
Abstract
Background
The identification of spotted leaf 50 (spl50), a novel lesion mimic mutant (LMM) in rice, provides critical insights into the mechanisms underlying programmed cell death (PCD) and innate immunity in plants.
Results
Based on ethyl methane sulfonate (EMS)-induced mutagenesis, the spl50 mutant mimics hypersensitive responses in the absence of pathogen by displaying spontaneous necrotic lesions after the tillering phase. SPL50, an ARM repeat protein essential for controlling reactive oxygen species (ROS) metabolism and boosting resistance to blast disease, was identified by map-based cloning techniques. This work also demonstrates the detrimental effects of spl50 on photosynthetic efficiency and chloroplast development. The crucial significance of SPL50 in cellular signaling and stress response is shown by its localization to the cytoplasm and constitutive expression in various plant tissues. In light of growing concerns regarding global food security, this study highlights the pivotal role of SPL50 in regulating programmed cell death (PCD) and enhancing the immune response in plants, contributing to strategies for improving crop disease resistance.
Conclusions
The novel identification of the SPL50 gene in rice, encoding an ARM repeat protein, reveals its pivotal role in regulating PCD and innate immune responses independently of pathogen attack.
Similar content being viewed by others


Introduction
Plant lesion mimics (LMMs) are a remarkable phenomenon in plant biology that reveal how plants can exhibit symptoms even when they are not experiencing disease or environmental stress (Zhu et al. 2020). These necrotic spots, varying in shapes and sizes, emerge spontaneously across various plant parts, including the leaves and leaf sheaths. The fact that this phenomenon develops independently of biotic (pathogen-related) or abiotic (environmental stress-related) factors raises the possibility that an underlying genetic or epigenetic mechanism is at work. Numerous plant species exhibit lesion mimic mutants, underscoring a widespread genetic foundation for this phenomenon. Examples include barley (Wolter et al. 1993), rice (Takahashi et al. 1999), Arabidopsis (Lorrain et al. 2003), maize (Mu et al. 2021), and wheat (Yu et al. 2023; Wang et al., 2023), among others. These mutant`s lesions bear a striking resemblance to those seen in plants after a hypersensitive response (HR), a well-documented form of programmed cell death (PCD) (Cai et al. 2021). The HR plays a crucial role in controlling the spread of diseases by rapidly eliminating contaminated cells and enclosing the afflicted area to stop the germs from moving further. A key component of the plant’s innate immune system, this fast cell death mechanism allows it to successfully fend off pathogen attacks without the involvement of adaptive immunity system, which are present in more complex species.
To date, an array of LMM genes has been elucidated, encoding proteins that span a diverse range of functions. These include the MEDIATOR SUBUNIT 16 (Zhang et al. 2023), the deubiquitinase OsLMP1 (Zou et al. 2023; Sun et al., 2021), receptor-like protein kinase SPL36 (Rao et al. 2021), translation factor OsWRKY19 (Du et al. 2021), ATP-citrate lyase SPL30 (Ruan et al. 2019), eukaryotic translation elongation factor 1 alpha (eEF1A)-like protein SPL33 (Wang et al. 2017), splicing factor 3b subunit SPL5 (Chen et al. 2012), and heat shock transcription factor SPL7 (Yamanouchi et al., 2002). Each of these genes plays a distinct and pivotal role in the intricate biological phenomena observed in plants, especially in underlining the mechanisms of disease resistance and PCD in rice.
The discovery of Armadillo (ARM) repeat proteins, initially identified in Drosophila melanogaster, signified a noteworthy progression in our comprehension of protein structure and function. These proteins are characterized by a highly conserved structural domain consisting of triplets of α-helices, each arranged in a repeating 42-amino acid pattern (Peifer et al. 1994). This unique structural design contributes to their adaptability and allows ARM repeat proteins to assume various conformations, adopting to different functional roles as required. The adaptability of ARM repeat proteins is a crucial aspect of their ability to interact with a wide range of protein partners. This flexibility facilitates their involvement in numerous essential cellular processes, underscoring their importance in maintaining cellular homeostasis and responding to internal and external signals. One critical role of ARM repeat proteins is in nucleo-cytoplasmic transport, which is fundamental to the proper functioning and regulation of cells (Wang et al. 2014). This process ensures that substances necessary for cellular function are correctly shuttled between the nucleus and the cytoplasm, allowing for the appropriate expression and regulation of genes. Additionally, ARM repeat proteins are vital in mediating cellular responses to environmental cues through signal transduction pathways (Kulich et al. 2020). These pathways are essential for the cell’s ability to detect and respond to changes in its environment, ensuring cellular adaptation and survival under various conditions. protein trafficking is another crucial process involving ARM repeat proteins (Yoshida et al. 2023). This mechanism is responsible for the correct transport and localization of proteins within the cell, ensuring that proteins delivered to where they are needed mos. Furthermore, ARM repeat proteins play a significant role in the ubiquitination processes, which tags proteins for degradation or functional modification (Wang et al., 2023; Lv et al. 2022).
ARM repeat proteins are pivotal across a vast array of organisms, playing particularly significant roles in plant biology where they regulate development, stress responses, and metabolic pathways. In Arabidopsis thaliana, the ARM domain-containing protein ARK1 interacts directly with RHD3 to modulate the architecture of the endoplasmic reticulum, a key factor in maintaining cellular integrity and function (Sun et al. 2020). This interaction underscores how ARM repeat proteins facilitate the organization of cellular structures. In terms of stress adaptation, ARM repeat proteins like OsPUB2 and its homolog OsPUB3 in rice significantly enhance the plant’s tolerance to cold stress, reflecting the functional adaptability of these proteins in response to environmental challenges (Byun et al. 2017). Additionally, ARM repeat proteins play integral roles in plant defense mechanisms. For example, OsIMα1a and OsIMα1b are crucial in defending rice against blast disease, thereby bolstering crop resilience and food security (Xu et al. 2022). The interaction of these proteins with pathogenic challenges illustrates the dynamic role of ARM repeat proteins in mobilizing plant defenses against external threats. Adding to the complexity, proteins like SPL11 and OsPUB15, which possess both ARM and U-box domains, are pivotal in the ubiquitin-mediated regulation of disease resistance mechanisms within rice (Zeng et al. 2004; Wang et al. 2015). Despite extensive research on ARM repeat proteins within the plant kingdom and their proven significance in multiple biological functions, the roles of many members within the ARM family remain largely unexplored. This gap in our knowledge underscores the necessity for ongoing research efforts to elucidate the complex mechanisms through which ARM repeat proteins influence cell death, disease resistance, and other critical processes. Unraveling these mechanisms is essential not only to advance our fundamental understanding of plant biology but also to exploit this knowledge in developing crops with enhanced resistance to stresses and diseases. Such research is crucial for promoting sustainable agriculture and ensuring food security amidst escalating global challenges.
To investigate the molecular mechanisms governing cell death and disease resistance in rice, we successfully isolated a novel LMM, named spotted leaf 50 (spl50), from ethyl methane sulfonate (EMS)-mutagenized Oryza sativa japonica cv. Wuyunjing 7 (WYJ). The spl50 mutant displayed distinct spotted leaves beginning at the tillering stage and continuing through the ripening phase, indicative of internal disruptions potentially linked to innate immunity mechanisms. To identify the genetic basis of the spl50 phenotype, we employed a map-based cloning strategy. This approach led us to discover that the mutation responsible for the spl50 characteristics occurred in a gene encoding an ARM repeat protein. ARM repeat proteins are known for their role in various cellular processes, including developmental regulation and stress response. The mutation in the SPL50 gene led to a notable accumulation of reactive oxygen species (ROS) in the mutant plants. Interestingly, despite the potential for damage, the increased ROS levels in spl50 mutants were also associated with enhanced resistance to disease. This observation suggests that SPL50 plays a dual role in maintaining ROS homeostasis and activating defense mechanisms against pathogens. This finding adds a new layer to our understanding of SPL50’s functions, positioning it as a crucial player in the plant’s innate immune response.
Results
Identification of the spl50 Mutant
The discovery of the spl50 mutant resulted from a meticulous screening process subsequent to the application of EMS (ethyl methane sulfonate) mutagenesis on the japonica rice cultivar, Wuyunjing 7 (WYJ). Under field conditions, the spl50 mutant does not exhibit significant differences in primary growth parameters such as height and primary leaf development at the earliest seedling stage, including the absence of visible lesions on its leaves (Fig. 1A-B). (Fig. 1A-B). As the plants matured from the tillering to the ripening stages, however, pronounced phenotypic differences began to emerge. Both aging and newly formed leaves of the spl50 mutants developed reddish-brown lesions, contrasting sharply with the vibrant green foliage observed in wild type plants (Fig. 1C-F). This shift in leaf coloration and the appearance of lesions are indicative of significant internal changes within the plant, potentially signaling a hypersensitive response or a self-initiated activation of cell death mechanisms, occuring in the absence of pathogen interaction. Such phenotypic changes suggest that the spl50 mutation may have profound effects on the plant’s vitality and stress response pathways. Moreover, the spl50 mutants exhibited considerable reductions in several agronomic traits compared to their wild type counterparts, such as plant height, grain size, 1000-grain weight, the number of productive panicles, the number of grains per panicle, primary and secondary panicle branches, grain yield per plant, total grain number per plant, and seed setting rate (Fig. S1).

Phenotypic comparison of WT and spl50 mutant plants. (A) Overall morphology of WT and spl50 mutant at 50 days post-germination. Scale bar = 10 cm. (B) Comparative leaf morphology of WT and spl50 at 50-day-old seedlings. (C) Morphological differences between WT and spl50 at the tillering stage. Scale bar = 10 cm. (D) Leaf morphology showcasing WT and spl50 at the tillering stage. (E) Comparison of WT and spl50 plant phenotypes at the maturity stage. Scale bar = 10 cm. (F) Leaf morphology contrast between WT and spl50 plants at the mature stage
Alterations in Reactive Oxygen Species (ROS) Metabolism in the spl50 Mutant
The appearance of lesion-mimic spots on the leaves of spl50 mutants serves as a visible indicator of the activation of cellular death pathways, accompanied by a notable accumulation of hydrogen peroxide (H2O2) within the cells. To confirm this phenomenon, we conducted a series of experiments utilizing 3,3’-diaminobenzidine (DAB) staining on both wild type and spl50 mutant leaves over an 8-hour period. The results revealed pronounced brown pigmentation exclusively in spl50 leaves, indicative of a significant buildup of H2O2, contrasting sharply with the unaltered appearance of wild type leaves (Fig. 2A). Furthermore, Trypan blue staining highlighted extensive cellular damage within spl50 leaves, diverging markedly from the unaffected wild type (Fig. 2B). Quantitative analyses further supported these observations, demonstrating elevated levels of H2O2 and superoxide anion (O2−) within the spl50 mutant, along with increased production of malondialdehyde (MDA), a marker signifying both cellular demise and lipid peroxidation (Fig. 2C-E). Enzymatic analyses revealed a decrement in catalase (CAT) activity within spl50, whereas peroxidase (POD) and superoxide dismutase (SOD) activities experienced significant elevation, underscoring an intensified oxidative stress response in the mutant (Fig. 2F-H). Concomitantly, transcriptomic analyses unveiled a significant upregulation of five senescence-associated genes, namely WRKY23, SGR, OsI85, Osh69, and RCCR1, in the spl50 mutant (Fig. 2I). This finding affirms the genetic response to oxidative stress and cellular senescence in spl50.

Molecular analysis of leaf senescence and ROS-associated parameters in WT and spl50 mutant lines. (A) DAB staining. (B) Trypan blue staining. (C-E) Quantification of H2O2, O2− and MDA levels in WT and spl50 leaves. (F-H) Enzymatic activity assays for CAT, POD, and SOD in leaves at the heading stage. (I) Relative gene expression levels of senescence-associated markers, WRKY23: WRKY DNA-binding protein 23. SGR: Stay-Green Rice. OsI85: Oryza sativa Inducer of 85 kDa protein. Osh69: Oryza sativa Homeobox 69. RCCR1: Red Chlorophyll Catabolite Reductase 1. Data are presented as mean values ± standard deviation (SD) for n = 3 biological replicates. Statistical significance was assessed using Student’s t-test (*P < 0.05, **P < 0.01)
To further investigate the cellular death mechanisms in spl50, we examined chromatin condensation within mature leaf cells. the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay can be used to identify the endonucleolytic fragmentation of nuclear DNA, which is the mechanism that is unmistakably associated with programmed cell death (PCD). This analysis unveiled a distinct disparity between the spl50 mutants and their wild type counterparts. Specifically, the spl50 mutants have a high frequency of TUNEL-positive nuclei, whereas the wild type has a very low frequency of these nuclei (Fig. 3).

TUNEL assay for DNA fragmentation in WT and spl50 mutants. (A-B) TUNEL staining depicting DNA fragmentation. (C-D) DAPI staining indicating total DNA. (E-F) Merged images of TUNEL and DAPI staining. Scale bar = 200 μm
Enhanced Innate Immune Responses in the spl50 Mutant
Magnaporthe oryzae, the fungus that causes rice blast disease, has caused a drastic decline in cereal yields over 30%, posing a significantly threat to global food security (Zhang et al. 2024). In light of this pressing issue, our study sought to assess on evaluating the resistance of the spl50 mutant against this formidable pathogen. After two weeks, specimens of both wild type and spl50 mutant plants, were inoculated with the M. oryzae strain R01-1. A week after inoculation, careful monitoring of disease symptoms was undertaken. Significantly, the spl50 mutant leaves showed noticeably less lesions than the wild type leaves, which displayed extensive necrotic lesions (Fig. 4A-B). To further substantiate the extent of fungal infiltration and proliferation within the leaf tissues, DNA was meticulously extracted from matched samples of both wild type and spl50 mutant leaves. The subsequent quantification of M. oryzae biomass using quantitative real-time PCR (qRT-PCR) unveiled a significantly diminished fungal biomass in the spl50 mutant compared to the wild type (Fig. 4C).

Disease resistance assessment and defense gene expression analysis in WT and spl50 mutant lines. (A) Pathogenicity assays conducted by spray inoculation with M. oryzae isolate RO1-1. (B) Quantification of relative lesion area caused by M. oryzae in WT and spl50 lines, Values are mean ± SD (n = 10). (C) Estimation of relative M. oryzae biomass within lesions of WT and spl50, Values are mean ± SD (n = 10). (D) Expression levels of defense signaling-related genes. Values are mean ± SD (n = 3). Significant differences determined by Student’s t-test (*P < 0.05, **P < 0.01)
Additionally, a comprehensive quantitative analysis of the expression levels of key defense genes, including PR1a, PR1b, PR10, PBZ1, and SL, was conducted via qRT-PCR. The results revealed a substantial upregulation of these genes in the spl50 mutant (Fig. 4D). This robust induction of these defense genes in the spl50 mutant provides compelling evidence for its enhanced resistance to the rice blast disease. Our findings underscore the potential of the spl50 mutation in bolstering the plant’s defense mechanisms against M. oryzae.
Impact of SPL50 Mutation on Chloroplast Development and Photosynthetic Efficiency
Lesion mimic phenomena are frequently associated with disruptions to chloroplast ontogeny, aberrant gene expression patterns related to chloroplast, and reduced levels of chlorophyll, all of which have a negative impact on photosynthetic efficiency (Cui et al. 2021; Wang et al. 2017). In an effort to determine if the spl50 mutant exhibits similar perturbations, extensive investigations were carried out on heading-stage wild type and spl50 plants. This involved quantifying the amount of chlorophyll and assessing the expression of protein linked to chloroplasts in their leaves. As compared to the wild type, our findings showed a considerable drop in the levels of chlorophyll a, chlorophyll b, and carotenoids in the spl50 mutant indicating a major impairment in chlorophyll biosynthesis and accumulation (Fig. 5A). Further, the spl50 mutation has a significant influence on the integrity and function of chloroplasts, as demonstrated by the increased degradation of several key proteins associated to chloroplast, including Tic110, CP47, RbcL, D1, and PsbO, as revealed by western blot analysis (Fig. 5B).

Analysis of chlorophyll content and expression of chloroplast-associated proteins in WT and spl50 mutant lines. (A) Comparative chlorophyll content in WT and spl50 leaves at the heading stage. Chl a, Chlorophyll a; Chl b, Chlorophyll b; Car, Carotenoids. Data are presented as means ± SD (n = 6). (B) Relative expression levels of chloroplast-associated proteins in WT and spl50 at the heading stage. (C) Photosynthetic parameter measurements in WT and spl50 leaves. Data are presented as means ± SD (n = 6), with statistical significance evaluated via Student’s t-test (*P < 0.05, **P < 0.01)
Subsequent examination of the photosynthetic efficiency within the flag leaves of both genotypes at the heading stage unveiled a marked decline in the net photosynthetic rate, stomatal conductance, and transpiration rate in the spl50 mutant (Fig. 5C). This reduction was accompanied by an elevated intracellular CO2 concentration, indicating a severe disruption in the photosynthetic machinery. These observations provide further evidence that the spl50 mutation adversely affects chloroplast functionality in rice, leading to a pronounced decrease in photosynthetic efficiency.
Map-Based Cloning of SPL50 Mutation on Chloroplast Development and Photosynthetic Efficiency
To conduct a genetic analysis of the spl50 mutant, we initiated a cross between the mutant phenotype and the wild type cultivar WYJ. Phenotypic examination of all progeny in the F1 generation revealed wild type characteristics, indicating the recessive nature of the mutation. In the F2 population, comprising 441 plants, 338 exhibited growth patterns similar to the wild type, while the remaining displayed the mutant phenotype. The observed segregation ratio of 3:1 (χ20.05 = 0.477 < 3.841) substantiates the hypothesis that the spl50 mutation is governed by a single recessive allele at a nuclear locus.
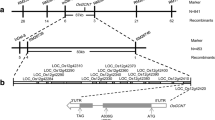
For the purpose of identifying the gene associated with the mutant phenotype, the spl50 mutant was crossed with an indica rice variety 9311. From this cross, 1345 F2 progeny manifesting the lesion-mimic spots phenotype were selected for further analysis. We employed a total of 183 simple sequence repeats (SSRs) and 41 sequence tagged sites (STSs), which are uniformly dispersed across the 12 chromosomes of rice, for genotyping. Initial mapping efforts positioned the mutant locus between markers B10-1 and B10-2 on chromosome 10, as determined by analyzing the 113 F2 plants (Fig. 6A). Subsequent fine mapping, utilizing a larger set of 1232 F2 mutants, refined the locus to a 112-kb interval flanked by markers P7 and P8 (Fig. 6B-C). According to data from the Rice Genome Annotation Project (RGAP, http://rice.plantbiology.msu.edu), this interval contains sixteen predicted open reading frames (ORFs) (Table S2). Sequencing of this specific region in the spl50 mutant identified a transversion mutation from T to A within an exon of LOC_Os10g05370, resulting in a Leu-to-His substitution at the 188th amino acid (Fig. 6D-G). LOC_Os10g05370 possesses 2 ARM repeats, closely mirroring the structure found in ARM-repeat-only (AMO) proteins (Fig. 6E), akin to ARO protein GS10 in rice (Chen et al. 2023). Thus, LOC_Os10g05370 has been identified as the candidate gene responsible for the observed mutant phenotype.

Map-based cloning of SPL50. (A) SPL50 preliminarily mapped between markers B10-1 and B10-8 on chromosome 10. (B-C) Fine mapping of SPL50. The SPL50 locus was mapped to a 122-kb region between markers P7 and P8. (D) Identification of sixteen candidate ORFs within the122-kb genomic interval. (E) Gene structure of LOC_Os10g05730. Orange-yellow boxes represent Armadillo-type fold domains (IPR016024) predicted by the InterPro database. (F) Comparative sequence analysis showing a T-to-A point mutation in spl50 relative to the wild type. (G) Amino acid substitution resulting from the mutation, replacing leucine with histidine in the SPL50 protein. (H) Rescue of the spl50 mutant phenotype through functional complementation
Confirmation of LOC_Os10g05370 Responsible for the Mutant Phenotype of spl50
To definitively confirm that SPL50 corresponds to LOC_Os10g05370, we meticulously inserted a 5.1 kb genomic fragment of LOC_Os10g05370 into the binary vector pCAMBIA1300, followed by its introduction into the spl50 mutant via transformation. This process yielded 23 independent T0 transformants. Detailed phenotypic analysis clearly demonstrated that each transformant effectively mitigated the spl50 mutant phenotype, as evidenced by an enhanced plant height and the elimination of lesion-mimic spots (Fig. 6H). Subsequent exposure of WT, spl50, and the complemented line pGSPL50-1 to the rice blast pathogen underscored the functional restoration of disease resistance. Notably, there were no significant differences in the extent of lesion formation and pathogen biomass accumulation between pGSPL50-1 and WYJ (Fig. 7A-C). In addition, a pronounced reduction in the expression levels of defense-related genes OsPR1a and OsPR1b in pGSPL50-1 compared to spl50 was observed, indicating a normalization of the defense response (Fig. 7D-E). Furthermore, TUNEL assay data from pSPL50-1 complementation lines confirmed that cell apoptosis rates also reverted to those observed in wild-type plants (Fig. S3). These findings provide compelling evidence supporting the hypothesis that the lesion mimic phenotype observed in the spl50 mutant is attributable to a single-base substitution mutation within the LOC_Os10g05370 gene of SPL50.

Resistance reactions to Maganaporthe oryzae isolates tested. (B) Disease symptoms on leaves of WT, spl50, and pGSPL50-1 after inoculation with M. oryzae. (B-C) Quantification of M. oryzae-infected leaf area and fungal biomass in WT, spl50, and pGSPL50-1 lines. (D-E) Expression analysis of defense-related markers via quantitative reverse transcription PCR (qRT-PCR) in WT, spl50, pGSPL50-1 and pGSPL50-2. Values are mean ± SD (n = 3). Statistical significance was determined using Student’s t-test (*P < 0.05, **P < 0.01)
Constitutive Expression of SPL50
Through qRT-PCR analysis, our investigation revealed the broad expression spectrum of SPL50 gene across diverse plant tissues, such as roots, culms, leaves, leaf sheaths, and panicles. Notably, SPL50 exhibited its highest expression level of in leaf tissue, while its lowest expression was observed within the panicle structures (Fig. 8A). To further elucidate the regulatory elements governing SPL50 expression, we engineered a construct by fusing a 2,052-bp genomic fragment, located upstream of SPL50’s start codon, with the β-glucuronidase (GUS) reporter gene. Subsequently, this recombinant DNA was introduced into the genome of wild type plants through transformation. Histochemical analysis of GUS activity in the progeny of these transgenic plants revealed enzymatic activity in roots, culms, leaves, and panicles (Fig. 8B). This pattern of GUS expression closely mirrors the expression profile of SPL50 as determined by qRT-PCR, thus corroborating the constitutive expression paradigm of SPL50 across the examined plant tissues.

Expression patterns of SPL50. (A) SPL50 transcript levels in various tissues of WT at booting stage, measured by qRT-PCR. Standard deviations are indicated (n = 3). (B-G) Localization of SPL50 promoter-driven GUS reporter gene activity in different tissues: roots (B), stem (C), leaf (D-E), sheath (F), and young panicles (G). Scale bar = 100 μm
Subcellular Localization of SPL50
To determine the subcellular localization of SPL50, plasmids encoding p35S::SPL50-GFP (the full-length cDNA of SPL50 fused to the N-terminus of GFP) and HSP70 (with cytoplasmic localization)-RFP were transformed into rice protoplasts or Nicotiana benthamiana epidermal cells. The results demonstrated that SPL50 is localized in the cytoplasm (Fig. 9A-F). Additionally, the co-localization of SPL50 with SDS2 (a plasma membrane marker) was examined, the findings indicated that SPL50 is predominantly distributed in the cytoplasm (Fig. 9G-I).

Subcellular localization of SPL50 (A-C) Localization of SPL50 protein in rice protoplast via GFP fusion protein assay. Scale bar = 50 μm. (C-I) Nicotiana benthamiana transient assay showing SPL50 subcellular distribution, with SPL50 tagged with GFP under the control of the 35 S promoter. HSP70-RFP serves as a cytoplasm marker, SDS2-RFP serves as a cytomembrane marker. Scale bar = 20 μm
Discussion
The novel identification and characterization of the SPL50 gene within Oryza sativa not only enriches our understanding of plant LMMs but also sheds light on the intricate regulatory networks governing PCD and innate immune responses in plants. SPL50, encoding an ARM repeat protein, emerges as a pivotal regulator orchestrating PCD and enhancing innate immune responses in rice. The spl50 mutant’s spontaneous development of necrotic lesions, independent of pathogen attack, underscores a self-activated PCD pathway, akin to an autoimmune response. This phenomenon is reflective of the mutant’s altered ROS metabolism, characterized by excessive accumulation of H2O2 and O2− (Fig. 2), hallmark indicators of oxidative stress and cell death (Mittler 2002).
Furthermore, the enhanced resistance of the spl50 mutant to rice blast disease (Fig. 4), underscores SPL50’s significant contribution to the plant’s immune defense. The observed upregulation of defense-related genes (e.g., PR1a, PR1b, PR10, PBZ1, and SL) (Fig. 4D) in the spl50 mutant suggests that SPL50 influences the transcriptional regulation of key components in rice’s defense signaling pathways.
The mechanisms by which SPL50 regulates PCD and immune response likely involve its ARM repeat domains, facilitating protein-protein interactions crucial for various cellular processes, including signal transduction, gene expression regulation, and multiprotein complex assembly (Yoshida et al. 2023; Wang et al., 2023; Lv et al. 2022; Kulich et al. 2020; Wang et al. 2014). In this study, it is plausible that its ARM domains interact with specific proteins involved in ROS metabolism and defense signaling pathways. The accumulation of ROS in the spl50 mutant (Fig. 2) may be attributed to SPL50’s potential role in modulating antioxidant enzyme activities, such as CAT, POD, and SOD. SPL50 might influence the expression or stability of these enzymes, thereby affecting ROS detoxification and the balance between ROS production and scavenging. SPL50’s contribution to enhanced disease resistance could involve the modulation of defense signaling pathways, possibly through interactions with key signal transducers or transcription factors involved in immune response activation. The upregulation of defense-related genes in the spl50 mutant (Fig. 4D) suggests that SPL50 may influence the transcriptional machinery directly or indirectly, perhaps by affecting the stability or activity of transcription factors, which play a central role in plant immune responses.
Moreover, SPL50 could alter the regulation of genes involved in chloroplast function and development, directly affecting protein synthesis within chloroplasts. The possibility that increased cell death in spl50 mutants could lead to a decline in chloroplast integrity and function, indirectly reducing the levels of chloroplast-associated proteins. SPL50-mediated regulation of chloroplast functionality and photosynthetic efficiency (Fig. 5) indicates a broader impact of SPL50 on plant physiology, connecting PCD and immune response to photosynthetic performance. This relationship underscores the interconnectedness of plant defense mechanisms with other physiological processes, highlighting the complexity of plant responses to environmental stresses.
In summary, the SPL50 gene exemplifies the sophisticated regulatory mechanisms plants employ to mediate PCD and innate immune responses. Through its ARM repeat domains, SPL50 likely interacts with a spectrum of proteins to modulate ROS metabolism and defense signaling pathways, playing a crucial role in rice’s ability to counteract pathogenic attacks while managing cellular homeostasis. Future studies should aim to elucidate the specific protein-protein interactions involving SPL50, further clarifying its role in plant PCD and immunity. Understanding these mechanisms not only contributes to our fundamental knowledge of plant biology but also offers potential avenues for enhancing crop resistance to diseases, a critical goal in the face of global food security.
Conclusions
The novel identification of the SPL50 gene in rice enriches our understanding of plant lesion mimic phenomena and unveils its pivotal role in regulating PCD and innate immune responses. Encoding an ARM repeat protein, SPL50 orchestrates PCD and enhances immune responses independently of pathogen attack. Its influence on ROS metabolism and defense signaling pathways underscores its significance in rice’s defense mechanisms. The upregulation of defense-related genes in the spl50 mutant suggests SPL50’s involvement in transcriptional regulation. Additionally, SPL50’s impact on chloroplast functionality and photosynthetic efficiency highlights its broader physiological effects. Through protein-protein interactions mediated by its ARM repeat domains, SPL50 likely modulates ROS metabolism and defense signaling pathways, contributing to both disease resistance and cellular homeostasis. Further research elucidating SPL50’s specific interactions and mechanisms promises insights into plant biology and avenues for crop improvement, vital for global food security challenges.
Materials and Methods
Plant Materials and Growth Conditions
This study was initiated with the isolation of an original spl50 mutant from EMS-induced mutagenesis in Wuyunjing 7 (Oryza sativa L. ssp. japonica), chosen for its distinctive phenotypic traits. Subsequently, an F2 mapping population was developed through hybridization between the spl50 mutant and an indica variety, 9311. To ensure comprehensive developmental observations, all plants were cultivated under natural field conditions in Fuyang (Zhejiang province, coordinates: 119°95’E, 30°05’N) and Lingshui (Hainan province, coordinates: 110°02’E, 18°48’N), exploiting the optimal growth periods across two distinct climatic zones during the rice growing season.
DAB Staining, H2O2 and MDA Determination and CAT Activity Detection
For the detection of ROS accumulation, leaves were subjected to DAB staining, employing a methodology adapted from Ruan et al. (2019). This technique facilitates the visual identification of H2O2 accumulation in situ. The quantification of H2O2 and MDA levels, alongside CAT activity assessments, were meticulously performed in accordance with protocols established by Zhao et al. (2023).
TUNEL Assay
Leaf tissues from both wild type, spl50, and pGSPL50-1 plants at the heading stage were collected for Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays. This assay was conducted following the detailed protocol by Huang and Zhou (2007), providing a quantitative and qualitative assessment of DNA fragmentation, a hallmark of PCD.
Inoculation with Magnaporthe Oryzae
The Magnaporthe oryzae strain R01-1 was propagated on oatmeal agar medium for approximately 10 days to facilitate enrichment culture. Spores were then resuspended in ddH2O and subsequently filtered through cheesecloth to collect a pure spore suspension. Spore density was accurately assessed using a hemocytometer (105 conidia ml-1), followed by the uniform application of the spore suspension (10μl) onto two-week-old plants of the WYG, spl50, and pGSPL50-1 varieties via a spray inoculation technique. Seven days after inoculation, photographic documentation of the infection was conducted. Lesion areas on WYG, spl50, and pGSPL50-1 were meticulously quantified using ImageJ software. Additionally, the biomass of Magnaporthe oryzae within the plant tissue was evaluated using qRT-PCR.
Measurement of Photosynthetic Rates
At approximately 10 a.m. during the summer season, the photosynthetic rates of twelve uniformly vigorous plants of both WYG and spl50 varieties, at their heading stages, were assessed using the LI-COR LI-6800 Portable Photosynthesis System. This evaluation was replicated three times for each measurement.
Western Blot Analysis
For total protein extraction, procedures were adapted from the methodology described (Rubio et al. 2005). Proteins were then resolved by 12% SDS-PAGE, followed by Western blot analysis in strict accordance with the protocol outlined (Kurien and Scofield 2006).
Map-Based Cloning and Complementation Test
For the map-based cloning of SPL50, a cohort of 1345 F2 progeny displaying the mutant phenotype was produced through hybridization between the spl50 mutant and the indica rice variety 9311. The gene prediction efforts targeted a precisely delineated 112-kb region on chromosome 10, utilizing resources from the Rice Genome Annotation Project (RGAP) database (http://rice.plantbiology.msu.edu/index.shtml) for comprehensive genomic analysis. A genomic fragment spanning 5121 base pairs (bp), encompassing the full SPL50 coding sequence, a 2052-bp upstream promoter region, and a 1086-bp downstream sequence, was meticulously amplified using the high-fidelity KOD Plus enzyme (Toyobo, Tokyo, Japan). This fragment was subsequently cloned into the binary vector pCAMBIA1300, preparing it for Agrobacterium-mediated transformation into spl50 mutant calli, following an established protocol (Toki et al. 2006). The specific primer sequences employed in this study are detailed in Supplementary Table S1.
Quantitative RT-PCR
Total RNA was isolated from various tissues including roots, leaves, leaf sheaths, culms, and panicles at the heading stage, utilizing TRIzol reagent (Invitrogen, Shanghai, China) according to the manufacturer’s instructions. Subsequent reverse transcription was performed employing SuperScript II reverse transcriptase with an integrated gDNA removal step (Invitrogen) to ensure the purity of the cDNA. The rice Actin 1 gene served as an endogenous reference for normalization. Expression data represent averages from three independent biological replicates. Statistical significance of the expression differences was evaluated using Student’s t-test. Specific primers utilized for gene amplification are detailed in Supplementary Table S1.
Histochemical GUS Assay
The promoter region of SPL50, extending 2052-bp upstream of the ATG start codon, was amplified from the genomic DNA of the WYJ cultivar. This fragment was subsequently cloned in-frame into the binary vector pCAMBIA1305, featuring a β-glucuronidase (GUS) reporter gene, to construct the recombinant plasmid. This plasmid was then transformed into WYJ calli via Agrobacterium-mediated transfer to generate transgenic rice plants. Various tissues from these transgenic lines, including roots, leaves, leaf sheaths, culms, and panicles, were subjected to GUS histochemical assays. These assays were conducted in accordance with the methodology described by Ren et al. (2018).
Subcellular Localization of SPL50
The cDNA sequence corresponding to SPL50, extracted from WYJ, was strategically cloned downstream of the green fluorescent protein (GFP) coding sequence under the control of the Cauliflower Mosaic Virus (CaMV) 35 S promoter. This recombinant construct was subsequently introduced into rice protoplasts and the leaf epidermal cells of Nicotiana benthamiana, utilizing the transformation methodology delineated by Ruan et al. (2019) and Zhao et al. (2022).
Data Availability
No datasets were generated or analysed during the current study.
Abbreviations
- LMMs:
-
Plant lesion mimics
- HR:
-
Hypersensitive response
- PCD:
-
Programmed cell death
- eEF1A:
-
Eukaryotic translation elongation factor 1 alpha
- ARM:
-
Armadillo
- CAT:
-
Catalase
- POD:
-
Peroxidase
- SOD:
-
Superoxide dismutase
- DAB:
-
3,3’-diaminobenzidine
- MDA:
-
Malondialdehyde
- EMS:
-
Ethyl methane sulfonate
- ROS:
-
Reactive oxygen species
- qRT-PCR:
-
Quantitative real-time PCR
- SSRs:
-
Simple sequence repeats
- STSs:
-
Sequence tagged sites
- GUS:
-
β-glucuronidase
- GFP:
-
Green fluorescent protein
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- RGAP:
-
Rice Genome Annotation Project
References
Byun MY, Cui LH, Oh TK, Jung YJ, Lee A, Park KY, Kang BG, Kim WT (2017) Homologous U-box E3 ubiquitin ligases OsPUB2 and OsPUB3 are involved in the positive regulation of low temperature stress response in Rice (Oryza sativa L). Front Plant Sci 8:16
Cai L, Yan M, Yun H, Tan J, Du D, Sun H, Guo Y, Sang X, Zhang C (2021) Identification and fine mapping of lesion mimic mutant spl36 in rice (Oryza sativa L). Breed Sci 71(5):510–519
Chen X, Hao L, Pan J, Zheng X, Jiang G, Jin Y, Gu Z, Qian Q, Zhai W, Ma BJMB (2012) SPL5, a cell death and defense-related gene, encodes a putative splicing factor 3b subunit 3 (SF3b3) in rice. Mol Breed 30(2):939–949
Chen E, Hou Q, Liu K, Gu Z, Dai B, Wang A, Feng Q, Zhao Y, Zhou C, Zhu J, Shangguan Y, Wang Y, Lv D, Fan D, Huang T, Wang Z, Huang X, Han B (2023) Armadillo repeat only protein GS10 negatively regulates brassinosteroid signaling to control rice grain size. Plant Physio 192(2):967–981
Cui Y, Peng Y, Zhang Q, Xia S, Ruan B, Xu Q, Yu X, Zhou T, Liu H, Zeng D, Zhang G, Gao Z, Hu J, Zhu L, Shen L, Guo L, Qian Q, Ren D (2021) Disruption of EARLY LESION LEAF 1, encoding a cytochrome P450 monooxygenase, induces ROS accumulation and cell death in rice. Plant J 105(4):942–956
Du D, Zhang C, Xing Y, Lu X, Cai L, Yun H, Zhang Q, Zhang Y, Chen X, Liu M, Sang X, Ling Y, Yang Z, Li Y, Lefebvre B, He G (2021) The CC-NB-LRR OsRLR1 mediates rice disease resistance through interaction with OsWRKY19. Plant Biotechnol J 19(5):1052–1064
Huang L, Sun Q, Qin F, Li C, Zhao Y, Zhou DX (2007) Down-regulation of a SILENT INFORMATION REGULATOR2-related histone deacetylase gene, OsSRT1, induces DNA fragmentation and cell death in rice. Plant Physiol 144(3):1508–1519
Kulich I, Vogler F, Bleckmann A, Cyprys P, Lindemeier M, Fuchs I, Krassini L, Schubert T, Steinbrenner J, Beynon J, Falter-Braun P, Längst G, Dresselhaus T, Sprunck S (2020) ARMADILLO REPEAT ONLY proteins confine rho GTPase signalling to polar growth sites. Nat Plants 6(10):1275–1288
Kurien BT, Scofield RH (2006) Western blotting. Methods 38(4):283–293
Lorrain S, Vailleau F, Balagué C, Roby D (2003) Lesion mimic mutants: keys for deciphering cell death and defense pathways in plants? Trends Plant Sci 8(6):263–271
Lv Q, Li X, Jin X, Sun Y, Wu Y, Wang W, Huang J (2022) Rice OsPUB16 modulates the ‘SAPK9-OsMADS23-OsAOC’ pathway to reduce plant water-deficit tolerance by repressing ABA and JA biosynthesis. PLoS Genet 18(11):e1010520
Mittler R (2002) Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci 7(9):405–410
Mu X, Li J, Dai Z, Xu L, Fan T, Jing T, Chen M, Gou M (2021) Commonly and specifically activated defense responses in Maize Disease Lesion mimic mutants revealed by Integrated Transcriptomics and Metabolomics Analysis. Front Plant Sci 12:638792
Peifer M, Berg S, Reynolds AB (1994) A repeating amino acid motif shared by proteins with diverse cellular roles. Cell 76(5):789–791
Rao Y, Jiao R, Wang S, Wu X, Ye H, Pan C, Li S, Xin D, Zhou W, Dai G, Hu J, Ren D, Wang Y (2021) SPL36 encodes a receptor-like protein kinase that regulates programmed cell death and defense responses in Rice. Rice 14(1):34
Ren D, Hu J, Xu Q, Cui Y, Zhang Y, Zhou T, Rao Y, Xue D, Zeng D, Zhang G, Gao Z, Zhu L, Shen L, Chen G, Guo L, Qian Q (2018) FZP determines grain size and sterile lemma fate in rice. J Exp Bot 69(20):4853–4866
Ruan B, Hua Z, Zhao J, Zhang B, Ren D, Liu C, Yang S, Zhang A, Jiang H, Yu H, Hu J, Zhu L, Chen G, Shen L, Dong G, Zhang G, Zeng D, Guo L, Qian Q, Gao Z (2019) OsACL-A2 negatively regulates cell death and disease resistance in rice. Plant Biotechnol J 17(7):1344–1356
Rubio V, Shen Y, Saijo Y, Liu Y, Gusmaroli G, Dinesh-Kumar SP, Deng XW (2005) An alternative tandem affinity purification strategy applied to Arabidopsis protein complex isolation. Plant J 41(5):767–778
Simeonova E, Sikora A, Charzyńska M, Mostowska AJP (2000) Aspects of programmed cell death during leaf senescence of mono and dicotyledonous plants. Protoplasma 214(1–2):93–101
Sun J, Zhang M, Qi X, Doyle C, Zheng H (2020) Armadillo-repeat kinesin1 interacts with Arabidopsis atlastin RHD3 to move ER with plus-end of microtubules. Nat Commun 11(1):5510
Sun J, Song W, Chang Y, Wang Y, Lu T, Zhang Z (2021) OsLMP1, encoding a deubiquitinase, regulates the Immune response in Rice. Front Plant Sci 12:814465
Takahashi A, Kawasaki T, Henmi K, Shi IK, Kodama O, Satoh H, Shimamoto K (1999) Lesion mimic mutants of rice with alterations in early signaling events of defense. Plant J 17(5):535–545
Toki S, Hara N, Ono K, Onodera H, Tagiri A, Oka S, Tanaka H (2006) Early infection of scutellum tissue with Agrobacterium allows high-speed transformation of rice. Plant J 47(6):969–976
Wang L, Ma H, Fu L, Yao J (2014) Kpna7 interacts with egg-specific nuclear factors in the rainbow trout (Oncorhynchus mykiss). Mol Reprod Dev 81(12):1136–1145
Wang J, Qu B, Dou S, Li L, Yin D, Pang Z, Zhou Z, Tian M, Liu G, Xie Q, Tang D, Chen X, Zhu L (2015) The E3 ligase OsPUB15 interacts with the receptor-like kinase PID2 and regulates plant cell death and innate immunity. BMC Plant Biol 15:49
Wang S, Lei C, Wang J, Ma J, Tang S, Wang C, Zhao K, Tian P, Zhang H, Qi C, Cheng Z, Zhang X, Guo X, Liu L, Wu C, Wan J (2017) SPL33, encoding an eEF1A-like protein, negatively regulates cell death and defense responses in rice. J Exp Bot 68(5):899–913
Wang K, Li S, Chen L, Tian H, Chen C, Fu Y, Du H, Hu Z, Li R, Du Y, Li J, Zhao Q, Du C (2023a) E3 ubiquitin ligase OsPIE3 destabilises the B-lectin receptor-like kinase PID2 to control blast disease resistance in rice. New Phytol 237(5):1826–1842
Wang Y, Abrouk M, Gourdoupis S, Koo DH, Karafiátová M, Molnár I, Holušová K, Doležel J, Athiyannan N, Cavalet-Giorsa E, Jaremko Ł, Poland J, Krattinger SG (2023b) An unusual tandem kinase fusion protein confers leaf rust resistance in wheat. Nat Genet 55(6):914–920
Wolter M, Hollricher K, Salamini F, Schulze-Lefert P (1993) The mlo resistance alleles to powdery mildew infection in barley trigger a developmentally controlled defence mimic phenotype. Mol Gen Genet 239(1–2):122–128
Xu X, Wang H, Liu J, Han S, Lin M, Guo Z, Chen X (2022) OsWRKY62 and OsWRKY76 interact with importin α1s for negative regulation of defensive responses in Rice Nucleus. Rice 15(1):12
Yamanouchi U, Yano M, Lin H, Ashikari M, Yamada K (2002) A rice spotted leaf gene, Spl7, encodes a heat stress transcription factor protein. Proc Natl Acad Sci U S A 99(11):7530–7535
Yoshida MW, Hakozaki M, Goshima G (2023) Armadillo repeat-containing kinesin represents the versatile plus-end-directed transporter in Physcomitrella. Nat Plants 9(5):733–748
Yu G, Matny O, Gourdoupis S, Rayapuram N, Aljedaani FR, Wang YL, Nürnberger T, Johnson R, Crean EE, Saur IM, Gardener C, Yue Y, Kangara N, Steuernagel B, Hayta S, Smedley M, Harwood W, Patpour M, Wu S, Poland J, Jones JDG, Reuber TL, Ronen M, Sharon A, Rouse MN, Xu S, Holušová K, Bartoš J, Molnár I, Karafiátová M, Hirt H, Blilou I, Jaremko Ł, Doležel J, Steffenson BJ, Wulff BBH (2023) The wheat stem rust resistance gene Sr43 encodes an unusual protein kinase. Nat Genet 55(6):921–926
Zeng LR, Qu S, Bordeos A, Yang C, Baraoidan M, Yan H, Xie Q, Nahm BH, Leung H, Wang GL (2004) Spotted leaf 11, a negative regulator of plant cell death and defense, encodes a U-box/armadillo repeat protein endowed with E3 ubiquitin ligase activity. Plant Cell 16(10):2795–2808
Zhang P, Ma X, Liu L, Mao C, Hu Y, Yan B, Guo J, Liu X, Shi J, Lee GS, Pan X, Deng Y, Zhang Z, Kang Z, Qiao Y (2023) MEDIATOR SUBUNIT 16 negatively regulates rice immunity by modulating PATHOGENESIS RELATED 3 activity. Plant Physiol 192(2):1132–1150
Zhang H, Yang J, Liu M, Xu X, Yang L, Liu X, Peng Y, Zhang Z (2024) Early molecular events in the interaction between Magnaporthe oryzae and rice. Phytopathol Res 6(1):9
Zhao H, Wang X, Jia Y, Minkenberg B, Wheatley M, Fan J, Jia MH, Famoso A, Edwards JD, Wamishe Y, Valent B, Wang GL, Yang Y (2018) The rice blast resistance gene ptr encodes an atypical protein required for broad-spectrum disease resistance. Nat Commun 9(1):2039
Zhao J, Liu X, Wang M, Xie L, Wu Z, Yu J, Wang Y, Zhang Z, Jia Y, Liu Q (2022) The miR528-D3 module regulates plant height in rice by modulating the gibberellin and abscisic acid metabolisms. Rice 15:27
Zhao J, Meng X, Zhang Z, Wang M, Nie F, Liu Q (2023) OsLPR5 encoding ferroxidase positively regulates the tolerance to salt stress in rice. Int J Mol Sci 24(9):8115
Zhu X, Ze M, Chern M, Chen X, Wang J (2020) Deciphering rice lesion mimic mutants to understand molecular network governing plant immunity and growth. Rice Sci 27(4):278–288
Zou T, Li G, Liu M, Liu R, Yang S, Wang K, Lu L, Ye Q, Liu J, Liang J, Deng Q, Wang S, Zhu J, Liang Y, Liu H, Yu X, Sun C, Li P, Li S (2023) A ubiquitin-specific protease functions in regulating cell death and immune responses in rice. Plant Cell Environ 46(4):1312–1326
Acknowledgements
Not applicable.
Funding
This work was supported by “Ten-thousand Talents Plan” of Zhejiang Province (2022R52027 to Y.Y.), the funds from the Natural Science Foundation of Zhejiang (LY23C130001 to B.R.), the National Natural Science Foundation of China (32370328 to L.W.) and the Science and Technology Project of Hangzhou (202203B03 to Y. Y).
Author information
Authors and Affiliations
Contributions
Y. Y, Q. Q, and B. R designed the experiments; B. R, H. W, Y. J, J. Q, F. C, Y. Q, M. T, Y. M, and L. W performed the experiments; Y. Y, Q. Q, B. R and H. W analyzed the data; B. R wrote the manuscript. L. W, Y. Y and Q. Q revised the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ruan, B., Wu, H., Jiang, Y. et al. SPL50 Regulates Cell Death and Resistance to Magnaporthe Oryzae in Rice. Rice 17, 51 (2024). https://doi.org/10.1186/s12284-024-00731-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-024-00731-x