
Abstract
Background
Recent comparative studies have brought to our attention how somatic mutation detection from next-generation sequencing data is still an open issue in bioinformatics, because different pipelines result in a low consensus. In this context, it is suggested to integrate results from multiple calling tools, but this operation is not trivial and the burden of merging, comparing, filtering and explaining the results demands appropriate software.
Results
We developed isma (integrative somatic mutation analysis), an R package for the integrative analysis of somatic mutations detected by multiple pipelines for matched tumor-normal samples. The package provides a series of functions to quantify the consensus, estimate the variability, underline outliers, integrate evidences from publicly available mutation catalogues and filter sites. We illustrate the capabilities of isma analysing breast cancer somatic mutations generated by The Cancer Genome Atlas (TCGA) using four pipelines.
Conclusions
Comparing different “points of view” on the same data, isma generates a unique mutation catalogue and a series of reports that underline common patterns, variability, as well as sites already catalogued by other studies (e.g. TCGA), so as to design and apply filtering strategies to screen more reliable sites. The package is available for non-commercial users at the URL https://www.itb.cnr.it/isma.
Similar content being viewed by others


Background
The identification of somatic mutations from Next Generation sequencing (NGS) data is a challenging task. Several studies compared the single nucleotide variations (SNVs) [1,2,3] and insertions/deletions (INDELs) [4, 5] detected by different computational tools and underlined relevant discrepancies. Therefore, it is recommended to analyse the same NGS data using multiple callers, like Mutect [6], SomaticSniper [7] and Varscan [8], which generate lists of mutations encoded in Variant Call Format (VCF) [9]. This way of facing conflicting predictions demands appropriate tools that harmonize different outputs and enable comparative analyses [4]. Indeed, for instance, mutation callers encode the same information in multiple ways (Table 1) and generate outputs with relevant qualitative (e.g. germline/somatic/loss-of-heterozygousity, SNVs/INDELs) and quantitative (number of sites found) differences. More generally if, in principle, the use of multiple callers is expected to reduce false positive findings, in practice, the resulting large and heterogeneous lists of mutation sites increase the complexity of the subsequent interpretations. Existing tools like myVCF [10], NGS-pipe [11], VariantTools [12], vcfR [13] and VCFTools [9], implement functions and pipelines to work with VCF files, but do not specifically address the problem of integrating and comparing the results of different mutation callers. A few tools exist to address this problem: Cake [14] (a bioinformatics pipeline implemented in perl) offers the opportunity to run multiple callers and applies customizable filtering steps to obtain a final unique list of single nucleotide variations (SNVs); BAYSIC [15] (implemented in perl) provides a bayesian method for combining SNVs from different variant calling programs.
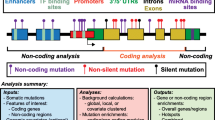
Here, we describe isma (integrative somatic mutation analysis), an R package that provides functions for the joint analysis of VCF files generated by somatic mutation callers from NGS data (Fig. 1). Differently from existing tools, beyond site integration and filtering, isma provides functions for a more in-depth analysis of mutation sites occurrence across subjects and tools, considering both SNVs and INDELs. The results generated by isma underline common patterns (e.g. recurrent calls, tool consensus in each subject), specificities (e.g. outlier samples, pipeline specific sites, genes enriched in calls from a single pipeline), as well as sites already catalogued by other studies (e.g. The Cancer Genome Atlas (TCGA) [16]), so as to design and apply filtering strategies to screen more reliable sites.

Overview of isma. Integrative analysis of somatic mutations detected by multiple pipelines
Implementation
The software isma is implemented in R. The package takes in input mutation sites encoded in VCF files or tab-delimited text files. isma extracts mutation site information from the output of multiple mutation callers by means of specific parsers and integrates sites into a unique data structure:
-
mut_sites <− pre_process (“config.txt”)
Most of the analyses can be easily carried out through a few wrapper functions, like site_analysis and gene_analysis for site- and gene-level analyses respectively. Nevertheless, many routines are available as part of the user interface to carry out custom analyses (Table 2). Gene-level analyses require mutation site annotation, for which isma relies on the R package VariantAnnotation [17] or, alternatively, on user-provided files. Computationally demanding analyses (e.g. the comparison among all-pairs of hundreds of subjects) are implemented in parallel, using the support provided by the R package parallel. The package isma contains a tutorial available as R vignettes:
-
vignette(“isma”)
Results
In this section, we will describe isma considering breast cancer (BC) mutations from TCGA, collected using the function get_TCGA_sites. In particular, we considered mutation profiles of 975 subjects detected by four variant callers: Mutect2, Varscan2, Muse and SomaticSniper (Additional file 1).
-
mut_sites <- get_TCGA_sites (tools = c("muse", "mutect2", "varscan2", "somaticsniper"), n_subjects = 975)
Note that these sites were already filtered by TCGA and are therefore less noisy than the corresponding initial variant caller outputs that would constitute the input of isma in a typical use scenario. Nevertheless, the exploratory analyses made possible by isma underlined interesting patterns even among such filtered calls from TCGA.
The analyses presented below can be easily run by means of site_analysis and gene_analysis wrapper functions and include quantification of site/gene occurrence across callers and subject, consensus among tools, detection of outlier subjects and tools, variation of detected sites at different cut-offs on alignment results (e.g. read depth) and integration of information from TCGA.
Site occurrence across callers and subjects
The co-occurrence of sites across tools and subjects is quantified by get_sites_statistics. This operation allows the user to quantify the fraction of tool-specific calls, the distribution of the sites across tools in each subject and tool consensus on sites. These results are used to detect and mark outlier features (subjects and tools), defined by the inter-quartile range (Tukey’s fences) (Table 3). The amount of shared sites between each pair of callers and subjects is calculated and organized, respectively, in callers-by-callers and subjects-by-subjects site co-occurrence matrices by the functions overlap_Tools and overlap_Subjects. Site co-occurrence matrices are used to summarize consensus and dispersion. Caller consensus relative to a subject is quantified by means of the consensus score (CS), defined as the sum of ratios between the amount of co-occurring sites (off-diagonal elements of the tools-by-tools site co-occurrence matrix) and tool-specific calls (diagonal elements) normalized by the total number of possible tool pairs:
where n is the number of tools, xi,j are the sites shared between tools i and j, and P(n, 2) is the number of permutations of tools in pairs.
The results of these analyses are summarized into consensus plots, co-occurrence matrices plot and a series of text files, like the summary table of outlier subjects. The overall consensus plot (Fig. 2) reports the total number of sites found by each tool and the fraction of calls shared among tools. Note how mutect2 found the highest number of sites, the 50% of which was not reported by other callers (Fig. 2). The consensus plot per subject shows the total number of unique sites, the fraction of sites found by each tool, the distribution of the consensus across subjects and the CS (Fig. 3). Note the presence of a few hypermutated subjects (i.e. A1XQ, A0U0, A08H, A1J5, A1NC and A25A) (Fig. 3a). Several subjects display an imbalance of calls among the pipelines (Fig. 3b). Further, there are subjects with a relevant (e.g. A1J5 and A0XR) or poor (e.g. AIKO and A0JC) proportion of sites supported by more than one caller (Fig. 3c). Lastly, note how CS underlines, by means of a unique score, subjects with issues in tool consensus, including imbalances in the number of sites or consensus among tools (Fig. 3d and Table 3).

Global consensus plot. Overall consensus among pipelines; results obtained on BC mutations detected by TCGA in 975 subjects

Detailed consensus plot. a Number of mutation sites. b Fraction of sites called by different pipelines. c Tool Consensus across subjects. d Consensus score (CS). a-d Asterisks indicate outliers. Results shown only for 50 subjects (out of 975), selected to include different types of outliers as well as samples without abnormal behaviours (Additional file 1)
Site co-occurrence between callers revealed that mutect2 detected up to 3 times more sites than other tools, while muse and varscan shared approximately the 60% of their sites (Fig. 4a). The mutation co-occurrence in each pair of subjects underlines similarities between mutation profiles; this information is completed with an estimation of the variability (coefficient of variation) of such co-occurrences due to the use of different callers (Fig. 4b). The package provides the possibility of calculating, for every gene, the fraction of subjects with at least one mutation, i.e. the gene mutation frequency across subjects (f), and its dispersion across callers. The corresponding plot, obtained on BC TCGA sites, underlined the presence of some genes, including known BC genes as GATA3 and CDH1, with a particularly higher variation of f (Fig. 4c): indeed, mutect2 and varscan2 detected much more sites than other callers in GATA3 and CDH1 (Fig. 4d).

Site co-occurrence plots and gene mutation frequency variability. a Total number of sites (diagonal), site co-occurrence among mutation callers (below diagonal) and corresponding similarity between callers (Jaccard index, above diagonal). b Total number of mutated genes (diagonal), mutation co-occurrence across subjects (below diagonal) and corresponding coefficients of variations (CVs) across pipelines (above diagonal). Asterisks indicate CVs greater than 1; grey colour indicates no mutation co-occurrence between two subjects. c Standard deviation of gene mutation frequency across pipelines; red: genes associated with BC [19,20,21]. d Number of subjects with mutations detected by each tool. a-b Results obtained on BC mutations from 50 subjects (Additional file 1); c-d results obtained on all 975 subjects (Additional file 1)
Called sites and sequencing results
The variation of caller output at different cut-offs on site-level quantities (e.g. minimum number of reads, allele frequency) is informative of caller performance and samples (subjects) specificities. This analysis can be done by the function:
-
ese1 <- ese_allsubj(mut_sites$sites, type = “Site”)
The pipelines used to call mutations in TCGA BC data show a different behaviour, especially at low tumor variant allele frequency (VAF). In fact, in this range, mutect2 calls more sites than other tools, SomaticSniper detects almost half of mutect2 sites, while muse and varscan2 show similar trend and are halfway between mutect2 and SomaticSniper (Fig. 5a). This global pattern is particularly relevant in some subjects (Fig. 5b-c).

Number of called sites at various filtering criteria. Number of mutation sites at varying tumor VAF for (a) the whole dataset (975 subjects) and (b) in single subjects. c Number of sites at varying number of reads supporting the alternative allele in four subjects. a-c Results obtained on BC mutations detected by TCGA
Collecting data from the TCGA
The function integrate_TCGA uses the R package TCGAbiolinks [18] to collected data from the TCGA. These data are used to support the mutation sites under analysis with the possible evidence of availability of the same sites among those already catalogued at TCGA, which would be an additional evidence of site reliability.
Conclusions
The R package isma provides functions for the integrative analysis of mutation sites detected by multiple pipelines. It quantifies the consensus between somatic mutation call pipelines, estimates pipeline variability and biological variability, and underlines outlier features (subject/tools) that may require further investigation. Indeed, an outlier subject may reflect a biological phenomenon (e.g. due to tumor genetic heterogeneity) and/or an experimental problem (e.g. poor biological sample, sequencing performance). The application of isma on BC mutations from TCGA underlined relevant variations among pipelines across subjects, with extreme cases characterized by a very poor consensus. Relevant imbalances among pipelines were also spotted at gene level, which implies a significant variability in the estimation of gene mutation frequency according to the pipeline used. In general, mutect2 reported a higher number of sites at low VAF in comparison to other callers.
In conclusion, the knowledge emerging from the analyses made possible by isma is useful to screen more reliable mutation sites, carry out comparative analysis among pipelines and, lastly, may suggest novel biological insights.
Availability and requirements
Project name: isma
Project home page: https://www.itb.cnr.it/isma
Operating system: Platform independent
Programming language: R (> = 3.3.3)
Other requirements: The R Project for Statistical Computing.
License: GNU General Public License (> = 2)
Any restrictions to use by non-academics: According to GNU General Public License (> = 2)
Abbreviations
- BC:
-
Breast cancer
- INDEL:
-
Insertions, deletions
- isma:
-
Integrative somatic mutation analysis
- NGS:
-
Next generation sequencing
- SNV:
-
Single nucleotide variations
- TCGA:
-
The cancer genome atlas
- VCF:
-
Variant Call Format
References
Cai L, Yuan W, Zhang Z, He L, Chou KC. In-depth comparison of somatic point mutation callers based on different tumor next-generation sequencing depth data. Sci Rep. 2016;6:36540.
Roberts ND, Kortschak RD, Parker WT, Schreiber AW, Branford S, Scott HS, Glonek G, Adelson DL. A comparative analysis of algorithms for somatic SNV detection in cancer. Bioinformatics. 2013;29:2223–30.
Wang Q, Jia P, Li F, Chen H, Ji H, Hucks D, Dahlman KB, Pao W, Zhao Z. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 2013;5:91.
Alioto TS, Buchhalter I, Derdak S, Hutter B, Eldridge MD, Hovig E, Heisler LE, Beck TA, Simpson JT, Tonon L, et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun. 2015;6:10001.
Krøigård AB, Thomassen M, Lænkholm AV, Kruse T, Larsen MJ. Evaluation of nine somatic variant callers for detection of somatic mutations in exome and targeted deep sequencing data. PLoS One. 2016;11(3):e0151664.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2011;28:311–7.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Danecek P, Auton A, Abecasis G, Albers C, Banks E, DePristo M, Handsaker R, Lunter G, Marth G, Sherry S, McVean G, Durbin R. 1000 genomes project analysis group. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Pietrelli A, Valenti L. myVCF: a desktop application for high-throughput mutations data management. Bioinformatics. 2017;33:3676–8.
Jochen Singer J, Ruscheweyh HJ, Hofmann AL, Thurnherr T, Singer F, Toussaint NC, Ng C, Piscuoglio S, Beisel C, Christofori G, et al. NGS-pipe: a flexible, easily extendable and highly configurable framework for NGS analysis. Bioinformatics. 2017;34:107–8.
Lawrence M, Gentleman R. VariantTools: an extensible framework for developing and testing variant callers. Bioinformatics. 2017;33:3311–3.
Knaus BJ, Grünwald NJ. vcfR: a package to manipulate and visualize variant call format data in R. Mol Ecol Resour. 2017;17:44–53.
Rashid M, Robles-Espinoza C, Rust AG, Adams JD. Cake: a bioinformatics pipeline for the integrated analysis of somatic variants in cancer genomes. Bioinformatics. 2013;29(17):2208–10.
Cantarel B, Weaver D, McNeill N, Zhang J, Mackey A, Reese J. BAYSIC: a Bayesian method for combining sets of genome variants with improved specificity and sensitivity. BMC Bioinformatics. 2014;15:104.
Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015;19(1A):A68–77.
Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, Morgan M. VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics. 2014;30:2076–8.
Colaprico A, Silva T, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, Ceccarelli M, Bontempi G, Noushmehr H. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015;44(8):e71.
Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The catalogue of somatic mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008;10:11.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–40.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Todd R, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–502.
Fan Y, Xi L, Hughes DST, Zhang J, Zhang J, Futreal PA, Wheeler DA, Wenyi Wang W. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016;17:178.
Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics. 2012;28:1811–7.
Acknowledgements
We would like to thank John Hatton (CNR-ITB) for proofreading the manuscript.
Funding
This work has been supported by: Italian Ministry of Education, University and Research [PON ELIXIR CNRBiOmics, INTEROMICS PB05, PRIN 2015 20157ATSLF]; Italian Ministry of Health [GR-2016-02363997]; and Lombardy Region Fondazione Regionale per la Ricerca Biomedica [LYRA 2015–0010]. None of the funding bodies had any role in the design of the study and collection, analysis and interpretation of data, and in writing the manuscript.
Availability of data and materials
The datasets analysed during the current study were collected from the GDC Data Portal [https://portal.gdc.cancer.gov] using isma R package (see Results and Additional file 1).
Author information
Authors and Affiliations
Contributions
NDN designed and implemented the software package, carried out the analyses and wrote the manuscript. MG and MM designed and implemented the computational environment, created the docker environment with isma package, revised the manuscript. LM designed the study and revised the manuscript critically. EM designed the study, implemented the software package, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
TCGA barcodes. List of TCGA barcodes used in this study. (TXT 33 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Di Nanni, N., Moscatelli, M., Gnocchi, M. et al. isma: an R package for the integrative analysis of mutations detected by multiple pipelines. BMC Bioinformatics 20, 107 (2019). https://doi.org/10.1186/s12859-019-2701-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-019-2701-0