the potential audience of users, bringing such analyses within the reach of non-specialist laboratories, and those from centres with limited funding available.
Similar content being viewed by others


Background
The generation of whole genome sequencing (WGS), whole exome sequencing (WES) or targeted gene panels, is now standard practice in biomedical research. On a large scale, international sequencing consortia study the genetic landscape of thousands of individuals. On an individual scale, sequencing data are also used in diagnostic medicine and so called Precision Medicine [1, 2], with the aim to tailor medical treatments to patient genetics. There are several practical challenges when processing next generation sequencing (NGS) data. For example, WGS data for one sample produced on the Illumina Hiseq X, one of the most popular sequencers, is about 100 gigabytes, allowing for a high depth of sequencing (average 40x), when stored using lossless compressed formats such as fastq.gz. Such large files are not easy to handle for the average non-specialised scientist or lab, since they require sophisticated tools, bioinformatics skills and high performance computing clusters for analysis. While such facilities are available in specialist, well-resourced centres in wealthy countries, they are not readily accessible in other settings. Cloud computing provides a solution for the computing aspect of the challenge, but not the cost or the specialist skills needed. Furthermore, privacy requirements, ownership policies, and lack of an adequate internet infrastructure can make their use impractical.
A further significant issue is the large number of bioinformatics tools available for NGS analysis. Omictools [3], a web database where most available tools are listed and reviewed, lists over 7000 bioinformatics NGS tools, and new ones are frequently released. Among these more than 100 analysis pipelines are listed, most of which do not cover the whole data analysis, annotation and visualisation process and are computationally more intensive. For example, SpeedSeq [4] and GATK Best Practise Workflow [5] (GATK BPW) are two of the most popular. While these pipelines guarantee a very high genotyping quality, their use requires high-performance computing facilities and specialized expertise. What is needed therefore is a single pipeline, able to be deployed by someone without training in bioinformatics, and able to run on readily available computing equipment, easily accessible to non-specialist labs in any part of the world.
Here we describe DNAscan, an extremely fast, accurate and computationally light bioinformatics pipeline for the analysis, annotation and visualisation of DNA next generation (short-reads) sequencing data. DNAscan is designed to provide a powerful and easy-to-use tool for applications in biomedical research and diagnostic medicine, at minimal computational cost. The pipeline can analyse 40x WGS data in 13 h using 4 threads and 16 Gb RAM and WES data in 1 h using 4 threads and 10.5 Gb of RAM, and detect SNVs, small indels, structural variants, repeat expansions and viral genetic material (or that of any other microbe, e.g. bacteria and fungi). Results are annotated using a variety of databases and made available for a local deployment of the gene.iobio platform for an on-the-fly visualisation. Additionally, user-friendly quality control and results reports are generated.
Material and methods
Pipeline description
The DNAscan pipeline consists of four stages: Alignment, Analysis, Annotation and Report generation, and can be run in three modes: Fast, Normal and Intensive, according to user requirements (Fig. 1 and Table 1). These modes have been designed to optimize computational effort without compromising performance for the type of genetic variant the user is testing (see mode recommendations in Table 2). The user can restrict the analysis to any sub-region of the human genome by proving either a region file in bed format, a list of gene names, or using the whole-exome option, reducing the processing time and generating region specific reports.

Pipeline overview. Central panel: DNAscan accepts sequencing data, and optionally variant files. The pipeline firstly performs an alignment step (details in the left panel), followed by a customisable data analysis protocol (details in the right panel). Finally, results are annotated and user-friendly QC and result reports are generated. The annotation step uses Annovar to enrich the results with functional information from external databases. Right panel: detailed description of the post alignment analysis pipeline. Aligned reads are used by the variant calling pipeline (Freebayes + GATK HC); both aligned and unaligned reads are used by Manta and ExpensionHunter (for which repeat description files have to be provided) to look for structural variants. The unaligned reads are mapped to a database of known viral genomes (NCBI database) to screen for their DNA in the input sequencing data. Left panel: Alignment stage description. Raw reads are aligned with HISAT2. Resulting soft-clipped and unaligned reads are realigned with BWA mem and then merged with the others using Samtools
Alignment
DNAscan accepts sequencing data in fastq.gz and as a Sequence Alignment Map (SAM) file (and its compressed version BAM). HISAT2 and BWA mem [6, 7] are used to map the reads to the reference genome (Fig. 1, left panel). This step is skipped if the user provides data in SAM or BAM formats. HISAT2 is a fast and sensitive alignment program for mapping next-generation sequencing reads to a reference genome. HISAT2 uses a new reference indexing scheme called a Hierarchical Graph FM index (HGFM) [8], thanks to which it can guarantee a high performance, comparable to state-of-the-art tools, in approximately one quarter of the time of BWA and Bowtie2 [9] (see Additional file 1).
Variant calling pipelines based on HISAT2 generally perform poorly on indels [10]. To address this issue, DNAscan uses BWA to realign soft-clipped and unaligned reads. This alignment refinement step is skipped if DNAscan is run in Fast mode.
Samblaster [11] is used to mark duplicates during the alignment step and Sambamba [12] to sort the aligned reads. Both the variant callers, Freebayes [13] and GATK Haplotype Caller (HC) [5] used in the following step, are duplicate-aware, meaning that they automatically ignore reads marked as duplicate. The user can optionally exclude it from the workflow according to the study design, e.g. when an intensive Polymerase Chain Reaction (PCR) amplification of small regions is required.
Analysis
Various analyses are performed on the mapped sequencing data (Fig. 1, right panel): SNV and small indel calling is performed using Freebayes, whose reliability is well reported [14, 15]. However, taking advantage of the documented better performance of GATK HC in small indel calling, we decided to add a customised indel calling step to DNAscan, called Intensive mode. This step firstly extracts the genome positions for which an insertion or a deletion is present on the cigar of at least one read, and secondly calls indels using GATK HC on these selected positions. The reduced number of positions where this occurs allows for a targeted use of GATK HC, limiting the required computational effort and time. The resulting SNVs and small indel calls with genotype quality smaller then 20 and depth smaller than 10 are discarded. The user can customize these filters according to their needs (see GitHub [16] for details and a complete list of available filters).
Two Illumina developed tools, Manta [17] and Expansion Hunter [18] are used for detecting medium and large structural variants (> 50 bp) including insertions, deletions, translocations, duplications and known repeat expansions. These tools are optimised for high speed and can analyse a 40x WGS sample in about one hour using 4 threads, maintaining a very high performance.
DNAscan also has options to scan the sequencing data for microbial genetic material. It performs a computational subtraction of human host sequences to identify sequences of infectious agents including viruses, bacteria or fungi, by aligning the non-human or unaligned reads to the whole NCBI database [19,20,21] of known viral, bacterial or any custom set of microbial genomes and reporting the number of reads aligned to each non-human genome, its length and the number of bases covered by at least one read.
Annotation
Variant calls are then annotated using Annovar [22]. The annotation includes the use of databases such as ClinVar [23], Exac [24], dbSNP [25] and dbNSFP [26] (more information about how to customise the annotation, e.g. by selecting alternative databases and/or focusing on specific genome regions, are available on GitHub).
Reports and visualization utilities
DNAscan produces a wide set of quality control (QC) and result reports and provides utilities for visualisation and interpretation of the results.
MultiQC [27] is used to wrap up and visualise QC results. FastQC [28], Samtools [29] and Bcftools [30] are used to perform QC on the sequencing data, its alignment and the called variants. An example is available on GitHub [31]. A tab delimited file including all variants found within the selected region is also generated [32]. This report would include all annotations performed by Annovar [22] in a format that is easy to handle with any Excel-like software by users of all levels of expertise.
Three iobio services (bam.iobio, vcf.iobio and gene.iobio) are locally provided with the pipeline allowing for the visualisation of the alignment file [33], the called variants [34] and for a gene based visualisation and interpretation of the results [35].
DNAscan benchmark
Benchmarking every DNAscan component is not needed since a range of literature is available [14, 15, 17, 36, 37]. However, to our knowledge, none exists assessing HISAT2 [8] (the short-read mapper used by the pipeline) either for DNA read mapping or as part of DNA variant calling pipelines. In this manuscript, we both assess the performance of HISAT2 with BWA and Bowtie2 [9] mapping 1.25 billion WGS reads sequenced with the Illumina Hiseq X and 150 million simulated reads (see Additional file 1), and compare our SNV/indel calling pipeline in Fast, Normal and Intensive modes with the GATK BPW [5] and SpeedSeq [4] over the whole exome sequencing of NA12878. Illumina platinum calls are used as true positives [38].
We also show how DNAscan represents a powerful tool for medical and scientific use by analysing real DNA sequence data from two patients affected by Amyotrophic Lateral Sclerosis (ALS) and of HIV infected human cells. For the ALS patients we use both a gene panel of 10 ALS-related genes, whose feasibility for diagnostic medicine has been previously investigated [2], sequenced with the Illumina Miseq platform, and the WGS data from the Project MinE sequencing dataset [39]. DNAscan was used to look for SNVs, small indels, structural variants, and known repeat expansions. The WGS of an HIV infected human cell sample [40] was used to test DNAscan for virus detection.
Variant calling assessment
To assess the performance of DNAscan in calling SNVs and indels, we used the Illumina Genome Analyzer II whole exome sequencing of NA12878. Illumina platinum calls [38] were used as true positives.
GATK BPW calls were generated using default parameters and following the indications on the GATK website [41] for germline SNVs and indels calling. These include the pre-processing and variant discovery steps for single sample, i.e. skipping the Merge and Join Genotype steps.
SpeedSeq calls were generated running the “align” and “var” commands as described on GitHub [42]. RTG Tools [43] (“vcfeval” command) was used to evaluate the calls. F-measure, Precision and Sensitivity are defined as in the following: \( Precision=\frac{T_p\ }{T_p+{F}_p} \), \( Sensitivity=\frac{T_p\ }{T_p+{F}_n} \) and \( F- measure=2\times \frac{Precision\times Sensitivity}{Precision+ Sensitivity} \), where Tp is true positives, Fp false positives and Fn false negatives.
ALS Miseq and whole genome sequencing test cases
Using DNAscan in Fast mode, we analysed real DNA sequence data from two ALS patients (case A and case B). Case A carries a non-synonymous mutation in the FUS gene [44] (variant C1561T, amino acid change R521C, variant dbSNP id rs121909670 [45]) known to be a cause of ALS (ClinVar id RCV000017611.25). A panel of 10 ALS related genes was sequenced with the Illumina Miseq platform for case A. The Miseq gene panel was designed and tested for diagnostic purposes [2]. For these 10 genes (BSCL2, CEP112, FUS, MATR3, OPTN, SOD1, SPG11, TARDBP, UBQLN2, and VCP), the full exon set was sequenced, generating over 825,000,222-base-long paired reads. DNAscan was used to call SNVs, indels, and structural variants on case A.
Case B has a confirmed C9orf72 expansion mutation on one allele, also known to be causative of ALS [46]. This expansion mutation is thousands of repeats long. and 40x WGS data was generated with the Illumina Hiseq X for case B. The WGS sample (paired reads, read length = 150, average coverage depth = 40) was sequenced as part of the Project MinE sequencing dataset [39]. For this sample we ran DNAscan on the whole genome. However, both for practical reasons and to simulate a specific medical diagnostic interest, we focused our analysis report on the 126 ALS related genes reported on the ALSoD webserver [47] and also looked for the C9orf72 repeat.
For both samples, we also reported variants linked to frontotemporal dementia, which is a neurodegenerative disease that causes neuronal loss, predominantly involving the frontal or temporal lobes, with a genetic and clinical overlap with ALS [48, 49].
C9orf72 repeat primed PCR
Pathological C9orf72 gene hexanucleotide repeat expansions were determined using repeat primed PCR (RP-PCR), as previously described [50].
Hardware
SpeedSeq was run on a single machine with 64 Gb of RAM and an Intel i7–670 processor. The other tests were performed using a machine with 16 Gb of RAM and an Intel i7–670 processor.
Results
Single nucleotide variant and small indels calling assessment
To assess the performance of the DNAscan variant calling pipeline with GATK BPW and SpeedSeq, we used the exome of the well-studied NA12878 sample and the Illumina platinum calls as a gold standard (our set of true calls). The SpeedSeq pipeline uses BWA for alignment, Sambamba [12] and Samblaster [11] to sort reads and to remove duplicates, and Freebayes [13] for variant calling. Considering the overlap in the software used by DNAscan and SpeedSeq, assessing their performance is therefore of interest. Figure 2a shows the results from this test. DNAscan in Fast mode performs comparably with both the GATK BPW and the SpeedSeq on SNVs. Their F-measures (Fm), a harmonic mean of precision and sensitivity defined in the Methods, are 0.92 (DNAScan), 0.91 (GATK BPW) and 0.93 (SpeedSeq).

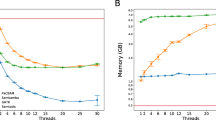
Variant calling assessment. Graph a shows the precision, sensitivity and F-measure of DNAscan in Fast, Normal and Intensive mode, SpeedSeq and GATK best practice workflow in calling SNVs and small indels over the whole exome sequencing of NA12878. Illumina platinum calls were used as true positives. The first three columns show the results for SNVs and the last three columns for indels. Graph b shows the time needed and the memory footprint for the same pipelines
In Normal mode DNAscan (Fm = 0.61) reaches an indel calling precision and sensitivity comparable to SpeedSeq (Fm = 0.62). The better performance of the Normal mode is driven by a major increase in sensitivity to 0.73 from 0.60. However, GATK BPW outperforms SpeedSeq on indels (GATK BPW Fm = 0.81). DNAscan, in Intensive mode, performs comparably to GATK BPW also on indels with an Fm of 0.82.
Figure 2b shows a comparison of the time needed by the tested pipelines and their memory usage. DNAscan in Fast mode completes the analysis in just 63 min while SpeedSeq takes over twice the time (132 min) and GATK BPW 5 times longer (310 min). DNAscan in both Normal and Intensive mode completes the analysis in a reasonable time (Normal 77 min, Intensive 98 min). DNAscan uses as little as 10.5 Gb RAM in Fast mode, and 12.9 Gb in Normal and Intensive mode, while GATK BPW uses 15 Gb and SpeedSeq over 25 Gb.
Screening of ALS patients
For Case A, using the Miseq DNA gene panel, DNAscan detected 13 SNVs reported to be related to ALS and 4 to frontotemporal dementia on ClinVar, 6 non-synonymous variants and 6 variants with a deleteriousness CADD phred score [51] equal to or higher than 13, meaning that they are predicted to be in the top 5% most deleterious substitutions (Table 3). The known pathogenic FUS SNV rs121909670 was detected. No structural variants were found. The whole analysis was performed in ~ 30 min using 4 threads.
On the WGS data of Case B, for the selected 126 genes, DNAscan identified 33 SNVs reported to be related to ALS and 3 to frontotemporal dementia on ClinVar, 64 non-synonymous variants, 748 variants with a deleteriousness CADD phred score equal to or higher than 13, one 60-base-pair insertion, 3 over 100,000-base-pair long deletions and 1 tandem duplication. DNAscan was also able to detect the known C9orf72 expansion (Table 3). The whole analysis was performed in ~ 8 h using 4 threads.
Virus scanning
We used DNAscan to detect the presence of viral genetic material in a whole genome sequencing sample of HIV infected human cells. The DNA sequencing data was produced using the Illumina Hiseq 2000 sequencer generating about 350 million 95-base length paired reads. Following the well-established approach of computational subtraction of human host sequences to identify sequences of infectious agents like viruses [52], the human reads (91%, Fig. 3a) were subtracted by mapping the sequencing data to the reference human genome using HISAT2. To screen our sequencing sample for the presence of known viral DNA, HISAT2 was then used to map the unmapped reads from the initial mapping phase of the pipeline (9%, Fig. 3a) to all the viral genomes available on the NCBI virus database.

Identification of non-human reads Panel a shows the proportion of human reads (blue), viral reads (red) and unknown reads (yellow). Panel b shows the proportion for viral reads belonging to HIV (blue), PhiX174 (red) and to other viruses (yellow). Human reads are defined as reads which aligned to the human reference genome, viral reads as the reads which did not align to the human reference genome but aligned to at least one of the NCBI viral genomes and unknown reads as the reads which did not align either to the human or to any viral reference genomes. In panel c we plotted the numbers of aligned reads in logarithmic scale, for the 20 non-human microbe genomes to which the highest number of reads was aligned
Figure 3c shows a logarithmic representation of the number of reads aligned to the viral genomes in descending order for the 20 viral genomes to which the highest number of reads were mapped. They show both the presence of HIV DNA and bacterial DNA in our sample. Indeed, 4,412,255 reads mapped to the human immunodeficiency virus (NCBI id NC_001802.1) and only the Escherichia virus phiX174 (NCBI id NC_001422.1), a bacterial virus, presented a comparable number of reads (4,834,017 reads). This phage sequence is commonly found in Illumina sequencing protocols [53] probably because of transfer from gut microbes into blood.
Also, a smaller (3–4 orders of magnitude) number of reads belonging to other viruses were found. The disproportion between the presence of the first two hits (phiX174 and HIV) and the rest of the viruses is also shown in Fig. 3b. The complete results with the list of the whole set of viruses (120 viruses) for which at least one read was aligned can be found on GitHub [54]. The whole screening was performed by DNAscan using 4 threads in 2 h.
Discussion and conclusion
DNAscan is an extremely fast, computationally efficient, easy to use pipeline for analysis, annotation and visualisation of next generation DNA sequencing data. It uses fast, but suboptimal tools to carry out first-line analysis, and optimal, but slower tools to refine the results. As a result, DNAscan is faster but not resource hungry, for example it is able to analyse 40x WGS data in 13 h and whole exome sequence data in one hour on a mid-range computer, performing as well as the widely used GATK BPW in terms of variant calling precision and sensitivity. Three different running modes, Fast, Normal and Intensive allow the pipeline to be tailored to specific needs while reducing time and RAM requirements compared to current standards, GATK BPW and SpeedSeq. It includes utilities for user friendly visualisation and interpretation of output. It is able to identify SNVs, structural variants, indels and expansion mutations, and favours use by non-specialists, and those with limited access to high performance computing facilities, for example, in less well-resourced countries or laboratories.
This comprehensive analysis approach aims to maximise the potential audience of users. However, NGS data can be used to investigate a very wide range of genetic variations which are impossible to enclose in only one analysis pipeline. DNAscan does not provide specific tools and protocols to detect whole classes of mutations, for example microsatellites, retrotransposons, novel or highly irregular repeat expansions and somatic variants. Moreover, it does not offer the adequate flexibility to allow different analysis approaches, for example consensus and meta variant calling that have been shown to be powerful strategies to detect SNVs and structural variants [55, 56] , or to analyse long-reads sequencing data such as PacBio and Nanopore [57, 58]. New implementations of DNAscan are already underway and will include new analysis protocols, including the detection of structural variants using long-read sequencing, the development of a webserver for an on-the-fly and a graphical user interface. However, the use of highly flexible and interactive analysis frameworks such as Seven-Bridges (www.sevenbridges.com), Galaxy [59], or ExScalibur [60] will remain a necessity for those users who need a higher degree of flexibility.
We also reported a few specific-use cases, such as the analysis of Miseq and WGS data of someone with ALS for diagnostic purposes, and the virus screening of HIV infected human cells. In the ALS test we showed how with MiSeq and Hiseq X WGS data, DNAscan detected a range of reported ALS-related variants in half an hour for the Miseq panel and 8 h for the WGS data (restricting the analysis to the 126 ALS genes), correctly reporting the presence of both the C9orf72 expansion and the rs121909670 SNV. In the HIV test, DNAscan detected the expected viral presence by finding both the HIV virus and a phage commonly present in Illumina next generation DNA sequencing data.
Cloud computing and storage services offer practically unlimited computational power and storage. However, this has a cost, and optimisation, in particular for large scale sequencing projects, is of primary importance. Amazon Web Services (AWS) is one of the most popular cloud computing services. Performing the alignment, variant calling and annotation using DNAscan Fast mode on an EC2 instance [61] would cost about $2.41 (13 h of usage of a t2.xlarge machine with 4 CPUs). The same analysis using SpeedSeq would cost about $18.72 (10 h of usage of a h1.8xlarge machine with 32 CPUs). These prices do not take into account the storage, were updated on the 29th of April 2019 and take into consideration the cheapest machines available in the US East (Ohio) region matching the pipeline computational requirements proposed by the authors (4 CPUs and 16 Gb RAM for DNAscan and 32 CPUs and 128 Gb RAM for SpeedSeq [4]).
DNAscan is also available as a Docker and a Singularity image. These allow the user to quickly and reliably deploy the pipeline on any machine where either of these programmes is available. Singularity also allows for the deployment of the pipeline on environments for which the user does not have root permission. This could be particularly useful for users working on shared high performance computing facilities.
Availability and requirements
Project name: DNAscan
Project home page: https://github.com/KHP-Informatics/DNAscan
Operating system(s): GNU/Linux based systems
Programming language: Python
Other requirements: https://github.com/KHP-Informatics/DNAscan#dependencies
License: MIT (https://github.com/KHP-Informatics/DNAscan/blob/master/LICENSE.txt)
Any restrictions to use by non-academics: no restrictions
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- FTD:
-
Frontotemporal dementia
- GATK BPW:
-
Genome analysis tool kit best practice workflow
- GATK:
-
Genome analysis tool kit
- HC:
-
Haplotype caller
- HPC:
-
High perfomance computing
- INDEL:
-
Insertion and deletion
- NGS:
-
Next generation sequencing
- PCR:
-
Polymerase chain reaction
- QC:
-
Quality control
- SNV:
-
Single nucleotide variant
- SV:
-
Structural variant
- WES:
-
Whole genome sequencing
- WGS:
-
Whole genome sequencing
References
Dong L, Wang W, Li A, Kansal R, Chen Y, Chen H, et al. Clinical next generation sequencing for precision medicine in Cancer. Curr Genomics. 2015;16(4):253–63.
Morgan S, Shoai M, Fratta P, Sidle K, Orrell R, Sweeney MG, et al. Investigation of next-generation sequencing technologies as a diagnostic tool for amyotrophic lateral sclerosis. Neurobiol Aging. 2015;36(3):1600. e5–8.
Henry VJ, Bandrowski AE, Pepin AS, Gonzalez BJ, Desfeux A. OMICtools: an informative directory for multi-omic data analysis. Database (Oxford). 2014;2014.
Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015;12(10):966–8.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Li H, Durbin R. Fast and accurate short read alignment with burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv e-prints [Internet]. 2013 March 1, 2013; 1303. Available from: http://adsabs.harvard.edu/abs/2013arXiv1303.3997L.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–9.
Sun Z, Bhagwate A, Prodduturi N, Yang P, Kocher JA. Indel detection from RNA-seq data: tool evaluation and strategies for accurate detection of actionable mutations. Brief Bioinform. 2016.
Faust GG, Hall IM. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014;30(17):2503–5.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–4.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. ArXiv e-prints [Internet]. 2012 July 1, 2012; 1207. Available from: http://adsabs.harvard.edu/abs/2012arXiv1207.3907G.
Sandmann S, de Graaf AO, Karimi M, van der Reijden BA, Hellstrom-Lindberg E, Jansen JH, et al. Evaluating variant calling tools for non-matched next-generation sequencing data. Sci Rep. 2017;7:43169.
Smith HE, Yun S. Evaluating alignment and variant-calling software for mutation identification in C. elegans by whole-genome sequencing. PLoS One. 2017;12(3):e0174446.
Dabbish L, Stuart C, Tsay J, Herbsleb J, editors. Social coding in GitHub: transparency and collaboration in an open software repository. Proceedings of the ACM 2012 conference on Computer Supported Cooperative Work; 2012: ACM.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–2.
Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. bioRxiv. 2017.
Brister JR, Ako-Adjei D, Bao Y, Blinkova O. NCBI viral genomes resource. Nucleic Acids Res. 2015;43(Database issue):D571–7.
Wheeler DL, Church DM, Lash AE, Leipe DD, Madden TL, Pontius JU, et al. Database resources of the National Center for biotechnology information. Nucleic Acids Res. 2001;29(1):11–6.
Coordinators NR. Database resources of the National Center for biotechnology information. Nucleic Acids Res. 2017;45(D1):D12–D7.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8.
Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45(D1):D840–D5.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32(8):894–9.
Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32(19):3047–8.
FastQC website [Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, Durbin R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics. 2016;32(11):1749–51.
QC sample report [Available from: https://goo.gl/MAjpqS.
Variant sample report [Available from: https://goo.gl/R8m5Rv.
Miller CA, Qiao Y, DiSera T, D'Astous B, Marth GT. Bam.Iobio: a web-based, real-time, sequence alignment file inspector. Nat Methods. 2014;11(12):1189.
Vcf.iobio platform [Available from: http://vcf.iobio.io.
Gene.iobio platform [Available from: http://gene.iobio.io.
Dolzhenko E, van Vugt J, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017;27(11):1895–903.
Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA, Grant GR. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat Methods. 2017;14(2):135–9.
Eberle MA, Fritzilas E, Krusche P, Kallberg M, Moore BL, Bekritsky MA, et al. A reference dataset of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. bioRxiv. 2016.
Project Min EALSSC. Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur J Hum Genet. 2018.
De Iaco A, Santoni F, Vannier A, Guipponi M, Antonarakis S, Luban J. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology. 2013;10:20.
GATK website [Available from: https://software.broadinstitute.org/gatk/.
Chiang C. SpeedSeq github repository [Available from: https://github.com/hall-lab/speedseq.
CJ G, Ross B, Kurt G, HB S, Stuart I, IS A, et al. Joint variant and De novo mutation identification on pedigrees from high-throughput sequencing data. J Comput Biol. 2014;21(6):405–19.
Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7(11):603–15.
Morgan S, Shatunov A, Sproviero W, Jones AR, Shoai M, Hughes D, et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain. 2017;140(6):1611–8.
Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, et al. The C9ORF72 expansion mutation is a common cause of ALS+/−FTD in Europe and has a single founder. Eur J Hum Genet. 2013;21(1):102–8.
Abel O, Powell JF, Andersen PM, Al-Chalabi A. ALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat. 2012;33(9):1345–51.
Synofzik M, Otto M, Ludolph A, Weishaupt JH. Genetic architecture of amyotrophic lateral sclerosis and frontotemporal dementia : overlap and differences. Nervenarzt. 2017;88(7):728–35.
Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59(7):1077–9.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of.human genetic variants. Nat Genet. 2014;46(3):310–5.
Daly GM, Leggett RM, Rowe W, Stubbs S, Wilkinson M, Ramirez-Gonzalez RH, et al. Host subtraction, filtering and assembly validations for novel viral discovery using next generation sequencing data. PLoS One. 2015;10(6).
Mukherjee S, Huntemann M, Ivanova N, Kyrpides NC, Pati A. Large-scale contamination of microbial isolate genomes by Illumina PhiX control. Stand Genomic Sci. 2015;10:18.
Iacoangeli A. DNAscan virus analysis report example [Available from: https://goo.gl/QiaYRo.
Hehir-Kwa JY, Marschall T, Kloosterman WP, Francioli LC, Baaijens JA, Dijkstra LJ, et al. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat Commun. 2016;7:12989.
Gezsi A, Bolgar B, Marx P, Sarkozy P, Szalai C, Antal P. VariantMetaCaller: automated fusion of variant calling pipelines for quantitative, precision-based filtering. BMC Genomics. 2015;16:875.
Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics. 2015;13(5):278–89.
Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, et al. Nanopore sequencing and assembly of a human genome with ultralong reads. Nat Biotechnol. 2018;36(4):338.
Afgan E, Baker D, Batut B, Van Den Beek M, Bouvier D, Čech M, et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46(W1):W537–W44.
Bao R, Hernandez K, Huang L, Kang W, Bartom E, Onel K, et al. ExScalibur: a high-performance cloud-enabled suite for whole exome germline and somatic mutation identification. PLoS One. 2015;10(8):e0135800.
EC2 A. AMAzon EC2 pricing website [Available from: https://aws.amazon.com/ec2/pricing/on-demand/.
Merkel D. Docker: lightweight linux containers for consistent development and deployment. Linux Journal. 2014;2014(239):2.
Kurtzer GM, Sochat V, Bauer MW. Singularity: scientific containers for mobility of compute. PLoS One. 2017;12(5):e0177459.
NCBI ftp server NA12878 [Available from: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/working/20101201_cg_NA12878/NA12878.ga2.exome.maq.raw.bam.
Illumina Platinum Calls ftp server [Available from: ftp://platgene_ro@ussd-ftp.illumina.com.
Kodama Y, Shumway M, Leinonen R. International nucleotide sequence database C. the sequence read archive: explosive growth of sequencing data. Nucleic Acids Res. 2012;40(Database issue):D54–6.
NCBI ftp server [Available from: ftp.ncbi.nlm.nih.gov/refseq/.
Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35(Database):D61–5.
Acknowledgements
We would like to thank people with MND/ALS and their families for their participation in this project.
Funding
This is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organisations under the aegis of JPND - www.jpnd.eu (United Kingdom, Medical Research Council (MR/L501529/1; MR/R024804/1) and Economic and Social Research Council (ES/L008238/1)) and through the Motor Neurone Disease Association. This study represents independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The work leading up to this publication was funded by the European Community’s Horizon 2020 Programme (H2020-PHC-2014-two-stage; grant agreement number 633413). Sequence data used in this research were in part obtained from the UK National DNA Bank for MND Research, funded by the MND Association and the Wellcome Trust. All funding bodies listed above did not play any role in the study or conclusions of this study.
Availability of data and materials
DNAscan is available on GitHub: https://github.com/KHP-Informatics/DNAscan. Docker [62] and Singularity [63] images are also available for fast deployment and reproducibility (see instructions on GitHub).
The whole exome sequencing of NA12878 can be downloaded from the NCBI ftp server [64]. The Illumina Platinum calls can be downloaded from the Illumina ftp site [65].
The WGS of HIV infected human cells used to test DNAscan, was generated as part of another study [40] and the complete high throughput sequencing dataset was downloaded from the Sequence Read Archive (SRA) [66] under accession number SRA056122.
DNAscan was also tested on the whole non-redundant NCBI database of complete viral genomes (9,334 genomes). These can be downloaded from the NCBI database ftp server [19, 67]. DNAscan can also be used to screen for the DNA of other organisms including bacteria or fungi by downloading the preferred database from the NCBI ftp [67, 68].
Author information
Authors and Affiliations
Contributions
AI designed and developed the method, performed the computational experiments and was the major contributor in writing the manuscript. AAC, SJN and RJD contributed in writing the manuscript. All authors read, revised and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Alignment assessment results. HISAT2, BWA and Bowtie2 were tested on 150 million simulated Illumina paired end human reads and 1.250 billion real Illumina paired end human reads. For the three aligners on the two dataset the table shows the time taken, their memory fingerprint and the percentage of aligned-one-or-more-times reads, aligned-only-once reads and properly pared. All tests were run using 4 threads. (DOCX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Iacoangeli, A., Al Khleifat, A., Sproviero, W. et al. DNAscan: personal computer compatible NGS analysis, annotation and visualisation. BMC Bioinformatics 20, 213 (2019). https://doi.org/10.1186/s12859-019-2791-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-019-2791-8