
Abstract
Background
Binding of transcription factors to transcription factor binding sites (TFBSs) is key to the mediation of transcriptional regulation. Information on experimentally validated functional TFBSs is limited and consequently there is a need for accurate prediction of TFBSs for gene annotation and in applications such as evaluating the effects of single nucleotide variations in causing disease. TFBSs are generally recognized by scanning a position weight matrix (PWM) against DNA using one of a number of available computer programs. Thus we set out to evaluate the best tools that can be used locally (and are therefore suitable for large-scale analyses) for creating PWMs from high-throughput ChIP-Seq data and for scanning them against DNA.
Results
We evaluated a set of de novo motif discovery tools that could be downloaded and installed locally using ENCODE-ChIP-Seq data and showed that rGADEM was the best-performing tool. TFBS prediction tools used to scan PWMs against DNA fall into two classes — those that predict individual TFBSs and those that identify clusters. Our evaluation showed that FIMO and MCAST performed best respectively.
Conclusions
Selection of the best-performing tools for generating PWMs from ChIP-Seq data and for scanning PWMs against DNA has the potential to improve prediction of precise transcription factor binding sites within regions identified by ChIP-Seq experiments for gene finding, understanding regulation and in evaluating the effects of single nucleotide variations in causing disease.
Similar content being viewed by others


Background
The sequence-specific binding of transcription factors to transcription factor binding sites (TFBSs) is key to the mediation of transcriptional regulation [1]. High throughput experimental methods for identifying TFBSs such as ChIP-Chip and ChIP-Seq identify a region of 100–1000 base pairs (b.p.) while the actual TFBS is a short region (typically 9–15 b.p.) within that region. Nonetheless, there is a small set of experimentally precisely validated functional transcription factor binding sites which are stored in reference databases such as PAZAR [2] and ORegAnno [3]. However this is an insignificant proportion of transcription factor binding sites in terms of the human genome. Hence there is a need for accurate computational prediction of transcription factor binding sites [4], for gene finding, understanding regulation and in applications such as evaluating the effects of single nucleotide variations (SNVs) in causing differential expression [4] and leading to disease [5].
Prediction of transcription factor binding sites is generally performed by scanning a DNA sequence of interest with a position weight matrix (PWM) for a transcription factor of interest [6, 7] and various pattern-matching tools have been developed for this purpose. These tools fall into two classes: those that predict clusters of transcription factor binding sites or those that predict individual sites.
Experimental identification of transcription factor binding sites
There are many in vitro and in vivo experimental approaches that have been used to identify transcription factor binding sites and these are reviewed briefly here.
In vitro methods include: (i) The Electro-Mobility Shift Assay (EMSA) [8] which exploits the ability of a non-denaturing polyacrylamide gel to act as a molecular sieve, separating protein-bound DNA from unbound DNA. (ii) The DNase I footprinting/protection assay combines the cleavage reaction of DNase I with EMSA [9]. A key problem with both EMSA and DNase I footprinting is the identification of unwanted protein-DNA interactions that result from non-specific DNA binding proteins [8]. (iii) Systematic Evolution of Ligands by EXponential enrichment (SELEX) [10] works by screening a large pool of short, random oligonucleotide probes which are recognized by a TFBS of interest [10]. A refinement, SELEX-seq, involves the selected dsDNAs being subjected to massively parallel sequencing [11].
There has been a recent shift towards in vivo approaches [4]. In the (iv) Chromatin ImmunoPrecipitation (ChIP) assay, a variation of the ‘pull down’ class of assay [12], the DNA-binding protein of interest is cross-linked to the DNA using formaldehyde. The DNA is then fragmented into small fragments of around 100–1000 b.p. and an antibody specific for a given transcription factor is then used to immunoprecipitate the DNA-protein complex. The cross-links are then reversed, releasing the DNA for PCR amplification [12]. High throughput versions of the ChIP assay involve hybridizing the resulting fragments to genomic tiling microarrays, an approach known as ChIP-chip [13], or the resulting DNA fragments can undergo massively parallel sequencing, an approach known as ChIP-Seq [14].
There are a number of advantages of using ChIP-Seq instead of ChIP-chip. Key improvements are in base pair resolution, avoiding non-linearity and saturation of ChIP-chip signal intensity, ability to analyze sequence repeat regions, and avoiding limitations from the limited selection of probes on a ChIP-chip array. Overall ChIP-Seq has a higher specificity and sensitivity compared with ChIP-chip [14, 15] and has largely superseded the ChIP-chip method. Consequently, ChIP-Seq is the current ‘gold standard’ for identifying protein/DNA interactions sites such as histone modifications as well as transcription factor binding sites [16]. A recent refinement to ChIP-Seq is ChIP-exo where the resulting fragments from the ChIP assay are trimmed using lambda exonuclease. This results in fragments that are shorter (∼50 b.p.), but still larger than the precise TFBS [17].
Position weight matrices (PWMs)
Position Weight Matrices (PWMs) are the most widely used approach to modelling TFBSs. In contrast to a consensus model (which simply gives the most common base(s) at each position of a binding motif), a matrix-based PWM model (which is simply a 4×n matrix of scores for each of the 4 bases across each position in the binding motif) accounts for the preference for each of the four nucleotides at each position in the motif [4, 6, 18].
The high-throughput techniques, particularly ChIP-Seq and SELEX-seq, provide an opportunity to identify and characterize protein-DNA binding events at a genome-wide level, contrary to the previous techniques that were only able to characterize a small number of protein-DNA binding events. Hu et al. [19] have suggested that PWMs derived from transcription factor binding sites detected by these methods will be more accurate than PWMs derived from techniques such as SELEX, or compilations of individual promoter assays that detect limited transcription factor binding site numbers. Further, the ChIP-Seq technique has been found to produce PWMs with greater accuracy than ChIP-chip owing to the superior resolution provided by the ChIP-Seq technique [19, 20].
PWMs can be obtained from a number of resources including the commercial database TRANSFAC [21] and the open access database JASPAR [20]. TRANSFAC PWMs are derived from experimental evidence obtained from the literature [21], but availability and application is limited by a commercial licence. The bulk of the PWMs in earlier versions of JASPAR were derived from SELEX experiments and individual promoter assays, but since 2014, updates to JASPAR [22] now include new PWMs derived from ChIP-Seq data using MEME for motif discovery. Other recent resources include HOCOMOCO [23], HOMER ([24] http://homer.salk.edu/homer/motif/HomerMotifDB/homerResults.html) and CIS-BP [25]. However, JASPAR is a well-established and widely-used resource that was employed by us in previous unpublished work and consequently was used in some of the work presented here.
de novo motif discovery
While large scale ChIP experiments allow the genome-wide identification of binding regions for a specific transcription factor, these regions are much longer than the actual binding site for a specific transcription factor meaning that the actual transcription factor binding sites still need to be identified [26, 27].
Various motif discovery methods have been developed and there have been several reviews of the approaches used ([28–34], for example). The most popular algorithms are either enumerative or probabilistic. Enumerative methods examine frequencies of all DNA strings forming a PWM from the over-represented strings that have been identified [1]. Probabilistic methods generate a local multiple alignment of sequences while learning the parameters of the PWM using approaches such as expectation-maximization [35], Gibbs sampling [36], or greedy approaches [37]. The advantage of enumerative methods is that there is less chance of them getting stuck in a local optimum, while probabilistic methods can cope with arbitrary motif model variations and hence remain unaffected by motif length [1]. For example, the well-known motif discovery program MEME [38] uses a probabilistic method with expectation-maximization [39].
De novo motif discovery has proved to be challenging when carried out on the binding regions resulting from the genome-wide techniques of ChIP-chip and ChIP-Seq using conventional motif discovery programs such as MEME, owing to the large volumes of data generated by these techniques; ChIP-Seq can generate over 10,000 sequences in a single run. Hence a common practice has been to use these tools on a subset of the sequences [19, 40, 41]. However, Hu et al. [19] have suggested that this practice will lead to inaccurate PWMs and consequently new tools have recently been developed that are able to handle the large volumes of data generated from these high-throughput technologies. These include the freely available software packages ChIPMunk [42], HOMER (Hypergeometric Optimization of Motif EnRichment) [24], rGADEM (Genetic Algorithm guided formation of spaced Dyads coupled with EM for Motif identification) [43] and MEME-ChIP [44, 45].
Evaluation of the performance of transcription factor binding site prediction tools and motif discovery tools
As well as high quality PWMs to model TFBSs, the computational prediction of TFBSs requires a pattern matching tool. A number of tools are available for this purpose which fall into two classes: those that predict clusters of sites and those that predict individual sites. Consequently the range of tools that can be used locally for motif discovery designed for use with high-throughput data and tools for identifying TFBSs using PWMs warrants an independent performance evaluation.
Approaches for scanning PWMs against DNA were reviewed by Hannenhalli [6] and by Bulyk [30], but the number of performance comparisons is limited. Most have been as part of authors’ evaluations of their own new tools ([46, 47], for example) although an independent assessment was performed by Roulet et al. [48] and a much more recent survey of online PWM scanning tools was performed by Tran and Huang [49].
A number of authors have performed comparisons of methods for motif discovery. These include work by Sandve and colleagues [32, 50, 51], McLeay and Bailey [52] and Orenstein et al. [53]. Kibet and Machanick [34] assessed the performance of matrices obtained from different sources, but did not directly assess the motif discovery tools. The most comprehensive evaluations of tools are those performed by Tompa et al. [39], Hu et al. [54], Medina-Rivera et al. [55] and, most recently, Weirauch et al. [56]. Tompa et al. [39] performed an independent assessment of the performance of 13 tools designed for discovery of novel regulatory elements with no a priori knowledge of the transcription factor involved. They made predictions across a number of species (fly, human, mouse and yeast) with known binding sites taken from TRANSFAC. Assessment was performed at a nucleotide level (i.e. whether individual bases were correctly identified as being part of a binding motif or not) and they concluded that, overall, Weeder [57] performed best. Hu et al. [54] performed another assessment around the same time. However, while Tompa et al. allowed the authors of tools to fine-tune parameters to achieve what they considered to be the best results, Hu et al. performed minimum intervention reflecting the approach likely to be taken by the average end user. They assessed five methods at different levels: nucleotide, binding site, sequence and motif. They also created a ‘consensus ensemble algorithm’ which exploits variations in predictions by stochastic methods to refine predictions. Neither Tompa et al. nor Hu et al. assessed the quality of any models (PWMs) generated from these motifs by applying them to search for TFBSs in DNA.
More recently, Kibet and Machanick [34] reviewed and evaluated different approaches and pointed out the difficulty in evaluating motif discovery tools by applying the PWMs to motif searching: annotation of precise true TFBSs in DNA, to use as a gold standard reference set, is limited. An assessment of motif discovery methods using binding site prediction for evaluation was performed by Medina-Rivera et al. [55]. They generated an assessment method that combines theoretical and empirical score distributions to assess reliability of PWMs for predicting TFBSs and used this to analyze PWMs for bacterial, yeast and mouse TFBSs. Weirauch et al. [56] evaluated 26 tools for motif discovery using in vitro data for 66 mouse TFBSs, looking at PWMs and more complex models such as dinucleotide matrices and secondary motifs. They added ChIPMunk [42] and MEME-ChIP [44, 45] for a further evaluation of performance on in vivo data using five mouse and four yeast TFBSs. During this comparison they found that ChIPMunk outperforms MEME-ChIP.
In this paper we conduct an independent assessment of a set of four motif discovery tools specifically designed for handling large datasets from high-throughput methods (including ChIPMunk and MEME-ChIP evaluated by Weirauch et al.), but using human ChIP-Seq data obtained from ENCODE [58]. Performance evaluation makes use of a gold standard reference set of experimentally-validated precise human transcription factor binding sites. We also evaluate a number of open source PWM scanning tools that are well documented and can be installed locally and are therefore more suitable for large scale analyses. These pattern matching tools represent both classes (individual and cluster).
Methods
Sources of experimentally validated TFBSs
To evaluate performance, we identified experimentally-validated TFBSs from resources that, rather than just providing PWMs or approximate regions to which TFBSs bind, provide precise validated binding sites for a limited set of genes. Three sources of such data are available: PAZAR [2], ORegAnno [3, 59] and TRANSFAC [21]. TRANSFAC was rejected because of its commercial licensing, while the data in PAZAR are a superset of ORegAnno and consequently, the PAZAR dataset was selected.
Selecting data from PAZAR
PAZAR contains some redundancy (multiple instances of the same TFBS annotated for a given gene), so any duplicate TFBSs were removed.
PAZAR contains 159 genes annotated with TFBSs that are contained in either JASPAR or ENCODE-ChIP-Seq data. This set contains data for 14 TFBSs with corresponding PWMs in JASPAR coming from a total of 156 human genes. This set is referred to below as ‘PAZAR-J’. The set also contains data for 12 transcription factors with binding data in the ENCODE-ChIP-Seq data which come from a total of 149 genes (‘PAZAR-E’). The PAZAR-J and PAZAR-E datasets overlap for 11 of the transcription factors (See Fig. 1 and Additional file 1 for details.)

Overlap of transcription factor data. The Venn diagram shows overlaps between known sites in PAZAR, the PWMs in JASPAR and those derived from the ENCODE-ChIP-Seq data used in this paper
Tool evaluation
Initial evaluation of the motif scanning tools (using PWMs from the 2010 release of JASPAR that we had used in earlier work, referred to here as JASPAR.2010) was performed for each of the 14 transcription factors in PAZAR-J by selecting the appropriate subset of the 156 genes in PAZAR-J having validated binding sites for the transcription factor in question.
Evaluation of the motif discovery tools was performed for each of the 12 transcription factors in PAZAR-E by selecting the appropriate subset of the 149 genes in PAZAR-E having validated binding sites for the transcription factor in question and using the motif discovery tool selected in the initial evaluation (FIMO).
Finally, re-evaluation of the motif scanning tools (using PWMs generated by rGADEM) was also performed for each of the 12 transcription factors in PAZAR-E by selecting the appropriate subset of the 149 genes in PAZAR-E having validated binding sites for the transcription factor in question.
DNA Data
TFBSs can occur in the promoter region, in introns and exons, and far upstream of genes [60, 61]. Consequently the complete gene sequence (i.e. both exons and introns), together with an upstream region of 10,000 b.p. of each of the genes was obtained from Biomart [62] using the biomaRt package in Bioconductor [63–65].
Performance Metrics
True positives (TP) were defined as predicted binding sites having a minimum overlap of 70 % of base pairs with known binding sites from PAZAR. Similarly, false positives (FP) were defined as predicted binding sites not having an overlap of at least 70 % with a known binding site and false negatives (FN) were defined as known binding sites that were not identified. Obtaining a true estimate of the total number of negative sites (and hence the number of true negatives, TN) is difficult and therefore we adopted the normal practice of avoiding performance measures that require true negative counts [66]. For cluster predictors, all predicted component TFBSs within a region must overlap with known sites by at least 70 % of base pairs for a prediction to be regarded as a true positive.
As a control, all the DNA sequences were scrambled using the ‘shuffleseq’ program from the EMBOSS suite (version 6.4.0) [67]. In this case there are no actual positives and therefore no true positives or false negatives. Any positive predictions are therefore classified as false positives and the number of actual negatives (AN=FP+TN) was defined as AN=L/l t where L is the length of the sequence and l t is the length of the PWM in question).
Performance was assessed by calculating sensitivity (Sn=TP/(TP+FN)), positive predictive value (PPV=TP/(TP+FP)) and geometric accuracy (\( {ACC}_{g} = \sqrt { {Sn}. {PPV}}\)) [66], averaged across the TFBS PWMs and genes analyzed. For the scrambled sequences, a false positive rate was calculated (FPR s =N p /AN, where N p is the number of predicted sites and AN is the number of actual negatives as defined above).
Derivation of PWMs
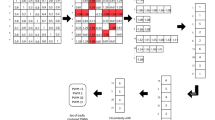
The methods used for deriving PWMs from the ENCODE-ChIP-Seq data are summarized in Fig. 2.

Flowchart summarising the methods used to derive PWMs from the ENCODE-ChIP-Seq data. See text for details
ChIP-Seq data for the human transcription factors were obtained from the ENCODE project (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhTfbs/) in FASTQ format. Only the ChIP-Seq data that had a corresponding control sample available were selected to help to control biases and artefacts that occur in the experimental protocol [14, 68]. ChIP-Seq control samples are obtained from a mock experiment without the specific antibody and were used during the peak calling process as recommended by Bardet et al. [68]. It is critical that the short reads arising from ChIP-Seq are aligned properly to the reference genome, otherwise false positives and false negatives would occur. Thus, low quality reads and adaptor sequences were identified using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and removed using the FASTX TOOLKIT (http://hannonlab.cshl.edu/fastx_toolkit/).
The reads were then mapped to the human genome version hg19 using Bowtie [69]. The resulting Sequence Alignment/Map format (SAM) files were converted to binary format (BAM) files and indexed using SAMtools [70]. This step reduces the file size and allows rapid access which is essential given the large size of the data.
After the reads were aligned to the reference genome, peak calling was performed by identifying statistically significant binding regions that are enriched in the ChIP-Seq sample compared with the control sample [14]. It has been suggested that peaks should be called using more than one peak caller and the intersection of peaks should then be taken [71]. Consequently peaks were called using both MACS [72] and the bioconductor package BayesPeak [63, 73, 74]. Common peaks were identified and replicates were pooled using the bioconductor package ChIPpeakAnno [63, 75]. A set of peak regions — centred on the summits of the peaks (±100 b.p.) in order to prevent bias towards longer peak regions [68] — were obtained in FASTA format. We refer to these filtered peak data as the ‘ENCODE-ChIP-Seq data’.
The TFBS motif discovery tools evaluated were MEME-ChIP [44, 45], HOMER [24], ChIPMunk [42] and rGADEM [43, 63] and these were tested using the 12 transcription factors in the PAZAR-E dataset. Since these programs are able to deal with large datasets, all peak regions were used. The motif discovery methods have various adjustable parameters and these were explored in 10 % steps.
Results and discussion
As shown in Fig. 1, the overlap between transcription factors having validated binding sites in PAZAR, the PWMs describing these TFBSs in JASPAR and the binding sites in the ENCODE-ChIP-Seq data is fairly small. Only 11 transcription factors (E2F1, ELK4, GATA2, GATA3, IRF1, MAX, NF- κB, STAT1, YY1, CTCF and NFYA) have validated TFBSs in PAZAR, PWMs in JASPAR and are also represented in the ENCODE-ChIP-Seq data. BRCA1 was also found in all datasets, but has recently been removed from JASPAR since its sequence specificity has been questioned [76]. While the ENCODE-ChIP-Seq data are actually more comprehensive than indicated, only ChIP-Seq datasets having no access restrictions and for which the transcription factor had a control ChIP-Seq sample available were chosen. Three additional transcription factors have data that overlap between JASPAR and the ENCODE-ChIP-Seq data (USF2, ZNF263 and JUND) and between PAZAR and JASPAR (ESR1, ESR2 and SP1) while one more has data that overlaps between PAZAR and the ENCODE-ChIP-Seq data (TAL1). Consequently the number of PWMs that could be used for the evaluations described below was limited to 11–14.
Logically it makes sense to evaluate motif discovery methods first and then to evaluate the tools available for matching the derived PWMs to DNA sequences. However the evaluation of the performance of motif discovery methods requires a tool to test the performance of the resulting PWMs. Therefore we needed to select a motif scanning tool for this purpose. In earlier work we had tested the performance of a number of PWM scanning tools using older JASPAR matrices (JASPAR.2010). These results are summarized below and the best performing tool was then used for evaluating the motif discovery methods. Finally, the performance of the scanning tools was reassessed using motifs from the best performing motif discovery method.
Selecting a PWM scanning tool for evaluation of motif discovery methods
As stated above, in order to evaluate motif discovery methods, we need to scan the motifs against DNA and compare the predictions with a gold-standard set of known precise TFBSs. In work done in 2011, we evaluated the performance of different PWM scanning tools using the older JASPAR.2010 matrices [20] which had been derived from SELEX and individual promoter assays. Consequently, we exploited that earlier analysis for this work. PWMs for 14 human transcription factors from JASPAR.2010 which are also present in PAZAR were selected (the ‘PAZAR-J’ dataset) and the performance of the scanning methods was evaluated on these using PAZAR as the gold standard.
TFBS cluster prediction tools chosen were MCast [77], Baycis [78], Cister [79], ClusterBuster [80] and Comet [81] while individual TFBS prediction tools chosen were FIMO [82], Clover [83], Matrix-Scan (part of the RSAT suite) [84], Patser (also part of RSAT) [84] and PossumSearch [85]. Note that Cister, Comet and ClusterBuster all come from the Weng laboratory, with ClusterBuster being their latest software. Consequently this analysis provides an interesting comparison to find out whether their latest software is indeed the best performing.
All tools having variable cutoffs for making predictions were evaluated to ensure the optimum cutoff was chosen by using 10 % steps for all parameters. In all cases, the default settings were found to give the best performance and were used for all future evaluations.
Table 1 shows that FIMO and MCAST are the best performing TFBS prediction tools for individual sites and clusters respectively and FIMO was therefore selected for evaluation of the motif finding methods. (Complete results for individual PWMs are provided in Additional file 2).
Evaluation of motif discovery methods
We chose to evaluate four methods for motif discovery that have been developed especially for working with large genome-wide datasets and that are open source and well documented: rGADEM [43], HOMER [24], ChIPMunk [42], and MEME-ChIP [44, 45]. For this purpose, TFBS PWMs were derived, using the protocol described above, for the 12 transcription factors in the PAZAR-E dataset.
The tools have parameters that can be adjusted for motif discovery and these were explored for all tools using a 10 % step size. It was found that the defaults produced PWMs that resembled well-established motifs for all tools with the exception of rGADEM where the e-value parameter had to be set to a value of 0.5 rather than the default value of 0.0. The motif discovery tools are also able to generate multiple possible motifs. During the exploration of parameters, it was found that the first PWM generated always best-resembled well-established motifs for the TFBSs used in this work, and consequently only the first PWM was used.
Performance was evaluated by using the FIMO motif scanning tool comparing predictions of TFBS locations with the PAZAR-E data as a gold standard. Table 2 shows that rGADEM has the best performance and MEME-ChIP the worst on all four performance metrics. (Complete results for individual PWMs are provided in Additional file 3 and sequence logos for the first PWM generated for the 12 TFBSs using each of the four motif discovery tools are provided in Additional file 4). We confirmed the finding of Weirauch et al. [56] that ChIPMunk outperforms MEME-ChIP, but showed that rGADEM outperforms both.
The PWMs obtained using the different methods were compared with each other and with those in JASPAR: both the older set derived from SELEX and individual promoter assays (JASPAR.2010) and the newer matrices obtained from ChIP-Seq data (JASPAR.2014). Normalized Euclidean distances between equivalent PWMs were calculated using the TFBSTools package (http://www.bioconductor.org/packages/release/bioc/html/TFBSTools.html) in Bioconductor. Reverse complement matrices were also checked and the minimum distances recorded. Results for each matrix set comparison were averaged across the PWMs used. The normalised Euclidean distance ranges from 0 to 1 where 0 denotes complete identity and 1 denotes complete dissimilarity. Results are shown in Table 3.
Comparing the PWMs generated in this work using different motif discovery tools, the best performing method (rGADEM) shows the largest difference in PWMs from the worst performing method (MEME-ChIP). Clearly there are small but significant differences in the PWMs generated by different motif discovery tools. However all the motif discovery methods applied to the ENCODE-ChIP-Seq data show even greater differences from the old JASPAR.2010 PWMs generated using SELEX or individual promoter assays.
Re-evaluation of PWM scanning tools
Having shown that rGADEM generates better PWMs than other motif-discovery methods, we returned to the evaluation of tools for scanning PWMs against DNA. We repeated this evaluation using PWMs generated from the ENCODE-ChIP-Seq data using rGADEM, and results are shown in Table 4. In general the tools predicting individual sites perform better than those predicting clusters. Because of the more stringent requirements for a true positive in predicting clusters (i.e. every predicted site within the cluster must have a 70 % overlap with a true site), it might be expected that the sensitivity for cluster predictors would be lowered, while the specificity would be improved. Indeed the sensitivity of cluster predictors is somewhat lower than the individual site predictors. Since we do not have the true negative count, we cannot calculate specificity, but surprisingly the false positive rate on scrambled sequences (FPR s ) for the cluster predictors is larger than that for single site predictors suggesting that the cluster predictors have lower specificity.
Using the JASPAR.2010 data, we had identified FIMO as the best tool for identifying individual TFBSs and MCast as the best cluster-based tool. Table 4 shows that these two tools still perform best using the PWMs derived here using rGADEM and ENCODE-ChIP-Seq data. (Complete results for individual PWMs are provided in Additional file 5). Indeed the overall ranking of all the tools remains the same:
MCast >Comet >ClusterBuster >Cister >Baycisfor cluster predictors and
FIMO >Patser >PossumSearch >Clover >Matrix-Scanfor individual predictors.
Cister, Comet and ClusterBuster all come from the same laboratory (published in 2001, 2002 and 2003 respectively). These results suggest that Comet from 2002 outperforms ClusterBuster from 2003, but both have made progress over their initial 2001 software. However MCast significantly outperforms all three methods.
Conclusions
As a comprehensive set of experimentally-characterized precise transcription factor binding sites is not available, having good reliable prediction methods is very important. While some experimental methods of identifying TFBSs are relatively accurate, identifying regions of around 10–20 b.p., methods such as ChIP-Chip, and more importantly the ‘gold standard’ ChIP-Seq method, identify DNA regions of 100–1000 b.p. which is much larger than the TFBS itself (typically 9–15 b.p.). Consequently, when these experimental methods are employed for identifying TFBSs, it is necessary to use a prediction tool to identify the TFBS within the much wider region. While the need for identifying TFBSs as an adjunct to gene prediction in the human genome has diminished, it is now much more important in order to have a full understanding of the regulation of gene expression and to be able to consider the potential phenotypic effects of mutations occurring in a TFBS.
Motif discovery
None of the ENCODE-ChIP-Seq data used to derive the PWMs for evaluating motif discovery tools overlapped the sequences obtained from genes present in PAZAR and consequently we know there is no overlap between the training and test sets. Table 2 clearly shows that PWMs derived using rGADEM outperform those derived using other motif discovery methods.
Alternative sources of binding data
The analysis here has focused on the use of data from ChIP-Seq experiments which, as described in the introduction, have largely superseded the earlier ChIP-chip approach; both of these are in vivo approaches. Another relatively new approach is the in vitro SELEX-seq [11] approach. To investigate whether SELEX-seq would be a useful addition to ChIP-Seq data, we used rGADEM with SELEX-seq data to derive a PWM for NF- κB, the only transcription factor for which SELEX-seq, ENCODE-ChIP-Seq data and PAZAR data are available.
The performance of the SELEX-seq derived PWM (Sn=0.913, PPV=0.810, ACC g =0.860, FPR s =0.004) is less than its counterpart derived from the ENCODE-ChIP-Seq data (Sn=0.937, PPV=0.831, ACC g =0.882, FPR s =0.002). However no firm conclusions can be drawn on the performance of SELEX-seq data in general on the basis of a single transcription factor.
Another recently developed technology is ChIP-exo [17]. Unfortunately no data are available from ChIP-exo for TFBSs that are present in the PAZAR gold standard dataset and consequently we cannot evaluate the performance of PWMs derived from these data.
Scanning tools
An inherent problem with TFBS prediction is their short and degenerate nature. The non-redundant vertebrate TFBS PWMs in JASPAR.2014 range from 5 b.p. (Pax4) to 30 b.p. (Prrx2), but with the majority being 9–15 b.p. (mean =12.2, σ=3.7). A naïve scanning of PWMs against a DNA sequence can therefore result in a high false positive rate. It is therefore essential to optimize the methods used to scan a PWM against a DNA sequence in order to minimize the false positive rate.
We have evaluated a set of transcription factor binding site prediction tools that could be downloaded and installed locally, identifying FIMO and MCAST as the best-performing tools for identifying individual TFBSs and clusters of TFBSs respectively. While it is possible that there is some inter-relationship between the choice of motif discovery method and the tool used to search those motifs against a DNA sequence, this seems unlikely to be significant. The ranking of tool performance was the same when used with the JASPAR.2010 PWMs (generated using MEME-based tools) and the PWMs generated in this work using rGADEM. Similarly, using FIMO (part of the MEME suite) as a search tool, PWMs generated using MEME-ChIP do not perform as well as PWMs generated using rGADEM (Table 2).
Alternatives to PWMs
Position Weight Matrices (PWMs) are the most widely used TFBS models, but are limited by the assumption of the model that positions within a binding site are independent, something which is not true in all cases [56]. There have therefore been several attempts to develop more complex alternatives to the PWM model that take into account nucleotide interdependencies [6, 18, 25]. Some examples include pair-correlation models [86], trees [87], non-parametric models [88], feature-based models [89], Markov chain optimization [90], maximal dependence decomposition [91], Hidden Markov Models [92], transcription factor flexible models [93] and Dinucleotide PWMs [94].
However it has been observed that classical PWM models tend to perform at least as well as more complex models [18] and that more complex models tend to be prone to learning noise. Consequently, it has been suggested that the PWM model may be the state of the art and that focus should be placed on optimizing the PWM model rather than developing more complex models [95].
While PWMs are not outperformed by more complex models for the majority of transcription factors, for a small number of individual transcription factors it has been found that more complex models do result in better performance [56]. For example, more complex models perform better for transcription factors AP-2A and REST, but not for HNF4A [94]. Thus, in future, it may be worth evaluating both PWMs and more complex models and selecting an appropriate model for each individual transcription factor.
Summary
While TFBS predictors which identify individual sites outperform those that identify clusters, the choice of the type of prediction tool depends on the context in which it is to be used. The evaluation used in this study was performed in the context of known TFBSs associated with genes. Consequently, if prior knowledge is available about the DNA sequence being scanned (i.e. the DNA sequence is that of a known protein coding gene) then using a predictor of individual TFBSs is probably a sensible strategy. When analyzing a stretch of DNA with no prior knowledge about the presence of a gene, it would be better to use a prediction tool that identifies clusters of TFBSs since the chance of a random match is much reduced [58, 96].
In conclusion, we have analyzed motif discovery tools for generating PWMs from ChIP-Seq data using experimentally-validated precise TFBSs from PAZAR as a gold standard. We found that rGADEM out-performed other tools. We then evaluated a number of tools for scanning PWMs against DNA, both for identifying individual TFBSs and clusters of TFBSs. We found that FIMO and MCAST performed best respectively. We also found that there appears to be no dependence between the tool used for motif discovery and the tool used for motif scanning — in other words, using (for example) a motif scanning tool from the MEME suite does not perform better when using PWMs generated using a motif discovery tool from the MEME suite than when using an unrelated motif discovery tool.
Abbreviations
- ChIP:
-
Chromatin immunoprecipitation
- EMSA:
-
Electro-mobility shift assay
- PWM:
-
Position weight matrix
- SNV:
-
Single nucleotide variant
- SELEX:
-
Systematic evolution of ligands by exponential enrichment
- TFBS:
-
Transcription factor binding site
References
Narlikar L, Ovcharenko I. Identifying regulatory elements in eukaryotic genomes. Brief Funct Genomics Proteomics. 2009; 8:215–30.
Portales-Casamar E, Arenillas D, Lim J, Swanson MI, Jiang S, McCallum A, Kirov S, Wasserman WW. The PAZAR database of gene regulatory information coupled to the ORCA toolkit for the study of regulatory sequences. Nucleic Acids Res. 2009; 37:54–60.
Griffith OL, Montgomery SB, Bernier B, Chu B, Kasaian K, Aerts S, Mahony S, Sleumer MC, Bilenky M, Haeussler M, Griffith M, Gallo SM, Giardine B, Hooghe B, Van Loo P, Blanco E, Ticoll A, Lithwick S, Portales-Casamar E, Donaldson IJ, Robertson G, Wadelius C, De Bleser P, Vlieghe D, Halfon MS, Wasserman W, Hardison R, Bergman CM, Jones SJM. Open Regulatory Annotation Consortium: ORegAnno: an open-access community-driven resource for regulatory annotation. Nucleic Acids Res. 2008; 36:107–13.
Worsley-Hunt R, Bernard V, Wasserman WW. Identification of cis-regulatory sequence variations in individual genome sequences. Genome Med. 2011; 3:1–14.
Jarinova O, Ekker M. Regulatory variations in the era of next-generation sequencing: Implications for clinical molecular diagnostics. Hum Mutat. 2012; 33:1021–30.
Hannenhalli S. Eukaryotic transcription factor binding sites–modeling and integrative search methods. Bioinformatics. 2008; 24:1325–31.
Garcia-Alcalde F, Blanco A, Shepherd A. An intuitionistic approach to scoring DNA sequences against transcription factor binding site motifs. BMC Bioinformatics. 2010; 11:551–64.
Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: Application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981; 9:3047–60.
Galas DJ, Schmitz A. DNase footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1978; 5:3157–170.
Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990; 249:505–10.
Riley TR, Slattery M, Abe N, Rastogi C, Liu D, Mann RS, Bussemaker HJ. SELEX-seq: a method for characterizing the complete repertoire of binding site preferences for transcription factor complexes. Methods Mol Biol. 2014; 1196:255–78.
Elnitski L, Jin VX, Farnham PJ, Jones SJM. Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res. 2006; 16:1455–64.
Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Genome-wide location and function of DNA binding proteins. Science. 2000; 290:2306–9.
Park PJ. ChIP-Seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009; 10:669–80.
Joshua H, Peter K, Nicolas N, Peter P. ChIP-chip versus ChIP-Seq: Lessons for experimental design and data analysis. BMC Genomics. 2011; 12:134–46.
Adli M, Bernstein BE. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-Seq. Nat Protoc. 2011; 6:1656–1668.
Rhee HS, Pugh BF. Chip-exo: A method to identify genomic location of DNA-binding proteins at near single nucleotide accuracy. In: Curr. Protoc. Mol. Biol. Hoboken: John Wiley & Sons, Inc.: 2012. Chap. 21. Unit 21.24.
Nguyen TT, Androulakis IP. Recent advances in the computational discovery of transcription factor binding sites. Algorithms. 2009; 2:582–605.
Hu M, Yu J, Taylor JMG, Chinnaiyan AM, Qin ZS. On the detection and refinement of transcription factor binding sites using ChIP-Seq data. Nucleic Acids Res. 2010; 38:2154–67.
Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010; 38:105–10.
Matys V, Kel-Margoulis O, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K. TRANSFAC ®;and its module TRANSCompel ®;: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006; 34:108–10.
Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen C-Y, Chou A, Ienasescu H, Lim J, Shyr C, Tan G, Zhou M, Lenhard B, Sandelin A, Wasserman WW. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014; 42:142–7.
Kulakovskiy IV, Medvedeva YA, Schaefer U, Kasianov AS, Vorontsov IE, Bajic VB, Makeev VJ. HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 2013; 41:195–202.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010; 38:576–89.
Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, Zheng H, Goity A, van Bakel H, Lozano JC, Galli M, Lewsey MG, Huang E, Mukherjee T, Chen X, Reece-Hoyes JS, Govindarajan S, Shaulsky G, Walhout AJM, Bouget FY, Ratsch G, Larrondo LF, Ecker JR, Hughes TR. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014; 158:1431–43.
Bailey TL, Machanick P. Inferring direct DNA binding from ChIP-Seq. Nucleic Acids Res. 2012; 40:128–8.
Worsley-Hunt R, Mathelier A, Del Peso L, Wasserman WW. Improving analysis of transcription factor binding sites within ChIP-Seq data based on topological motif enrichment. BMC Genomics. 2014; 15:472. doi:10.1186/1471-2164-15-472.
Hannenhalli S, Levy S. Promoter prediction in the human genome. Bioinformatics. 2001; 17 Suppl 1:90–6.
Wasserman WW, Krivan W. In silico identification of metazoan transcriptional regulatory regions. Naturwissenschaften. 2003; 90:156–66.
Bulyk ML. Computational prediction of transcription-factor binding site locations. Genome Biol. 2003; 5:201–1.
Pavesi G, Mauri G, Pesole G. In silico representation and discovery of transcription factor binding sites. Brief Bioinform. 2004; 5:217–36.
Sandve GK, Drabløs F. A survey of motif discovery methods in an integrated framework. Biol Direct. 2006; 1:11–11.
Das MK, Dai HK. A survey of DNA motif finding algorithms. BMC Bioinformatics. 2007; 8 Suppl 7:21–1.
Kibet CK, Machanick P. Transcription factor motif quality assessment requires systemartic comparative analysis. F1000Res. 2015; 4(ISCB Comm J). doi:10.12688/f1000research.7408.2.
Cardon LR, Stormo GD. Expectation maximization algorithm for identifying protein-binding sites with variable lengths from unaligned DNA fragments. J Mol Biol. 1992; 223:159–70.
Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993; 262:208–14.
Hertz GZ, Hartzell GW, Stormo GD. Identification of consensus patterns in unaligned DNA sequences known to be functionally related. Comput Appl Biosci (CABIOS). 1990; 6:81–93.
Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Intl Conf Intell Syst Mol Biol ISMB. 1994; 2:28–36.
Tompa M, Li N, Bailey TL, Church GM, de Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, Kent WJ, Makeev VJ, Mironov AA, Noble WS, Pavesi G, Pesole G, Régnier M, Simonis N, Sinha S, Thijs G, van Helden J, Vandenbogaert M, Weng Z, Workman C, Ye C, Zhu Z. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005; 23:137–44.
Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008; 36:5221–31.
Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods. 2008; 5:829–34.
Kulakovskiy IV, Boeva V, Favorov A, Makeev V. Deep and wide digging for binding motifs in ChIP-Seq data. Bioinformatics. 2010; 26:2622–3.
Mercier E, Droit A, Li L, Robertson G, Zhang X, Gottardo R. An integrated pipeline for the genome-wide analysis of transcription factor binding sites from ChIP-Seq. PLoS ONE. 2011; 6:16432.
Ma W, Noble WS, Bailey TL. Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat Protoc. 2014; 9:1428–50.
Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011; 27:1696–7.
Terai G, Mizuno T, Takagi T. Evaluation of a method for predicting transcription factors using motif-search programs. JSBi Genome Inform. 1999; 10:249–50.
del Val C, Pelz O, Glatting KH, Barta E, Hotz-Wagenblatt A. PromoterSweep: a tool for identification of transcription factor binding sites. Theor Chem Acc. 2010; 125:583–91.
Roulet E, Fisch I, Junier T, Bucher P, Mermod N. Evaluation of computer tools for the prediction of transcription factor binding sites on genomic DNA. In Silico Biol. 1998; 1:21–8.
Tran NTL, Huang CH. A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data. Biol Direct. 2014; 9:4. doi:10.1186/1745-6150-9-4.
Sandve GK, Abul O, Walseng V, Drabløs F. Improved benchmarks for computational motif discovery. BMC Bioinformatics. 2007; 8:193–3.
Klepper K, Sandve GK, Abul O, Johansen J, Drablos F. Assessment of composite motif discovery methods. BMC Bioinformatics. 2008; 9:123–3.
McLeay RC, Bailey TL. Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics. 2010; 11:165–5.
Orenstein Y, Linhart C, Shamir R. Assessment of algorithms for inferring positional weight matrix motifs of transcription factor binding sites using protein binding microarray data. PLoS ONE. 2012; 7:46145–6145.
Hu J, Li B, Kihara D. Limitations and potentials of current motif discovery algorithms. Nucleic Acids Res. 2005; 33:4899–913.
Medina-Rivera A, Abreu-Goodger C, Thomas-Chollier M, Salgado H, Collado-Vides J, van Helden J. Theoretical and empirical quality assessment of transcription factor-binding motifs. Nucleic Acids Res. 2011; 39:808–24.
Weirauch MT, Cote AG, Norel R, Annala M, Zhao Y, Riley TR, Saez-Rodriguez J, Cokelaer T, Vedenko A, Talukder S, DREAM5 Consortium, Bussemaker HJ, Morris QD, Bulyk ML, Stolovitzky G, Hughes TR. Evaluation of methods for modeling transcription factor sequence specificity. Nat Biotechnol. 2013; 31:126–34.
Pavesi G, Mereghetti P, Mauri G, Pesole G. Weeder Web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res. 2004; 32:199–203.
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74.
Montgomery SB, Griffith OL, Sleumer MC, Bergman CM, Bilenky M, Pleasance ED, Prychyna Y, Zhang X, Jones SJM. ORegAnno: an open access database and curation system for literature-derived promoters, transcription factor binding sites and regulatory variation. Bioinformatics. 2006; 22:637–40.
Farnham PJ. Insights from genomic profiling of transcription factors. Nat Rev Genet. 2009; 10:605–16.
Cline MS, Karchin R. Using bioinformatics to predict the functional impact of SNVs. Bioinformatics. 2011; 27:441–8.
Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A. BioMart — biological queries made easy. BMC Genomics. 2009; 10:22–34.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:80.
Durinck S, Moreau Y, Kasprzyk A, Davis S, de Moor B, Brazma A, Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005; 21:3439–440.
Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009; 4:1184–91.
Sand O, Valéry Turatsinze J, van Helden J. Evaluating the prediction of cis-acting regulatory elements in genome sequences In: Frishman D, Valencia A, editors. Modern Genome Annotation. New York: Springer: 2008. p. 55–90.
Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000; 16:276–7.
Bardet AF, He Q, Zeitlinger J, Stark A. A computational pipeline for comparative ChIP-Seq analyses. Nat Protoc. 2012; 7:45–61.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:25–35.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–9.
Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in ChIP-Seq peak detection. PLoS ONE. 2010; 5:11471.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:137–46.
Spyrou C, Stark R, Lynch A, Tavaré S. BayesPeak: Bayesian analysis of ChIP-Seq data. BMC Bioinformatics. 2009; 10:299–316.
Cairns J, Spyrou C, Stark R, Smith ML, Lynch AG, Tavaré S. BayesPeak — an R package for analysing ChIP-Seq data. Bioinformatics. 2011; 27:713–4.
Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, Green MR. ChIPpeakAnno: a Bioconductor package to annotate ChIP-Seq and ChIP-chip data. BMC Bioinformatics. 2010; 11:237–47.
Gorski JJ, Savage KI, Mulligan JM, McDade SS, Blayney JK, Ge Z, Harkin DP. Profiling of the BRCA1 transcriptome through microarray and ChIP-chip analysis. Nucleic Acids Res. 2011; 39:9536–48.
Bailey TL, Noble WS. Searching for statistically significant regulatory modules. Bioinformatics. 2003; 19:16–25.
Lin T, Ray P, Sandve GK, Uguroglu S, Xing EP. BayCis: a Bayesian hierarchical HMM for cis-regulatory module decoding in metazoan genomes In: Vingron ML, Wong L, editors. Research in Computational Molecular Biology: Proceedings of the 12th Annual International Conference on Research in Computational Molecular Biology (RECOMB 2008); Lecture Notes in Computer Science, vol. 4955. Berlin, Heidelberg: Springer: 2008. p. 66–81.
Frith MC, Hansen U, Weng Z. Detection of cis-element clusters in higher eukaryotic DNA. Bioinformatics. 2001; 17:878–89.
Frith MC, Li MC, Weng Z. Cluster-Buster: Finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003; 31:3666–8.
Frith MC, Spouge JL, Hansen U, Weng Z. Statistical significance of clusters of motifs represented by position specific scoring matrices in nucleotide sequences. Nucleic Acids Res. 2002; 30:3214–24.
Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–8.
Frith MC, Fu Y, Yu L, Chen JF, Hansen U, Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004; 32:1372–81.
Turatsinze JV, Thomas-Chollier M, Defrance M, van Helden J. Using RSAT to scan genome sequences for transcription factor binding sites and cis-regulatory modules. Nat Protoc. 2008; 3:1578–88.
Beckstette M, Homann R, Giegerich R, Kurtz S. Fast index based algorithms and software for matching position specific scoring matrices. BMC Bioinformatics. 2006; 7:389–414.
Zhou Q, Liu JS. Modeling within-motif dependence for transcription factor binding site predictions. Bioinformatics. 2004; 20:909.
Barash Y, Elidan G, Friedman N, Kaplan T. Modeling dependencies in protein-DNA binding sites In: Vingron ML, Istrail S, Pevzner P, Waterman M, editors. Proceedings of the Seventh Annual International Conference on Research in Computational Molecular Biology (RECOMB 2003). New York: Association for Computational Machinery (ACM): 2003. p. 28–37.
King OD, Roth FP. A non parametric model for transcription factor binding sites. Nucleic Acids Res. 2003; 31:116–24.
Sharon E, Lubliner S, Segal E. A feature-based approach to modeling protein–DNA interactions. PLoS Comput Biol. 2008; 4:1000154.
Ellrott K, Yang C, Sladek FM, Jiang T. Identifying transcription factor binding sites through Markov chain optimization. Bioinformatics. 2002; 18:100–9.
Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997; 268:78–94.
Thijs G, Lescot M, Marchal K, Rombauts S, de Moor B, Rouze P, Moreau Y. A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics. 2001; 17:1113–22.
Mathelier A, Wasserman WW. The next generation of transcription factor binding site prediction. PLoS Comput Biol. 2013; 9:1003214.
Kulakovskiy IV, Levitsky V, Oshchepkov D, Bryzgalov L, Vorontsov IE, Makeev VJ. From binding motifs in ChIP-Seq data to improved models of transcription factor binding sites. J Bioinform Comput Biol. 2013; 11:1340004.
Fazius E, Shelest V, Shelest E. SiTaR: a novel tool for transcription factor binding site prediction. Bioinformatics. 2011; 27:2806–11.
Graur D, Zheng Y, Price N, Azevedo RBR, Zufall RA, Elhaik E. On the immortality of television sets: ‘function’ in the human genome according to the evolution-free gospel of ENCODE. Genome Biol Evol. 2013; 5:578–90.
Acknowledgements
None.
Funding
NJ thanks the UCL Impact Studentship scheme for funding. The funding body had no role in the design of the study, collection, analysis, and interpretation of data or in writing the manuscript.
Availability of data and materials
The data sets supporting the results of this article are included within the article and its additional files, or from referenced sources.
Authors’ contributions
NJ performed the analyses and produced an initial draft of the paper. An evaluation of online tools was performed by DU who also contributed to the manuscript. ACRM directed the project and completed the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1
The PAZAR Reference Dataset. Entries from PAZAR, with the numbers of each TFBS that they contain. The spreadsheet also indicates whether the TFBSs are found in JASPAR and/or the ENCODE-ChIP-Seq data. (XLS 54 kb)
Additional file 2
Evaluation of search tools using JASPAR.2010 PWMs. Sensitivity, Positive predictive value, Geometric accuracy and False positive rate for the 14 TFBSs that are found in the PAZAR-J dataset. Separate sheets are provided for each of the search tools. (XLS 31.5 kb)
Additional file 3
Motif Discovery Tool Performance. Complete results for individual PWMs generated using different motif discovery tools and scanned against PAZAR-E data using the FIMO motif scanning tool. (XLS 14 kb)
Additional file 4
Sequence Logos. Sequence logos for the first PWM generated for the 12 TFBSs using each of the four motif discovery tools. (PDF 29.5 kb)
Additional file 5
Evaluation of search tools using PWMs generated from the ENCODE-ChIP-Seq data using rGADEM. Sensitivity, Positive predictive value, Geometric accuracy and False positive rate for the 12 TFBSs that are found in the PAZAR-E dataset. Separate sheets are provided for each of the search tools. (XLS 29.5 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jayaram, N., Usvyat, D. & R. Martin, A.C. Evaluating tools for transcription factor binding site prediction. BMC Bioinformatics 17, 547 (2016). https://doi.org/10.1186/s12859-016-1298-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-016-1298-9