
Abstract
Background
Body weight (BW) is an economically important trait in the broiler (meat-type chickens) industry. Under the assumption of polygenicity, a “large” number of genes with “small” effects is expected to control BW. To detect such effects, a large sample size is required in genome-wide association studies (GWAS). Our objective was to conduct a GWAS for BW measured at 35 days of age with a large sample size.
Methods
The GWAS included 137,343 broilers spanning 15 pedigree generations and 392,295 imputed single nucleotide polymorphisms (SNPs). A false discovery rate of 1% was adopted to account for multiple testing when declaring significant SNPs. A Bayesian ridge regression model was implemented, using AlphaBayes, to estimate the contribution to the total genetic variance of each region harbouring significant SNPs (1 Mb up/downstream) and the combined regions harbouring non-significant SNPs.
Results
GWAS revealed 25 genomic regions harbouring 96 significant SNPs on 13 Gallus gallus autosomes (GGA1 to 4, 8, 10 to 15, 19 and 27), with the strongest associations on GGA4 at 65.67–66.31 Mb (Galgal4 assembly). The association of these regions points to several strong candidate genes including: (i) growth factors (GGA1, 4, 8, 13 and 14); (ii) leptin receptor overlapping transcript (LEPROT)/leptin receptor (LEPR) locus (GGA8), and the STAT3/STAT5B locus (GGA27), in connection with the JAK/STAT signalling pathway; (iii) T-box gene (TBX3/TBX5) on GGA15 and CHST11 (GGA1), which are both related to heart/skeleton development); and (iv) PLAG1 (GGA2). Combined together, these 25 genomic regions explained ~ 30% of the total genetic variance. The region harbouring significant SNPs that explained the largest portion of the total genetic variance (4.37%) was on GGA4 (~ 65.67–66.31 Mb).
Conclusions
To the best of our knowledge, this is the largest GWAS that has been conducted for BW in chicken to date. In spite of the identified regions, which showed a strong association with BW, the high proportion of genetic variance attributed to regions harbouring non-significant SNPs supports the hypothesis that the genetic architecture of BW35 is polygenic and complex. Our results also suggest that a large sample size will be required for future GWAS of BW35.
Similar content being viewed by others
Background
Poultry meat represents a major component of human nutrition [1]. At the beginning of 2020, the production and consumption of poultry meat surpassed those of pork and it is expected that in the next decade, poultry meat will account for nearly half of the additional produced meat [1, 2]. Body weight (BW) is one of the most economically important traits in the broiler industry. Traditional broiler breeding programs have achieved an increase in meat production efficiency of ~ 3.3% per year and selection on body weight has contributed to this result [3]. However, in spite of the importance of this trait, relatively little is known about the genetic variants that underlie the variance observed in body weight.
Knowledge of the genetic variants that underlie the variance observed in traits can amplify the breeding efficiency. For example, accuracy of genomic prediction can be increased by using markers that are strongly linked to the causative loci in genomic prediction models. Furthermore, information on the genetic architecture that underlies growth in meat type poultry will help to unravel the genes and pathways that are involved and enhance our understanding on such complex developmental processes.
Quantitative trait loci (QTL) mapping and genome-wide association studies (GWAS) have been used to improve BW in chicken [4,5,6,7,8,9]. However, although significant associations have been detected, their practical value to breeding programs is limited. Typically, the associations were not finely mapped and encompassed broad chromosome regions. Furthermore, the populations used were often F2 or advanced inter-crosses between lines of chicken that have been selected for egg laying, which are relatively slow growing, and fast growing broiler lines [4, 10, 11]. Relatively few publications on GWAS of body weight are available for commercially relevant lines [9, 12, 13].
Moreover, a large part of the genetic variance might be due to rare variants, or variants that are highly correlated/linked with other variants [14]. If this is the case, then a GWAS with a large sample size is required to detect those variants [15,16,17,18]. There is theoretical [19] and empirical evidence [16, 20, 21] that the power of GWAS increases as the size of the dataset increases. For example, a series of studies with datasets of increasing size for human height discovered 180 significant associations with a dataset of 183,727 individuals [15], 697 significant associations with a dataset of 253,288 individuals [16], and recently, another 83 new significant associations not previously detected were identified with a dataset of 711,428 individuals [21]. Analogous results are reported for studies on type 2 diabetes [22] and Crohn’s disease [20] in humans. If BW is a complex polygenic trait [23, 24], a large number of small-effect variants might regulate its expression. Hence, a large GWAS (in terms of sample size and number of markers analysed) is required to discover such variants.
The routine use of genomic selection in broiler breeding makes large GWAS possible. As part of the routine implementation of genomic selection over the past decade, Aviagen has accumulated both single nucleotide polymorphisms (SNP) array genotype and phenotype data for BW on 157,674 individuals from one of its lines.
Our objective was to conduct a GWAS, with a large sample size, for BW measured at 35 days of age (BW35) in broilers, which is a typical age at which broilers are slaughtered for meat production. After editing routinely collected data from a commercial broiler line, we analysed a dataset consisting of 137,343 broilers with phenotypes and 595,299 imputed SNPs.
Methods
Data
In total, we used 157,674 broilers spread across 15 generations of a pedigree for which BW35 and SNP array genotype data were collected as part of the routine commercial broiler breeding program (Aviagen Ltd, Newbridge, UK). The line used in this study was a female line (maternal side). The birds were genotyped with SNP arrays of different densities: 600 k SNPs for 1690 birds, 50 k SNPs for 59,773 birds, 42 k SNPs for 1507 birds, 3 k SNPs for 72,221 birds and 384 SNPs for 2152 birds. The development of these arrays is described in detail in [25]. Of these 632,439 SNPs, 52,408 are proprietary to Aviagen. We included all the SNPs in the analysis but do not show the base pair positions of the proprietary significant SNPs in our results, which represent 18 of the 96 significant SNPs). To unify the data from the different arrays and reach the highest density of 600 k, we imputed the genotypes of all broilers’ to the 600 k Affymetrix Axiom chip with the AlphaImpute software v1.9 [26, 27]. Broilers with more than 10% missing SNP genotypes were excluded from the analysis. Quality control of the SNPs was carried out using the PLINK v1.07 software [28]; SNPs with a call rate higher than 0.95 and a minor allele frequency higher than 0.01, that showed no extreme deviation from the Hardy–Weinberg proportions (P < 0.000001), and that were located on the Gallus gallus (GGA) autosomes 1 to 28 (except GGA16) were retained. After quality control, 137,343 birds and 392,255 SNPs remained for the analysis. The Galgal4 assembly in the Ensembl Genome Browser (version 85) was used to map the SNP positions on the genome (www.ensembl.org).
Statistical analysis
Pedigree genetic analysis
Variance component and heritability estimates were based on a pedigree-based model using ASReml.v3 [29]:
where \(\mathbf{y}\) is a vector of BW35 records; \(\mathbf{b}\) is a vector of fixed non-genetic effects (sex, mating group (the average genetic level of the parents) with 325 levels, the pen effects and hatch week with 381 levels); \(\mathbf{a}\) is a vector of random additive genetic effects; \(\mathbf{e}\) is a vector of random residuals; \(\mathbf{X}\) and \(\mathbf{Z}\) are design matrices linking phenotypes to effects. The model assumptions were \(\mathbf{a}\sim N(\mathbf{0},\mathbf{A}{\upsigma }_{\mathrm{a}}^{2})\) and \(\mathbf{e}\sim N(\mathbf{0},\mathbf{I}{\upsigma }_{\mathrm{e}}^{2})\), where \(\mathbf{A}\) is the pedigree relationship matrix, \(\mathbf{I}\) is an identity matrix and \({\upsigma }_{\mathrm{a}}^{2}\) and \({\upsigma }_{\mathrm{e}}^{2}\) are the additive genetic and residual variances, respectively. Heritability was estimated as the ratio of \({\upsigma }_{\mathrm{a}}^{2}\) to the total phenotypic variance (\({\upsigma }_{\mathrm{a}}^{2}+{\upsigma }_{\mathrm{e}}^{2}\)).
Genome-wide association study
The GWAS was conducted by single SNP regression while simultaneously correcting for the background polygenic effect using the GEMMA software [30]:
where \(\mathbf{y}\) is a vector of BW35 records pre-corrected for the non-genetic effects of sex, mating group, pen, and hatch; μ is intercept, \(\mathbf{w}\) is a column vector of genotypes for the SNP of interest with the corresponding allele substitution effect \(\mathbf{b}\); \(\mathbf{g}\) is a vector of random additive genomic (polygenic) effects; and \(\mathbf{e}\) is a vector of random residuals. The model assumptions were \(\mathbf{g}\sim N(\mathbf{0},\mathbf{G}{\upsigma }_{\mathrm{g}}^{2})\) and \(\mathbf{e}\sim N(\mathbf{0},\mathbf{I}{\upsigma }_{\mathrm{e}}^{2})\), where \(\mathbf{G}\) is the genomic relationship matrix calculated following the first method of VanRaden [31] and \({\upsigma }_{\mathrm{g}}^{2}\) and \({\upsigma }_{\mathrm{e}}^{2}\) are, respectively, the additive genomic and residual variances. Matrix \(\mathbf{G}\) was constructed and eigen decomposed using an in-house Python script. The eigenvalues and eigenvectors were subsequently used in GEMMA via flag -d and -u, respectively. A false discovery rate (FDR; Benjamini and Hochberg) of 1% was adopted to account for multiple testing when declaring significant SNPs [32]. Manhattan and quantile–quantile (Q–Q) plots of the GWAS results were drawn in R [33] with the qqman package [34]. Annotation of all the significant SNPs was performed with the variant effect predictor (https://www.ensembl.org/Tools/VEP) program using the Ensembl database and the Galgal4 assembly. Moreover, genes located 1 Mb up/downstream of the top SNP in each genomic region that contained significant SNPs were annotated using the BioMart tool of the Ensembl database and the Galgal4 assembly (http://www.ensembl.org/biomart/martview/).
Genetic variance partitioning by genomic region
Based on the GWAS results, the genome was partitioned into different regions that harboured significant and non-significant SNPs. Regions that contained significant SNPs were defined by considering the region 1 Mb upstream and 1 Mb downstream from the SNP with the highest p-value in each region. Due to closely located GWAS signals on GGA13 (13a and 13b) and GGA14 (14a and 14b), the two regions on each of these chromosomes were merged. To reduce the computational cost, we used all the significant SNPs from the 600 k Affymetrix Axiom chip and among the non-significant SNPs only those that overlap between the 50 k and 600 k Affymetrix Axiom chips. To estimate the variance explained by each region, a Bayesian ridge regression model was implemented using AlphaBayes [35] and the same inputs as for the GWAS, but analysing all the SNPs simultaneously. Posterior samples for SNP effects for each region were obtained from 50,000 Markov-chain Monte Carlo (MCMC) iterations with a burn-in period of 10,000 iterations. For each region and each iteration, breeding values were calculated from SNP effects and SNP genotypes, the variance of these regional breeding values was calculated and divided by the variance of the breeding values for the whole genome to estimate the proportion of the (additive) genetic variance explained per genomic region, accounting for linkage-disequilibrium within and between regions [36].
Results
Descriptive statistics and pedigree genetic parameters
The summary statistics and variance components of the raw data are presented in Fig. 1. The average BW35 in the full dataset was 1840 g, and ranged from 1080 to 2740 g, while the estimated pedigree heritability was 0.44 (0.01).

Descriptive statistics, additive genetic variance (\({\sigma }_{a}^{2}\)), and heritability (\({h}^{2}\)) estimates from the pedigree model
Genome-wide associations
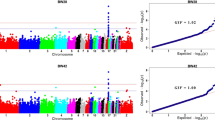
A summary of the results of the GWAS is in Table 1 and Figs. 2, 3 and 4. In total, 96 SNPs were significant at 1% FDR and distributed across 25 genomic regions on 13 chromosomes (GGA1 to 4, 8, 10 to 15, 19, and 27). Of these, 45 SNPs showed a very high significance and were located on GGA4 at ~ 65.86 Mb (Figs. 2 and 3). Details on these 96 significant SNPs including their chromosome and base pair location, their minor allele frequency and their effects and variances are in Additional file 1: Table S1. Of these 96 significant SNPs, 18 are proprietary to Aviagen and thus their base pair location is not shown in Additional file 1: Table S1.

Manhattan plot of P-values for the genome-wide association study. A 1% false discovery rate was adopted to declare significance

Manhattan plots of P-values for the genome-wide association study on Gallus gallus autosome 4. A 1% false discovery rate was adopted to declare significance

Manhattan plots of P-values for the genome-wide association studies on Gallus gallus autosomes (GGA) 1 to 3, 8, 10 to 15, 19 and 27. A 1% false discovery rate was adopted to declare significance
On GGA1, four regions harboured significant SNPs, denoted 1a to 1d, at positions ~ 54.68 (1a), ~ 134.49 (1b), ~ 184.46 (1c) and ~ 193.81 Mb (1d). On GGA2, two regions harboured significant SNPs, denoted 2a and 2b, at ~ 103.15 (2a), ~ 111.28 Mb (2b) (Fig. 4). On GGA4, the most significantly associated SNP was located at ~ 65.86 Mb (\(P=6.47\times {10}^{-42}\)). The large region between ~ 65.67 and ~ 66.31 Mb (4e) contained 45 significant SNPs. Four more regions were significant on GGA4, at ~ 44.84 (4a), ~ 49.80 (4b), ~ 52.73 (4c) and ~ 59.55–63.00 Mb (4d) (Fig. 3). On GGA8, three significant SNPs were detected at ~ 23.00 (8a), ~ 27.23 (8b) and ~ 28.20 Mb (8c). At the tail of GGA13, two neighbouring regions were detected at ~ 16.33–16.48 Mb (13a) and ~ 16.71–16.87 Mb (13b). On GGA14, two closely located regions contained nine significant SNPs at ~ 13.14–13.94 Mb (14a) and ~ 14.50–15.06 Mb (14b). On GGA15, two SNPs were detected at ~ 11.61 (15a) and ~ 12.34 Mb (15b). On GGA27, three SNPs were significant at ~ 4.11–4.96 Mb. Moreover, five chromosomes had only one significant SNP: GGA3 (~ 17.00 Mb), GGA10 (~ 1.87 Mb), GGA11 (~ 16.45 Mb), GGA12 (~ 1.84 Mb) and GGA19 (~ 8.62 Mb) (Fig. 4).
Partitioning of the genetic variance by genomic region
The proportion of the genetic variance over the total genetic variance for each genomic region that harboured significant SNPs is in Table 2. Combined together, the 25 genomic regions that harboured the 96 significant SNPs explained ~ 30% of the total genetic variance. Region 4e (GGA4 at ~ 65.67–66.31 Mb) that contained 45 significant SNPs explained the highest portion of the total genetic variance (4.37%). Regions with significant SNPs that explained the next largest amount of the total genetic variance were on GGA13 and GGA14, which each explained ~ 2.5%. All the other regions with significant SNPs each explained less than 2% of the total genetic variance. GGA4 explained more of the total genetic variance than any other chromosome. Taken together, the regions that harboured significant SNPs on GGA4 explained ~ 8.6% of the total genetic variance.
Discussion
Previous studies have shown that the genomic architecture of BW is age- and population-dependent [8, 9, 37]. We focused on BW measured at 35 days of age, which is a typical age at which commercial broilers are slaughtered. In total, we found 96 significant SNPs with a 1% FDR that were located in 25 genomic regions across 13 chromosomes and explained ~ 30% of the genetic variance. We identified several candidate genes that might affect BW35 in broilers, and encode e.g. growth factors and the leptin receptor, and are involved in the JAK/STAT signalling pathway (Table 3). Furthermore, inspection of the Q–Q plot (Fig. 2) provided additional evidence of true associations, with an extreme departure observed at the tail of the distribution. We have divided the Discussion section into seven sub-sections: (i) summary of the QTL already known for BW35, (ii) growth factors, (iii) the leptin receptor overlapping transcript (LEPROT)/leptin receptor (LEPR) locus, (iv) the JAK/STAT signalling pathway, (v) the T-box genes (TBX3/TBX5), (vi) other candidate genes for BW35, and (vii) implications for breeding programs.
Summary of the QTL already known for BW35
A search on the animal QTL database (QTLdb; http://www.animalgenome.org/QTLdb) revealed a number of QTL for BW35 on GGA1 to 5, 7, 10–11, 15, 18, 20 and 27 (https://www.animalgenome.org/cgi-bin/QTLdb/GG/traitmap?trait_ID=2151). In particular, there is strong evidence in the literature for QTL associated with BW35 on GGA1 to 4.
Previous studies on F2 crosses between broiler and layer lines have identified QTL that are associated with a variety of carcass characteristics, such as BW measured at day 35, 41 and 63 [6, 7], and carcass and breast muscle yield [38, 39]. Nevertheless, such QTL studies detected only large chromosomal regions. The most significant chromosomal regions were mainly located on GGA1 to 4, but also GGA7 to 9, and GGA13, and on the Z chromosome [4, 5, 39].
Several GWAS have been conducted in broilers for a variety of carcass and growth traits, using commercial lines [9, 13], birds from experimental stations [7, 37, 40], or field data [41], and a variety of SNP array densities that ranged from ~ 44,000 [7,8,9] to 470,486 [41]. In spite of this, previous GWAS that scanned the entire genome, were still limited in terms of statistical power because the size of the samples was usually only a few hundreds of birds.
Growth factor pathways
Growth factors, such as the transforming growth factor-β (TGF-β) and the insulin-like growth factor-1 (IGF1) are known to be key regulators of several traits related to body composition, growth, and development in chicken [42, 43]. Our analysis detected seven regions that include genes coding for growth factors, i.e. regions 1a and 1b, 4a, 4c, 4d, 13b, 14a (Table 3). More precisely, the thioredoxin reductase 1 gene (TXNRD1; at ~ 54.74–54.77 Mb) is located in region 1a. The second most significant SNP detected in this region is located within TXNRD1 (54,756,840 bp). Although TXNRD1 is not considered as a growth factor, studies on salivary adenoid cystic carcinoma [44] have found a synergistic action between TXNRD1 and TGF-β. The insulin like growth factor 1 (IGF1; at ~ 55.43–55.48 Mb) gene is located within a 1-Mb region from TXNRD1. IGF1 has a major role in the body size of dogs [45, 46], with a single allele causing a small size. It also affects body size in mice [47] and height in humans [48, 49]. In broilers, increased IGF1 levels have been related with increased BW [43], growth of muscle [50], and IGF1 levels have been shown to differ between lines that are divergently selected for growth [51]. In a recent GWAS on a F2 chicken population, TXNRD1 and IGF1 have been associated with BW35 and BW41, respectively [52]. Moreover, the IGFBP4 (insulin like growth factor binding protein 4) gene has been identified as a candidate gene for broiler BW in a study that analysed a subset of the population used in our work [13]. This gene is in close proximity (~ 0.5 Mb) to the signal we detected on GGA27. Another insulin-like growth factor modulating protein, namely that encoded by the IGFALS (insulin-like growth factor binding protein, acid labile subunit; ~ 13.2 Mb) gene, is located ~ 8 kb downstream of the region 14a. In addition, on GGA1, near the region 1b (~ 134.49 Mb), we identified the TGFBRAP1 (transforming growth factor beta receptor associated protein 1; ~ 134.45–134.48 Mb) gene.
On GGA4, the region 4a (~ 44.84 Mb) contains two genes with growth factor activity: FGF5 (fibroblast growth factor 5; ~ 44.74 Mb) and BMP3 (bone morphogenetic protein 3; ~ 44.86 Mb). The FGF20 gene, which is a key regulator of skin development in chicken [53], is also located on the same chromosome (region 4d; ~ 62.87 Mb), and the FGF2 gene at ~ 53.00 Mb is close to the 4c region (~ 52.73 Mb). We detected another component of the growth factor pathway on GGA8, with the SNP in region 8a being located within the intronic region of the EPS15 (epidermal growth factor receptor pathway substrate 15; ~ 22.96–23.0 Mb) gene. The region 13b is located between NDF1P1 (Nedd4 family interacting protein 1; ~ 16.65–16.66 Mb) and another growth factor, FGF1 (fibroblast growth factor 1; ~ 16.72–16.73 Mb).
The leptin receptor overlapping transcript (LEPROT) and leptin receptor (LEPR)
Leptin (LEP) is a well-known hormone that is strongly related to appetite, through regulation of the brain satiety centres [54]. Its effect on reducing weight results from its interaction with another protein, the leptin receptor (LEPR) [55,56,57]. Several studies have investigated the association of the LEP and LEPR genes with obesity [58, 59], feed intake [60, 61], growth and fat traits [62, 63], and their cardio-metabolic implications [64, 65] in a variety of species. However, the results are controversial, especially in the chicken literature which currently reports evidence against the leptin system being involved in body weight control in birds [59, 66,67,68]. In our GWAS, a significant association in region 8b was located within the 3ʹ untranslated region of the LEPROT (leptin receptor overlapping transcript; ~ 27.22–27.23 Mb) gene. Moreover, the leptin receptor (LEPR; ~ 27.24–27.27 Mb) and the JAK1 (Janus kinase 1; ~ 27.10–27.13 Mb) genes are located near this genomic region. It should be noted that LEPROT is not in any way homologous to the leptin receptor gene, but its expression has been associated with muscle development in turkey [69] and it is thought to regulate the expression of cytokine receptor, growth hormone receptor (GHR) and LEPR genes.
The JAK/STAT signalling pathway
Although leptin activation of the JAK/STAT pathway in birds may not be important, as it is in other animals [70], this JAK/STAT pathway has a role in the mediation of many cytokine signals, such as those through GHR, and therefore for the growth of poultry. The JAK/STAT pathway is related to the generation of spermatogonial stem cells in chicken [71]. The association signal on GGA27 at ~ 4.96 Mb) (a gene dense region) was located near the STAT3 (signal transducer and activator of transcription 3; ~ 4.90–4.91 Mb) gene, and more precisely is within an intron of the ATP6V0A1 (ATPase, H+ transporting, lysosomal V0 subunit a1; ~ 4.93–4.96 Mb) gene (Table 3). The association between the STAT3/STAT5B locus and BW in chicken confirms the findings of [13]. We also detected several other genes related to reproduction. The association signal in region 4c (~ 52.73 Mb) is located near the spermatogenesis associated 5 (SPATA5; ~ 52.76–52.98 Mb) gene. The ADAD1 (adenosine deaminase domain containing 1; ~ 53.3 Mb) and TRPC3 (short transient receptor potential channel 3; ~ 53.15 Mb) genes are located in the same region, and are involved in spermatid development and single fertilization. Interestingly, another spermatogenesis linked gene, SPATA18 (~ 65.75–65.77 Mb), is located on GGA4 in the region with significant associations in our study (4e; ~ 65.67–66.31 Mb), that harbors two significant SNPs (at ~ 65.75 Mb) downstream of the gene (Table 3).
Apart from the effect of LEPR on the JAK/STAT pathway, Hou and Luo [64] have suggested a relationship between the leptin, JAK/STAT and mitogen-activated protein kinases (MAPK) signal pathways with an effect on cardiovascular diseases. Interestingly, the association on GGA12 was within an intron of the MAPKAPK3 (mitogen-activated protein kinase-activated protein kinase 3; ~ 1.84–1.88 Mb) gene. The results of an experimental study in pigs that compared the expression level of MAPKAPK3 in mini and large-type Diannan small-ear pigs, indicated that MAPKAPK3 might have an important role in growth and development [72]. Interestingly, Tarsani et al. [13] suggested LEMD2 (LEM domain containing 2), located on GGA26, as a strong candidate gene for BW and it is considered to have an important role during embryonic development in mice by regulating the MAPK signalling pathway [73].
T-box genes (TBX5 and TBX3)
Several studies have implicated the effect of the T-box genes (TBX4 and TBX5) in the development of the chicken limb, heart and embryo [74,75,76,77]. The significant SNP on GGA15 was included in the intron of TBX5 (T-box 5; ~ 12.31–12.35 Mb). The TBX3 gene is adjacent to TBX5. TBX4 is located on GGA19 at ~ 7.59–7.61 Mb but the region that we detected on this chromosome is located further down at ~ 6.62 Mb, in the intron of the SYNRG (synergin, gamma) gene. In the same region on GGA19 (within 1 Mb) are found the TBX2 (~ 7.63–7.64 Mb) and HNF1B (HNF1 homeobox B; ~ 8.64–8.66 Mb) genes. Interestingly, Tarsani and colleagues [13] have recently reported several candidate genes for BW35 in broilers, among which the TBX21 gene and several members of the homeobox family (HOXB1-9 and HOXB13), and Moreira et al. [52] have shown an association of HOXB2,4,7,9 and HOXB13 with BW35 in an F2 cross. However, it should be noted that all these genes are located on GGA27 at more than one Mb from our top SNP on that chromosome. In our analysis, another limb morphogenesis gene was detected in region 1a, with the SNP at 54,681,614 bp being within an intron of the CHST11 (carbohydrate (chondroitin 4) sulfotransferase 11; ~ 54.54–54.74 Mb) gene (Table 3), which has been suggested to be involved in the elongation of limb buds and bone formation [78]. CHST11 has also been recently reported as a candidate gene for BW35 [52].
The zinc finger protein PLAG1
PLAG1, together with the LCORL-NCAPG locus, has been associated with body weight and height in a variety of species (human, cattle, horses, pigs and dogs) [79]. In cattle, a QTL for growth and development is located on bovine chromosome 14 (at ~ 25 Mb) and a cluster of four genes on the same chromosome, namely, PLAG1, LYN (v-yes-1 Yamaguchi sarcoma viral related oncogene homolog), RPS20 (ribosomal protein S20), and CHCHD7 (coiled-coil-helix-coiled-coil-helix domain containing 7) [80,81,82,83,84], is thought to be involved in this trait. Recently, a mutation in PLAG1 has been reported to have a major contribution to stature in modern cattle [85]. More importantly, PLAG1 is also known to affect body weight and milk characteristics [86] and to regulate several growth factors, such as IGF2 [87, 88]. The large region 2b contains the PLAG1, LYN, and RPS20 genes. Our top SNP on GGA2 was located 3.5 kb downstream of the IMPAD1 (inositol monophosphatase domain containing 1) gene, but the second most significant SNP in the region was a synonymous variant in PLAG1 (Table 3). Altogether, these findings mark PLAG1 as a very good candidate for BW35 in broilers.
Other candidate genes for BW35
On GGA4 (peak at ~ 65.86 Mb)
In line with previous studies [8, 89, 90], we found that GGA4 contains genes that play a role in body weight in chicken. Among these, the CCKAR (cholecystokinin A receptor) gene is located at ~ 72.8 Mb on GGA4. Decreased expression of this gene is associated with increased BW and growth in chicken and it has been suggested that modern chicken breeds might have been selected during the early domestication process for the high-growth haplotype [91]. Other studies have reported the LCORL-NCAPG locus, at ~ 74.0 Mb [12, 89], which explains part of the variance in stature in many species [92,93,94,95,96] and is associated with carcass traits in beef cattle [97]. However, the region identified in our study is located at ~ 65.86 Mb, which is quite far away (~ 7 Mb) from CCKAR and even further away from the LCORL-NCAPG locus. Whereas a long-range enhancer might be involved, a more conservative hypothesis indicates another gene. The top SNP in the region 4e detected here was ~ 400 bp upstream of the CWH43 (cell wall biogenesis 43 C-terminal homolog) gene, which encodes the PGAP2-interacting protein. This SNP also had the highest positive effect, indicating an effect that increases BW35 (see Additional file 1: Table S1). Eight other SNPs in this region 4e are located in CWH43, among which four are in introns and two are synonymous variants. Forty-one SNPs in this region 4e were mapped to eight additional genes, some of these being located in their introns (Table 3), namely: SPATA18, SGCB (sarcoglycan beta), DCUN1D4 (DCN1, defective in cullin neddylation 1, domain containing 4), OCIAD1 (OCIA domain containing 1), SLAIN2 (SLAIN motif family member 2), TEC (tec protein tyrosine kinase), NFXL1 (nuclear transcription factor, X-box binding like 1), and CORIN (corin, serine peptidase). In the same region, five more genes are present, namely FRYL (FRY like transcription coactivator), SLC10A4 (solute carrier family 10 member 4), TXK (tyrosine kinase), NIPAL1 (NIPA like domain containing 1) and CNGA1 (cyclic nucleotide gated channel alpha 1). Among all these genes, SLAIN2 was recently reported as a strong candidate gene regulating BW in broilers [13]. Moreover, several members of the general solute carrier gene family have been associated with BW35 and BW41 [52].
Concerning the second region 4b on GGA4, we identified the group-specific component (GC) gene, which is located ~ 19 kb from the region (Table 3). GC (a vitamin D-binding protein) belongs to the general albumin family, involved in vitamin transportation, and vitamin D, lipids and lipoproteins metabolism and is expressed in all vertebrates [98]. In humans, it is one of the major determinants of the status in vitamin D, as assessed by measuring the circulating concentrations of 25 hydroxyvitamin D (25(OH)D) [99]. The role of vitamin D in the maintenance of skeletal health has been known for over a century but there is now growing evidence that vitamin D plays an important role also in the health of non-skeletal tissues. The linkage between GC and BW35 is consistent with recent studies that found that 25(OH)D supplementation increases breast meat yield in broilers [100]. This observation highlights the importance of understanding how key vitamin D metabolism pathways regulate physiological processes relevant to production in farm animals. In dairy cattle, the same gene was recently associated with complex traits such as mastitis and milk traits [101, 102].
On GGA1 to 3
Although the important role of GGA1 was previously reported in the literature, we detected only weak associations in our study, significant at 1% FDR. The top SNP in region 1b (~ 134.49) was located within the 3ʹ untranslated region of the C2orf49 gene (~ 134.48–134.50 Mb), which is near the region containing the C2orf40 gene. The SNP in region 1c was located within the MTMR2 (myotubularin related protein 2; ~ 184.42–184.48 Mb) gene. In region 1d, several genes are mapped. The significant SNP located in region 1c was in an intron of the STIM1 gene (stromal interaction molecule 1; ~ 193.79–193.83 Mb), which is close to DGAT2 (diacylglycerol O-acyltransferase homolog 2; ~ 193.95–193.97 Mb), a gene that is related to fatty acid metabolism and associated with changes in carcass and meat quality characteristics in domestic pigeons [103].
On GGA2 two significant regions were detected, namely at 103.15 and 111.28 Mb. The closest genes in region 2a were IMPACT (impact RWD domain protein; ~ 103.03–103.05) and HRH4 (histamine receptor H4; ~ 103.07) genes. On GGA3, the significant SNP was located within the FBX028 (F-box protein 28; ~ 16.95–16.97 Mb) gene.
On chromosomes other than GGA1, 2, 3 and 4
We detected significant associations on nine other chromosomes (GGA8, 10, 11, 12, 13, 14, 15, 19 and 27). The association on GGA10 points to the PPCDC (phosphopantothenoylcysteine decarboxylase; ~ 1.86–1.88 Mb) gene, which is involved in the biosynthesis of coenzyme A. Coenzyme A is essential for energy production of the body. The association on GGA11 was located between the CRISPLD2 (cysteine-rich secretory protein LCCL domain containing 2; ~ 16.37–16.40 Mb) and HNF4beta (hepatic nuclear factor 4beta; ~ 16.46–14.47 Mb) genes. On GGA13, two regions were identified and because of the “two-peak” pattern in the Manhattan plot (Fig. 3), they were considered as two different regions. The top SNP in region 13a is within the SEPT8 gene (septin 8; ~ 16.31–16.40 Mb). On GGA14, in region 14b the significant SNP was located within the PDIA2 gene (protein disulfide-isomerase A2-like; ~ 14.49–14.52 Mb). Finally, of particular interest are the signals on GGA27, on which apart from the STAT3/STATB5 locus, we detected the CACNB1 (~ 4.11 Mb; calcium channel, voltage-dependent, beta 1 subunit) gene, which has been reported as a strong candidate for BW35 in broilers [13] and for which skeletal muscle-specific isoforms are reported in humans [104].
Implications for breeding programs
Combined together, the 25 genomic regions that contain 96 significant SNPs explained ~ 30% of the total genetic variance. This implies that the genetic architecture of BW35 is polygenic and complex, and therefore genomic prediction (using all available genomic data), rather than targeting specific genes via marker-assisted selection, will be more effective to improve BW35 in broilers. However, the region that contains significant SNPs and explains the largest proportion of the total genetic variance, is region 4e (~ 65.67–66.31 Mb), which explains 4.368% of the total genetic variance, and taken together the regions with significant SNPs on chromosome GGA4 explain ~ 8.6% (Table 2), which suggests that genomic prediction models that upweight regions of the genome known to harbour significant SNPs [105] may be effective. Moreover, information about the relevant genes identified in this paper could be included in the design of future SNP arrays.
Conclusions
To the best of our knowledge, this is the largest GWAS that has been conducted for BW in chicken to date. Our analysis revealed 25 genomic regions that harbour 96 significant SNPs on 13 Gallus gallus autosomes, which combined together explain ~ 30% of the total genetic variance. Although the region on GGA4 at ~ 65.67–66.31 Mb explains 4.37% of the total genetic variance, the high proportion of genetic variance attributed to regions that harbour non-significant SNPs supports the hypothesis that the genetic architecture of BW35 is polygenic and complex. The significant SNPs and associated genes identified here could be used in future experimental designs targeting specific genes and biological pathways, and in the design of future SNP arrays as well as in statistical models of genomic prediction using prior biological knowledge of genome regions known to affect the traits of interest. Our results also illustrate the importance of a large sample size for future GWAS of BW35.
References
OECD/FAO. OECD-FAO agricultural outlook 2019–2028. Rome: OECD Publishing, Paris/Food and Agriculture Organization of the United Nations. https://doi.org/10.1787/agr_outlook-2019-en. Accessed 23 Aug 2021.
United States Department of Agriculture/USDA GMA. Livestock and poultry: world markets and trade. 2020. https://downloads.usda.library.cornell.edu/usda-esmis/files/73666448x/sb397r25n/0z709c25b/livestock_poultry.pdf. Accessed 23 Aug 2021.
Hill WG. Is continued genetic improvement of livestock sustainable? Genetics. 2016;202:877–81.
Sewalem A, Morrice DM, Law A, Windsor D, Haley CS, Ikeobi CO, et al. Mapping of quantitative trait loci for body weight at three, six, and nine weeks of age in a broiler layer cross. Poult Sci. 2002;81:1775–81.
Ambo M, Moura ASAMT, Ledur MC, Pinto LFB, Baron EE, Ruy DC, et al. Quantitative trait loci for performance traits in a broiler by layer cross. Anim Genet. 2009;40:200–8.
Tercic D, Holcman A, Dovc P, Morrice DR, Burt DW, Hocking PM, et al. Identification of chromosomal regions associated with growth and carcass traits in an F(3) full sib intercross line originating from a cross of chicken lines divergently selected on body weight. Anim Genet. 2009;40:743–8.
Gu X, Feng C, Ma L, Song C, Wang Y, Da Y, et al. Genome-wide association study of body weight in chicken F2 resource population. PLoS One. 2011;6:e21872.
Liu R, Sun Y, Zhao G, Wang F, Wu D, Zheng M, et al. Genome-wide association study identifies loci and candidate genes for body composition and meat quality traits in Beijing-You chickens. PLoS One. 2013;8:e61172.
Reyer H, Hawken R, Murani E, Ponsuksili S, Wimmers K. The genetics of feed conversion efficiency traits in a commercial broiler line. Sci Rep. 2015;5:16387.
Schreiweis MA, Hester PY, Settar P, Moody DE. Identification of quantitative trait loci associated with egg quality, egg production, and body weight in an F2 resource population of chickens. Anim Genet. 2006;37:106–12.
Rubin C-J, Zody MC, Eriksson J, Meadows JRS, Sherwood E, Webster MT, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–91.
Wolc A, Arango J, Jankowski T, Dunn I, Settar P, Fulton JE, et al. Genome-wide association study for egg production and quality in layer chickens. J Anim Breed Genet. 2014;131:173–82.
Tarsani E, Kranis A, Maniatis G, Avendano S, Hager-Theodorides AL, Kominakis A. Discovery and characterization of functional modules associated with body weight in broilers. Sci Rep. 2019;9:9125.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53.
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–8.
Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–86.
Visscher PM. Sizing up human height variation. Nat Genet. 2008;40:489–90.
Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–5.
Spencer CCA, Su Z, Donnelly P, Marchini J. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5:e1000477.
Lee JC, Parkes M. Genome-wide association studies and Crohn’s disease. Brief Funct Genomics. 2011;10:71–6.
Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, et al. Rare and low-frequency coding variants alter human adult height. Nature. 2017;542:186–90.
Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–45.
Carlborg Ö, Kerje S, Schütz K, Jacobsson L, Jensen P, Andersson L. A global search reveals epistatic interaction between QTL for early growth in the chicken. Genome Res. 2003;13:413–21.
Le Rouzic A, Álvarez-Castro JM, Carlborg Ö. Dissection of the genetic architecture of body weight in chicken reveals the impact of epistasis on domestication traits. Genetics. 2008;179:1591–9.
Wolc A, Kranis A, Arango J, Settar P, Fulton JE, O’Sullivan NP, et al. Implementation of genomic selection in the poultry industry. Anim Front. 2016;6:23–31.
Hickey JM, Kinghorn BP, Tier B, van der Werf JH, Cleveland MA. A phasing and imputation method for pedigreed populations that results in a single-stage genomic evaluation. Genet Sel Evol. 2012;44:9.
Antolín R, Nettelblad C, Gorjanc G, Money D, Hickey JM. A hybrid method for the imputation of genomic data in livestock populations. Genet Sel Evol. 2017;49:30.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Gilmour AR, Gogel RBJ, Cullis BR, Thopmson R. ASReml user guide release 3.0. Hemel Hempstead: VSN International Ltd.; 2009.
Zhou X, Stephens M. Genome-wide efficient mixed model analysis for association studies. Nat Genet. 2012;44:821–4.
VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300.
R Core Team RC. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2013.
Turner SD. qqman: an R package for visualizing GWAS results using QQ and manhattan plots. bioRxiv. 2014. https://doi.org/10.1101/005165.
Hickey JM, Tier B. AlphaBayes (Beta): software for polygenic and whole genome analysis. User manual. Armidale: University of New England; 2009.
de C Lara LA, Pocrnic I, Gaynor RC, Gorjanc G. Temporal and genomic analysis of additive genetic variance in breeding programmes. bioRxiv. 2020. https://doi.org/10.1101/2020.08.29.273250.
Pértille F, Guerrero-Bosagna C, da Silva VH, Boschiero C, da Silva Nunes JdR, Ledur MC, et al. High-throughput and cost-effective chicken genotyping using next-generation sequencing. Sci Rep. 2016;6:26929.
Baron EE, Moura ASAMT, Ledur MC, Pinto LFB, Boschiero C, Ruy DC, et al. QTL for percentage of carcass and carcass parts in a broiler x layer cross. Anim Genet. 2011;42:117–24.
Ikeobi CON, Woolliams JA, Morrice DR, Law A, Windsor D, Burt DW, et al. Quantitative trait loci for meat yield and muscle distribution in a broiler layer cross. Livest Prod Sci. 2004;87:143–51.
Van Goor A, Bolek KJ, Ashwell CM, Persia ME, Rothschild MF, Schmidt CJ, et al. Identification of quantitative trait loci for body temperature, body weight, breast yield, and digestibility in an advanced intercross line of chickens under heat stress. Genet Sel Evol. 2015;47:96.
Psifidi A, Banos G, Matika O, Desta TT, Bettridge J, Hume DA, et al. Genome-wide association studies of immune, disease and production traits in indigenous chicken ecotypes. Genet Sel Evol. 2016;48:74.
Li H, Deeb N, Zhou H, Mitchell AD, Ashwell CM, Lamont SJ. Chicken quantitative trait loci for growth and body composition associated with transforming growth factor-beta genes. Poult Sci. 2003;82:347–56.
Goddard C, Wilkie RS, Dunn IC. The relationship between insulin-like growth factor-1, growth hormone, thyroid hormones and insulin in chickens selected for growth. Domest Anim Endocrinol. 1988;5:165–76.
Jiang Y, Feng X, Zheng L, Li S, Ge X, Zhang J-G, et al. Thioredoxin 1 mediates TGF-β-induced epithelial-mesenchymal transition in salivary adenoid cystic carcinoma. Oncotarget. 2015;6:25506–19.
Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, et al. A single IGF1 allele is a major determinant of small size in dogs. Science. 2007;316:112–5.
Boyko AR, Quignon P, Li L, Schoenebeck JJ, Degenhardt JD, Lohmueller KE, et al. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010;8:e1000451.
Liu J-P, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 1993;75:59–72.
Kant SG, Wit JM, Breuning MH. Genetic analysis of short stature. Horm Res. 2003;60:157–65.
Walenkamp MJE, Wit JM. Genetic disorders in the growth hormone—insulin-like growth factor-I axis. Horm Res. 2006;66:221–30.
Duclos MJ, Wilkie RS, Goddard C. Stimulation of DNA synthesis in chicken muscle satellite cells by insulin and insulin-like growth factors: evidence for exclusive mediation by a type-I insulin-like growth factor receptor. J Endocrinol. 1991;128:35-NP.
Guernec A, Berri C, Chevalier B, Wacrenier-Cere N, Bihan-Duval EL, Duclos MJ. Muscle development, insulin-like growth factor-I and myostatin mRNA levels in chickens selected for increased breast muscle yield. Growth Horm IGF Res. 2003;13:8–18.
Moreira GCM, Poleti MD, Pértille F, Boschiero C, Cesar ASM, Godoy TF, et al. Unraveling genomic associations with feed efficiency and body weight traits in chickens through an integrative approach. BMC Genet. 2019;20:83.
Wells KL, Hadad Y, Ben-Avraham D, Hillel J, Cahaner A, Headon DJ. Genome-wide SNP scan of pooled DNA reveals nonsense mutation in FGF20 in the scaleless line of featherless chickens. BMC Genomics. 2012;13:257.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32.
Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–9.
Stephens TW, Basinski M, Bristow PK, Bue-Valleskey JM, Burgett SG, Craft L, et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. 1995;377:530–2.
Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6.
Rosenbaum M, Leibel RL. Leptin: a molecule integrating somatic energy stores, energy expenditure and fertility. Trends Endocrinol Metab. 1998;9:117–24.
Bender N, Allemann N, Marek D, Vollenweider P, Waeber G, Mooser V, et al. Association between variants of the leptin receptor gene (LEPR) and overweight: a systematic review and an analysis of the CoLaus study. PLoS One. 2011;6:e26157.
Denbow DM, Meade S, Robertson A, McMurtry JP, Richards M, Ashwell C. Leptin-induced decrease in food intake in chickens. Physiol Behav. 2000;69:359–62.
Kuo AY, Cline MA, Werner E, Siegel PB, Denbow DM. Leptin effects on food and water intake in lines of chickens selected for high or low body weight. Physiol Behav. 2005;84:459–64.
Mackowski M, Szymoniak K, Szydlowski M, Kamyczek M, Eckert R, Rozycki M, et al. Missense mutations in exon 4 of the porcine LEPR gene encoding extracellular domain and their association with fatness traits. Anim Genet. 2005;36:135–7.
Ros-Freixedes R, Gol S, Pena RN, Tor M, Ibáñez-Escriche N, Dekkers JCM, et al. Genome-wide association study singles out SCD and LEPR as the two main loci influencing intramuscular fat content and fatty acid composition in Duroc pigs. PLoS One. 2016;11:e0152496.
Hou N, Luo JD. Leptin and cardiovascular diseases. Clin Exp Pharmacol Physiol. 2011;38:905–13.
Koh KK, Park SM, Quon MJ. Leptin and cardiovascular disease: response to therapeutic interventions. Circulation. 2008;117:3238–49.
Friedman-Einat M, Seroussi E. Quack leptin. BMC Genomics. 2014;15:551.
Friedman-Einat M, Cogburn LA, Yosefi S, Hen G, Shinder D, Shirak A, et al. Discovery and characterization of the first genuine avian leptin gene in the Rock dove (Columba livia). Endocrinology. 2014;155:3376–84.
Boswell T, Dunn IC. Regulation of the avian central melanocortin system and the role of leptin. Gen Comp Endocrinol. 2015;221:278–83.
Nierobisz LS, Sporer KRB, Strasburg GM, Reed KM, Velleman SG, Ashwell CM, et al. Differential expression of genes characterizing myofibre phenotype. Anim Genet. 2012;43:298–308.
Adachi H, Takemoto Y, Bungo T, Ohkubo T. Chicken leptin receptor is functional in activating JAK–STATpathway in vitro. J Endocrinol. 2008;197:335–42.
Zhang L, Zuo Q, Li D, Lian C, Ahmed KE, Tang B, et al. Study on the role of JAK/STAT signaling pathway during chicken spermatogonial stem cells generation based on RNA-Seq. J Integr Agric. 2015;14:939–48.
Yonggang L. A novel porcine gene, MAPKAPK3, is differentially expressed in the pituitary gland from mini-type Diannan small-ear pigs and large-type Diannan small-ear pigs. Mol Biol Rep. 2010;37:3345–9.
Tapia O, Fong LG, Huber MD, Young SG, Gerace L. Nuclear envelope protein Lem2 is required for mouse development and regulates MAP and AKT kinases. PLoS One. 2015;10:e0116196.
Logan M, Simon HG, Tabin C. Differential regulation of T-box and homeobox transcription factors suggests roles in controlling chick limb-type identity. Development. 1998;125:2825–35.
Rodriguez-Esteban C, Tsukui T, Yonei S, Magallon J, Tamura K, Belmonte JCI. The T-box genes Tbx4 and Tbx5 regulate limb outgrowth and identity. Nature. 1999;398:814–8.
Takeuchi JK, Koshiba-Takeuchi K, Matsumoto K, Vogel-Höpker A, Naitoh-Matsuo M, Ogura K, et al. Tbx5 and Tbx4 genes determine the wing/leg identity of limb buds. Nature. 1999;398:810–4.
Khalil A, Tanos R, El-Hachem N, Kurban M, Bouvagnet P, Bitar F, et al. A HAND to TBX5 explains the link between thalidomide and cardiac diseases. Sci Rep. 2017;7:1416.
Cortes M, Baria AT, Schwartz NB. Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development. 2009;136:1697–706.
Takasuga A. PLAG1 and NCAPG-LCORL in livestock. Anim Sci J. 2016;87:159–67.
Karim L, Takeda H, Lin L, Druet T, Arias JAC, Baurain D, et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011;43:405–13.
Littlejohn M, Grala T, Sanders K, Walker C, Waghorn G, Macdonald K, et al. Genetic variation in PLAG1 associates with early life body weight and peripubertal weight and growth in Bos taurus. Anim Genet. 2012;43:591–4.
Hoshiba H, Setoguchi K, Watanabe T, Kinoshita A, Mizoshita K, Sugimoto Y, et al. Comparison of the effects explained by variations in the bovine PLAG1 and NCAPG genes on daily body weight gain, linear skeletal measurements and carcass traits in Japanese Black steers from a progeny testing program. Anim Sci J. 2013;84:529–34.
Nishimura S, Watanabe T, Mizoshita K, Tatsuda K, Fujita T, Watanabe N, et al. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012;13:40.
Fortes MRS, Kemper K, Sasazaki S, Reverter A, Pryce JE, Barendse W, et al. Evidence for pleiotropism and recent selection in the PLAG1 region in Australian Beef cattle. Anim Genet. 2013;44:636–47.
Utsunomiya YT, Milanesi M, Utsunomiya ATH, Torrecilha RBP, Kim E-S, Costa MS, et al. A PLAG1 mutation contributed to stature recovery in modern cattle. Sci Rep. 2017;7:17140.
Fink T, Tiplady K, Lopdell T, Johnson T, Snell RG, Spelman RJ, et al. Functional confirmation of PLAG1 as the candidate causative gene underlying major pleiotropic effects on body weight and milk characteristics. Sci Rep. 2017;7:44793.
Voz ML, Agten NS, de Ven WJMV, Kas K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000;60:106–13.
Van Dyck F, Declercq J, Braem CV, de Ven VWJM. PLAG1, the prototype of the PLAG gene family: versatility in tumour development (Review). Int J Oncol. 2007;30:765–74.
Pértille F, Zanella R, Felício AM, Ledur MC, Peixoto JO, Coutinho LL. Identification of polymorphisms associated with production traits on chicken (Gallus gallus) chromosome 4. Genet Mol Res. 2015;14:10717–28.
Pértille F, Moreira GCM, Zanella R, da Silva de Nunes JR, Boschiero C, Rovadoscki GA, et al. Genome-wide association study for performance traits in chickens using genotype by sequencing approach. Sci Rep. 2017;7:41748.
Dunn IC, Meddle SL, Wilson PW, Wardle CA, Law AS, Bishop VR, et al. Decreased expression of the satiety signal receptor CCKAR is responsible for increased growth and body weight during the domestication of chickens. Am J Physiol-Endocrinol Metab. 2013;304:E909–21.
Kuehn C, Widmann P, Pfuhl R, Weikard R. Evaluation of the NCAPG I442M locus, a major gene for bovine prenatal growth, for effects on postnatal development compared to a disruptive mutation in the myostatin encoding gene GDF8. J Dairy Sci. 2010;93:540.
Glenske K, Brandt H, Erhardt G. Association of the Ile-442-Met substitution in NCAPG with birth weight in German Angus and German Simmental cattle (Brief Report). Arch Anim Breed. 2011;54:104–6.
Makvandi-Nejad S, Hoffman GE, Allen JJ, Chu E, Gu E, Chandler AM, et al. Four loci explain 83% of size variation in the horse. PLoS One. 2012;7:e39929.
Sovio U, Bennett AJ, Millwood IY, Molitor J, O’Reilly PF, Timpson NJ, et al. Genetic determinants of height growth assessed longitudinally from infancy to adulthood in the Northern Finland birth cohort 1966. PLoS Genet. 2009;5:e1000409.
Metzger J, Schrimpf R, Philipp U, Distl O. Expression levels of LCORL are associated with body size in horses. PLoS One. 2013;8:e56497.
Lindholm-Perry AK, Sexten AK, Kuehn LA, Smith TP, King DA, Shackelford SD, et al. Association, effects and validation of polymorphisms within the NCAPG-LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011;12:103.
Anwar S, Iqbal MP, Zarina S, Bhutta ZA. Evolutionary journey of the Gc protein (vitamin D-binding protein) across vertebrates. Intrinsically Disord Proteins. 2013;1:e27450.
Jiang X, O’Reilly PF, Aschard H, Hsu YH, Richards JB, Dupuis J, et al. Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat Commun. 2018;9:260.
Vignale K, Greene ES, Caldas JV, England JA, Boonsinchai N, Sodsee P, et al. 25-hydroxycholecalciferol enhances male broiler breast meat yield through the mTOR pathway. J Nutr. 2015;145:855–63.
Olsen HG, Knutsen TM, Lewandowska-Sabat AM, Grove H, Nome T, Svendsen M, et al. Fine mapping of a QTL on bovine chromosome 6 using imputed full sequence data suggests a key role for the group-specific component (GC) gene in clinical mastitis and milk production. Genet Sel Evol. 2016;48:79.
Dadousis C, Biffani S, Cipolat-Gotet C, Nicolazzi EL, Rossoni A, Santus E, et al. Genome-wide association of coagulation properties, curd firmness modeling, protein percentage, and acidity in milk from Brown Swiss cows. J Dairy Sci. 2016;99:3654–66.
Mao HG, Dong XY, Cao HY, Xu NY, Yin ZZ. Association of DGAT2 gene polymorphisms with carcass and meat quality traits in domestic pigeons (Columba livia). Br Poult Sci. 2017;59:149–53.
Powers PA, Liu S, Hogan K, Gregg RG. Skeletal muscle and brain isoforms of a beta-subunit of human voltage-dependent calcium channels are encoded by a single gene. J Biol Chem. 1992;267:22967–72.
Tiezzi F, Maltecca C. Accounting for trait architecture in genomic predictions of US Holstein cattle using a weighted realized relationship matrix. Genet Sel Evol. 2015;47:24.
Acknowledgements
This work was supported by the Innovative UK project “Precision Breeding: Broilers from Sequence to Consequence (49521-361188)”, the BBSRC Strategic Programme to The Roslin Institute (BBS/E/D/30002275), and the University of Edinburgh’s Data-Driven Innovation Chancellor’s fellowship.
Author information
Authors and Affiliations
Contributions
JH and AK conceived the idea and supervised the project. AS and PG helped in data preparation and imputation. AW and DW developed the Python script to construct the \(\mathbf{G}\) matrix and its eigen decomposition. CD conducted the analysis and wrote the first draft of the paper. MJ helped with the gene annotation. GG conceived the idea of genome partitioning analysis and contributed to discussions about the statistical analysis. LB performed the partitioning analysis. JJI, MJ, RM, DH, and ICD contributed with fruitful discussions on gene functionality. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
List of all significant SNPs sorted by significance1. SNP = name of the single nucleotide polymorphism; GGA = Gallus gallus chromosome; location (bp) = position of the SNP on the chromosome in base pairs on the Galgal4 assembly; MAF = minor allele frequency; EFF = allele substitution effect; SE = standard error of the effect; P-value = P-values from the single SNP regression while simultaneously correcting for the background polygenic effect; P-valueFDR = false discovery rate P-value; LOG = the −log10 of the P-value; VSNP = variance due to the SNP (calculated as 2pqb2, where p is the frequency of major allele, q = 1 − p is the frequency of the minor allele and b is the allele substitution effect); VA = additive genetic variance; VP = phenotypic variance; VAsnp (%) = percentage of additive genetic variance explained by each SNP; VPsnp (%) = percentage of phenotypic variance explained by each SNP. 1Of the 96 significant SNPs 18 are proprietary to Aviagen and therefore their base pair location has been excluded.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dadousis, C., Somavilla, A., Ilska, J.J. et al. A genome-wide association analysis for body weight at 35 days measured on 137,343 broiler chickens. Genet Sel Evol 53, 70 (2021). https://doi.org/10.1186/s12711-021-00663-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-021-00663-w