
Abstract
Background
High yield and quality determine the commercial potential of rice variety. Brown rice rate (BRR) is a key factor ensuring grain yield and quality in rice. So far, there were few reports about the genes that directly controlled the BRR in rice.
Therefore, dissecting the genetic mechanism of the BRR genes can facilitate improving effective rice supply or edible grain yield.
Results
A double haploid population derived from the cross between Taichung Native 1 (TN1) (an indica variety) and Chunjiang 06 (CJ06) (a japonica variety) was used to investigate the genetic basis of grain milling and appearance traits affecting the BRR. By using a constructed molecular linkage map, four quantitative trait loci (QTLs) for the BRR were detected on chromosomes 1, 8, 9, and 10, respectively. In addition, three QTLs for appearance traits, including grain weight and grain length/width ratio, were detected on chromosomes 6, 9 and 10, respectively. Chromosome segment substitution lines (CSSLs) were established at the qBRR-10 locus. Finally, the qBRR-10 was narrowed to a 39.5 kb region on chromosome 10. In this region, two candidate genes, LOC_Os10g32124 and LOC_Os10g32190, showed significantly differential expression in TN1 and CSSL1-2 compared with CJ06. Histocytological analysis suggested that cell size and hull thickness may be important factors for the BRR.
Conclusion
In the study, the qBRR-10 affected the BRR and was finally located to a region between two markers, P13 and P14. Two candidate genes were selected based on the expression difference between two parents, which facilitated the further cloning of the qBRR-10 gene and largely contributed to improve the grain yield and quality in rice.
Similar content being viewed by others


Background
Rice is a major food staple for human. Currently, food security is becoming an ever more serious problem due to the pressure from continuous population growth and the increasing competition for arable land between food and energy crops (Gao et al., 2013). Therefore, it is urgent to secure grain production, which can be achieved by increasing grain yield and quality (Rosegrant and Cline, 2003). In most cases, the weight of brown rice is directly related to yield. Although rice hulls play a vital role in grain yield, they cannot serve as food for human. Therefore, BRR is an important trait in rice, which is also suggested by the milling quality effect (Tan et al., 2000; Aluko et al., 2004; Wan et al., 2008).
Based on molecular marker linkage maps and genomic sequencing, a large number of genes or quantitative trait loci (QTLs) related to yield traits have been successfully identified and applied in rice production. Dissection and characterization of these genes or QTLs have largely promoted the understanding of underlying mechanisms regulating yield traits in rice (Xing and Zhang, 2010; Guo and Ye, 2014), and have provided very helpful targets for improving grain yield and quality (Xing and Zhang, 2010; Wang et al., 2015b). GRAIN SIZE 3 (GS3) encodes a putative transmembrane protein which has major impacts on grain length and weight as well as minor impacts on grain width and thickness in rice (Fan et al., 2006; Zhang et al., 2014). GRAIN SIZE 5 (GS5) encodes a putative serine carboxypeptidase which controls grain size in rice by regulating grain width, filling and weight (Li et al., 2011). Grain length QTL (qGL3/GL3.1) encodes a putative protein phosphatase with Kelch-like repeat domain which can be used to significantly increase grain yield in rice (Qi et al., 2012; Zhang et al., 2014). GRAIN WIDTH8 (GW8/OsSPL16) encodes a positive regulator of cell proliferation which can be used to promote cell division and grain filling (Wang et al., 2012). GRAIN LENGTH7/GRAIN WIDTH7 (GL7/GW7) encodes a putative microtubule-binding protein which can regulate gain length and grain size diversity (Wang et al., 2015a; Wang et al., 2010). Therefore, function analysis of the genes related to seed traits is very important and can help to understand the genetic mechanisms regulating the BRR.
Generally, japonica varieties have higher BRRs than indica varieties. Wide variations of BRR are evident in rice germplasms, and most modern elite rice varieties show higher BRR than particular landraces (Luo and Yang 1998). Zhang and Peng (2012) reported that there was no significantly difference in the BRR, milled rice rate and head rice rate between reciprocal crosses. Although the factors determining the BRR have been studied for decades, no consensus has been reached. Chen et al. (1998) considered that BRR was mainly controlled by grain direct additive effects and maternal additive effect, and the latter is dominant. These results are in accordance with the additive-dominance model, and indicate that high BRR shows complete dominance or partial dominance over low BRR. However, cytoplasm effect is not significant. Besides, BRR is controlled by nuclear genes but not by maternal effects and cytoplasm effects. However, little attention is focused on the genetic basis of BRR in previous studies. It is reported that hybrid BRR, head rice rate, as well as the male parent and mid-parent values have significantly positive correlations (Tan et al., 2000; Luo et al., 2014). Using a double haploid (DH) population derived from the cross O. sativa × O. glaberrima, Aluko detected three QTLs for the BRR on chromosomes 1, 7, and 8, respectively (Aluko et al., 2004).
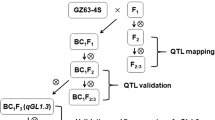
In the present study, a genetic analysis of QTLs determining BRR was carried out using a DH population derived from the cross between the indica rice cultivar Taichung Native 1 (TN1) and the japonica variety Chunjiang 06 (CJ06). Using the developed Chromosome segment substitution lines (CSSLs) population, qBRR-10 was identified and finally narrowed down to a 39.5 kb region between two sequence-tagged site (STS) markers on chromosome 10. Two candidate genes in this region showed significant expression differences in TN1 and CSSL1-2 compared with CJ06. They might be responsible for the BRR. The genetic basis for other grain traits was also tested. The BRR related genes or QTLs identified in this study may be helpful for dissecting the genetic basis of grain yield and quality characteristics, which can facilitate the development of strategies to improve milling quality and grain yield.
Results
Phenotypic Variation of BRR in the DH Population
The BRRs of the DH population and its parents were shown in Fig. 1. Significant differences in the BRR were found between two parents. The Parent CJ06 had a higher BRR reaching 83.9 %. However, the BRR of the parent TN1 was as low as 75.8 %. The BRR showed a continuous distribution from 66 % to 84 % among 120 DH lines, with an apparent peak at 78 %. Transgressive variation was also found in the DH population (Fig. 1).

The distribution of brown rice rate in the DH population
QTL Analysis for BRR
The whole genome was scanned for detecting QTLs using Mapmaker/ QTL1.1B with a LOD threshold of 2.4. The analysis of the DH population identified four QTLs for the BRR located on chromosomes 1, 8, 9 and 10, respectively (Fig. 2, Additional file 1: Table S1). The QTL located in the interval RM258-RM1108 on chromosome 10 showed the largest effect on the BRR trait with an LOD score of 5.95, and accounted for 23.1 % of the phenotypic variances (Fig. 3). The effects of the other three QTLs were much smaller. The alleles in all of the four QTLs from CJ06 contributed to the increased BRR.

Locations of the QTLs for appearance and milling quality traits of rice grains. Only those portions of the linkage maps where QTLs were detected are shown

Molecular linkage map of rice chromosome 10 showing the location of qBRR-10. The genetic distance (Kosambi, centiMorgans) and marker name are shown on the left and right of the chromosome, respectively
Correlation Analysis
Correlation analysis between the BRR and other seed traits was performed using the software SAS 8.0. Other seed traits included grain length, grain width, grain length/width ratio, grain thickness, grain weight, brown rice length, brown rice width, brown rice length/width ratio, brown rice thickness and brown rice weight. The distribution of these traits in the DH population was shown in Additional file 1: Figure S1. Our results suggest that BRR is positively correlated with the brown rice weight, brown rice width, and has no relationship with other seed traits. Moreover, there were extremely significant relationships between the BRR and brown rice weight, grain thickness and brown rice thickness (Table 1), and our measurement data also supported it (Additional file 1: Figure S2). Meanwhile, the QTLs for other seed traits were also investigated using the same linkage map. Three QTLs were detected: one for grain length/width ratio and two for grain weight, and the intervals of two grain weight QTLs were near from those of BRR QTLs (Fig. 2, Additional file 1: Table S2).
Substitution Mapping of qBRR-10
Three lines from the DH population were selected to backcross with CJ06 for 4 rounds. According to the QTL analysis and a previous genetic linkage map, the SSR markers RM271 and RM304 were used in marker-assisted selection for segregating the progenies carrying qBBR-10 during every generation of backcross. After 4 rounds of backcrosses with CJ06, the BC4F1 and BC4F2 generations were scanned with a set of 71 SSR markers, which were uniformly distributed on a previous linkage map (Additional file 1: Table S4). The plant CSSL1-2 containing a small amount of TN1 DNA was selected. It carried a homozygous introgression across the entire qBRR-10 region, but without any introgression across qBRR-1, qBRR-8 and qBRR-9 regions on chromosomes 1, 8, and 9, respectively (Fig. 4a and b). The BRR of CSSL1-2 was significantly lower than that of its recurrent parent CJ06, but was much more similar to that of TN1 (Fig. 4c).

The development of CSSL. a QTL analysis for brown rice rate in DH population. Circle centers indicate positions of QTLs on the rice chromosomes. Circle sizes indicate contributions to explained variation for brown rice rate. b Graphical genotype of CSSL1-2 (a substitution line of chromosome 10. Black bar indicates the genome fragment from TN1; the other parts were from CJ06. c The brown rice rate for CJ06, TN1 and CSSL1-2
Fine Mapping of qBRR-10 and Analyzing Candidate Genes
The simple sequence repeat (SSR) markers RM271 and RM304 across the qBRR-10 target region were used to determine the recombination breakpoints in segregating progenies derived from the cross between CSSL1-2 and CJ06. A total of 235 selected plants from 910 BC4F2 progenies were cultivated in the paddies to gain enough seeds for the BRR evaluation and further study. These plants could be divided into two subgroups based on the detected BRR values. One group was with higher BRR, while the other group was with lower BRR. A total of 41 plants with extremely low BRRs (BRR < =72.6) were selected for fine mapping and the use of heterozygous plants or confounding plants was avoided. Then 9 and 7 recombination events between the marker and qBRR-10 were identified based on analyses using RM271 and RM304, respectively. The STS markers P1, P2 and P3 revealed 6, 5 and 3 recombinants, respectively; the markers P11, P10 and P9 indicated 4, 3 and 2 recombinants, respectively (Fig. 5). Therefore, the qBRR-10 locus was finally located to a region between two STS markers, P13 and P14 on the BAC OSJNBa0013H08 (Fig. 5 and Additional file 1: Table S3). By comparison, it was found that the interval was 39.5 Kb. The target region contains 6 predicted genes (LOC_Os10g32124, LOC_Os10g32140, LOC_Os10g321250, LOC_Os10g32160, LOC_Os10g32170 and LOC_Os10g32190) based on Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/cgi-bin/gbrowse/rice/). We performed a quantitative reverse transcription-PCR (qPCR) analysis to determine the candidate genes, and the analysis results showed that no significant difference was detected in LOC_Os10g32140, LOC_Os10g32150, LOC_Os10g32160 and LOC_Os10g32170 among CJ06, TN1 and CSSL1-2 (Fig. 6 and Additional file 1: Table S3). On the contrary, LOC_Os10g32124 and LOC_Os10g32190 showed significantly different expression levels in TN1 and CSSL1-2 compared with CJ06 (Fig. 6 and Additional file 1: Table S3). Therefore, LOC_Os10g32124 and LOC_Os10g32190 were likely to be the candidate genes for qBRR-10.

Fine mapping of qBRR-10

The qPCR analysis of predicted genes in CJ06, TN1 and CSSL1-2
Histocytological Analysis
Because the hull percentage is negatively correlated with milling quality (Jongkaewwattana and Geng 2001), we examined the hulls of CJ06, TN1 and CSSL1-2 using paraffin section and scanning electron microscopy (SEM) to reveal the impacts of hull effect on the BRR. Hulls are comprised of four layers: silicified cells, fibrous sclerenchyma, spongy parenchymatous cells and nonsilicified cells from outside to inside (Ren et al., 2013). The hulls of TN1 and CSSL1-2 showed larger nonsilicified and silicified cells compared to CJ06 (Fig. 7a, b, e, f, I and j). SEM analysis showed that TN1 and CSSL1-2 hulls were thicker than those of CJ06 (Fig. 7c, g and k, and Additional file 1: Figure S2), which was consistent with the results of paraffin sections. These results suggest that qBRR-10 is involved in regulating hull development, and CJ06 variety has higher BRR and provides more grain productivity since the percentage of hull is negatively correlated with the BRR. A higher cell density (i.e., protrusions ) was also observed in the outer surface of CJ06 hulls than those of TN1 and CSSL1-2, which was consistent with the observation that the grains of CJ06 were longer than those of TN1 (Figs. 7d, h and l). Hulls control the vital exchanges of gas, water and nutrients with environment, as well as affect seed development and grain filling (Javelle et al., 2011). These results provide valuable background information on the organization of hull tissues, which facilitate the understanding of molecular mechanisms determining BRR.

Histocytological analysis of hulls in CJ06, TN1 and CSSL1-2. a paraffin section of hull in CJ06. b partial magnification of black box region in a. c hull thickness in CJ06. d Epidermal surface of hull in CJ06. e paraffin section of hull in TN1. f partial magnification of black box region in e. g hull thickness in TN1. h Epidermal surface of hull in TN1. i paraffin section of hull in CSSL1-2. j partial magnification of black box region in i. k, hull thickness in CSSL1-2. l Epidermal surface of hull in CSSL1-2. Bars = 200 μm in a, b, e, f, i, j and 20 μm in c, d, g, h, k, l
Discussion
Rice grain consists of an inner brown rice (edible part) and a hull (inedible part) reciprocally interlocked by the lemma and palea (Yang, 2005). Converting photosynthetic products to people edible part or brown rice as far as possible was one of the effective methods to increase the “output” of rice. Therefore, increasing the grain the BRR or decreasing the hull percentage played a vital role in improvement of the actual grain yield. However, BRR was related to many seed traits, including grain length, grain width, grain weight, brown rice weight and hull weight etc. Generally, grain length, grain width, grain length/width ratio and the translucency of endosperm directly determined grain appearance and quality (Tan et al., 2000; Koutroubas et al., 2004; Wang et al., 2015a; Xing and Zhang, 2005; Li et al., 2014). Currently, a lot of QTLs and genes related to these traits had been cloned, such as GRAIN WIDTH2 (GW2), GS3, qGL3/GL3.1, GW8/OsSPL16, SMALL AND ROUND SEED 3 (SRS3), POSITIVE REGULATOR OF GRAIN LENGTH 1 (PGL1) and GL7/GW7 (Fan et al., 2006; Song et al., 2007, Kanako et al., 2010, Heang and Sassa 2012, Wang et al., 2012, Zhang et al., 2014, Wang et al., 2012; Wang et al., 2015b). These QTLs and genes controlled the brown rice size and weight by regulation of cell proliferation and expansion of the hull, and further promoted the application of molecular breeding technology. These results had significant implications for the improvement of effective rice supply or edible grain and that the hull may be involved in regulation of brown rice yield or BRR. So far, there were few reports about the genes that directly controlled the BRR in rice. Therefore, dissecting the genetic mechanism of the BRR genes can facilitate to improve effective rice supply or edible grain yield.
In this study, the fine mapped qBRR-10, a major potential QTL controlling the BRR, was reported. The major QTL for BRR detected in the present study on chromosome 10 was different from the results of previous studies (Aluko et al., 2004; Kepiro et al., 2008). However, there was a QTL locus for rice bran on chromosome 10 (Aluko et al., 2004), which was located in the same region with the qBRR-10. Perhaps the major QTL on chromosome 10 controlled both BRR and rice bran. The other three BRR loci including the qBRR-1, qBRR-8 and qBRR-9 had minor effects. Finally, we fine mapped the qBRR-10 gene between STS markers P13 and P14 on the long arm of chromosome 10 with a 39.5 kb physical distance in Nipponbare. In this region, there were six open reading frames and no known genes or QTLs were reported, so the qBRR-10 gene was considered as a novel gene. Two candidate genes, LOC_Os10g32124 and LOC_Os10g32190, encoding proteins with unknown functions, were identified based on their differences in gene transcription level. Thus more work, such as complementary test, was needed to examine whether LOC_Os10g32124 or LOC_Os10g32190 was the target gene for the qBRR-10.
Hulls, main parts of grains, affected grain size and BRR which were the main factors affecting the actual grain yield (Yang, 2009; Song et al., 2007; Xing and Zhang, 2010; Luo et al., 2014; Song et al., 2015). GW2, a major grain width and weight QTL, increased grain width and weight by enlarging hulls. The NIL (GW2) hull showed more cells than that of FAZ1, and resulted in a higher brown rice yield and BRR (Song et al., 2007). ABNORMAL FLOWER AND DWARF1 (AFD1) determined plant height, floral development and grain yield. The thicker hulls in the afd1 mutant caused reduced brown rice yield than that of the wild type. But, no differences were observed in the brown rice size and weight between the wild type and afd1 mutant when parts of both hulls were removed (Ren et al., 2015). Meantime, our data also showed that brown rice of CJ06 was thicker than that of TN1 and CSSL1-2 (Additional file 1: Figure S2). These findings suggested that the rice hull and brown rice thickness may be involved in regulation of brown rice yield and BRR. In the study, paraffin sections and SEM analysis revealed that the TN1 hulls had larger nonsilicified and silicified cells, and thicker layers. The results indicated that cell size and hull layers may be important factors affecting the BRR. It provides very valuable information for cloning and functional analysis of the qBRR-10 gene. Moreover, three more traits were detected in the present study, i.e., one for brown grain length/width ratio (qGL/W-6), and two for grain weight (qGW-9 and qGW-10). The QTL on chromosome 6 had significant impact on grain length/width ratio, which had not been reported before. Thus, these QTLs identified in this study are very useful for studying the BRR and grain weight. The identification of qBRR-10, qGL/W-6, qGW-9, and qGW-10 will receive much attention because of their importance on improving milling quality and rice yield.
In rice production, many factors can affect the BRR, such as processing equipments and environmental conditions. The content of inorganic elements can also affect the BRR. The study performed by Juliano and Villarea (1993)) suggests that the grains containing more minerals have higher brittleness, thus are more easily processed and lead to a lower BRR. The results from Zhang also showed that high iron and zinc contents caused a lower BRR in rice (Zhang and Peng, 1997). Therefore, more work is needed to determine the target gene of the qBRR-10 and understand the genetic mechanisms controlling the BRR, which will provide a new opportunity for increasing the BRR and thus further improving grain yield and quality.
Conclusion
In this study, using the DH population, 4 QTLs were detected in Hangzhou and Hainan to control the BRR. With the BC4F2 progenies derived from the cross between CSSL1-2 and CJ06, a new major QTL (qBRR-10) for the BRR, was fine mapped within 39.5 kb physical interval on chromosome 10. Two candidate genes were finally selected based on the difference in the transcriptional expression. The cloning and genetic mechanism study of the qBRR-10 will facilitate increasing the BRR and further improving rice yield and milling quality.
Methods
DH Population
We constructed the DH population to detect BRR related QTLs in our lab (Ma et al. 2009a). The indica cultivar ‘TN1’ and the japonica cultivar ‘CJ06’ of rice (Oryza sativa L.) were used as parents to generate hybrids. TN1 had a lower BRR, while CJ06 was with higher BRR. The anthers from F1 plants were collected and cultured on the inducing medium SK3. After natural doubling or treatment with colchicines, DH plants were obtained, and have been successfully used for studying whitebacked planthopper resistances and ligule length (Sogawa et al., 2005; Sogawa et al., 2004; Zeng et al., 1997; Su et al., 2011). The DH population and its parents were transplanted in a density of 20 cm × 20 cm at rice growing season in the experimental farms at China National Rice Research Institute, Hangzhou, or in Lingshui County, Hainan Province. Each DH line was planted in six rows, and each row contained six plants in the year 2010 and 2011.
The Development of CSSLs
To obtain CSSLs, the plants carrying TN1 genotype at the flanking region of qBRR-10 were selected to backcross with CJ06 for 4 rounds. The SSR markers RM304 and RM271 were simultaneously used to identify the plants containing TN1 genotype in backcross lines. A set of 71 SSR markers (Additional file 1: Table S4) uniformly distributed on a previous linkage map (Sogawa et al., 2005; Sogawa et al., 2004; Zeng et al., 2009) were used to select the individual plants containing the least TN1 DNA in BC4F1 lines. CSSL1-2 was selected to fine map the qBRR-10 genes. It contained a small amount of TN1 DNA in its genetic background, and carried a homozygous introgression across the entire qBRR-10 region and without any introgression across qBRR-1, qBRR-8 and qBRR-9 regions on chromosomes 1, 8, and 9 respectively.
DNA Extraction and PCR Analysis
Total DNA was extracted from fresh rice leaves via the CTAB method described by Murray and Thompson (1980) with slight modifications. PCR was performed in 20 μL reaction mixture containing 0.2 mM of each primer, 200 mM dNTP mix, 50 mM KCl, 10 mM TRIS-HCl, pH 8.3, 1.5 mM MgCl2, 0.1 % Triton X-100 and 1 unit of Taq polymerase. The PCR protocol was: initial denaturation at 94 °C for 5 min, followed by 35 cycles performed at 94 °C for 1 m, 55 °C for 45 s and 72 °C for 50 s, and then a final extension at 72 °C for 10 m. The PCR reaction was performed in PTC-225 tetrad (MJ Research, Watertown, MA) (Su et al., 2011). The PCR products were separated via electrophoresis on a 4.0-5.0 % (w/v) agarose gel and stained with nucleic acid dye.
Marker Development
Primers were designed around qBRR-10 on chromosome 10 to distinguish CJ06 and TN1 (Additional file 1: Table S3). The STS markers were developed based on the sequence differences between the indica var. 93-11 and the japonica var. Nipponbare (http://www.ncbi.nlm.nih.gov) (Additional file 1: Table S3) (Su et al., 2011). This strategy was helpful because of the close genetic relationship between CJ06 and Nipponbare and the relative sequence similarity between TN1 and 93-11. The sequences were aligned using the SeqMan program of DNA star (Gene-Codes). Meanwhile, insertions and deletions were identified. Primers flanking the indels were designed using the software Primer Premier 5.0 and tested on the parents.
Grain Phenotyping
Rice grains were harvested and stored at room temperature for at least 3 months before processing. For each rice line, the hulls of 50 g rough grains were removed by a huller (manufactured by Jiading Food and Oil Machinery Factory, Shanghai, China) according to the National Standards NY 147-88. The whole and broken kernels obtained after milling were added to define the total milling yield. BRR was calculated with dividing total grain weight by brown rice weight.
The quality of appearance is determined by grain length, width, width/length ratio, size and shape, and translucency of endosperm (Juliano and Villareal., 1993; Unnevehr et al., 2006). Several appearance-related traits were also used to test whether they have any relationship with BRR. The length and width of 10 fully formed grains from each DH line were measured using a vernier caliper. The length/width ratio of the grains was calculated with dividing grain width by grain length. It reflects the shape of grains. Ten randomly selected unbroken brown rice grains of each DH line were lined up to measure grain length, width, and length/width ratio in the same way as described above. The thickness of 10 grains and 10 brown rice grains in each line were also measured using a vernier caliper.
Data Analysis and QTL Mapping
The linkage map used in this study was established by the China National Rice Research Institute (Zeng et al., 2009). Recently, this linkage map has been widely used in QTL mapping (Ma et al. 2009b; Zeng et al., 2009). Analysis of variance for all phenotypic characters was performed using the JMP statistical package, version 7.0 for Windows (SAS Institute Inc., Cary, NC) (Su et al., 2011). QTL mapping for BRR was conducted using Mapmaker/QTL 1.1. The presence of QTL was claimed when LOD score was larger than 2.4. The genetic variances explained by each QTL and QTL additive effect were calculated. Moreover, the discovered QTLs were named according to the standard nomenclature, as suggested by McCouch et al. (1997).
Paraffin Sections and Microscopic Analysis
The hulls of CJ06 and TN1 were collected at heading stage and fixed overnight at 4 °C in the solution containing 50 % ethanol, 0.9 M glacial acetic acid and 3.7 % formaldehyde, then dehydrated by a graded ethanol series, infiltrated with xylene, and embedded in paraffin (Sigma) (Ren et al. 2013). The 8 um-thick sections were transferred onto poly-L-Lys-coated glass slides, deparaffinized in xylene, and dehydrated through an ethanol series. The sections were sequentially stained with 1 % safranine (Amresco) and 1 % Fast Green (Amresco), then dehydrated through an ethanol series, infiltrated with xylene, and finally mounted beneath a coverslip (Ren et al., 2013). Light microscopy was performed using a Nikon SMZ1500 microscope. The samples were also examined using a Hitachi S-3400 scanning electron microscope.
RNA Isolation and Qpcr Analysis
Total RNA was isolated using the RNeasy Plant Mini Kit from Watson. The first strand of complementary DNA was synthesized from 2 mg total RNA using oligo(dT)18 primers in a 25 mL reaction mixture from the SuperScript III Reverse Transcriptase Kit (Invitrogen). Reverse-transcribed RNA (0.5 mL) was used as PCR template for gene-specific primers (Additional file 1: Table S3) (Ren et al., 2013). The qPCR analysis was performed using SYBR Supermix Kit (Bio-Rad) in ABI Prism 7000 Sequence Detection System. At least three replicates were performed, and the mean expression level for each gene was calculated.
Abbreviations
- BRR:
-
Brown rice rate
- QTLs:
-
Quantitative trait loci
- CSSLs:
-
Chromosome segment substitution lines
- DH:
-
Double haploid
- STS:
-
Sequence-tagged site
- SSR:
-
Simple sequence repeat
- SEM:
-
Scanning electron microscopy
References
Aluko G, Martinez C, Tohme J, Castano C, Bergman C, Oard JH (2004) QTL mapping of grain quality traits from the interspecific cross Oryza sativa x O. glaberrima. Theor Appl Genet 109:630–639
Chen JG, Song GQ, Qu SH, Zhou Y (1998) Direct and maternal genetic effects on grain quality characters in early hybrid rice. Chinese J of Rice Sci 12:79–84
Fan C, Xing Y, Mao H, Lu T, Han B, Xu C et al (2006) GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112:1164–1171
Gao ZY, Zhao SC, He WM, Guo LB, Peng YL, Wang JJ et al (2013) Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. Proc Natl Acad Sci U S A 110:14492–14497
Guo LB, Ye GY (2014) Use of major quantitative trait loci to improve grain yield of rice. Rice Sci 21:65–82
Heang D, Sassa H (2012) Antagonistic actions of HLH/bHLH proteins are involved in grain length and weight in rice. Plos One 2:e31325
Javelle M, Vernoud V, Rogowsky PM, Ingram GC (2011) Epidermis: the formation and functions of a fundamental plant tissue. New Phytol 189:17–39
Jongkaewwattana S, Geng S (2001) Inter-relationships amongst grain characteristics, grain-filling parameters and rice (Oryza sativa L.) milling quality. J Agron Crop Sci 187:223–229
Juliano B, and Villarea C (1993) Grain quality evaluation of world rices. Int. Rice Res. Inst, Manila, Philippines,p 205.
Kanako K, Shigeru K, Katsuyuki O, Yuki A, Tsuyu A, Izumi K et al (2010) A novel kinesin 13 protein regulating rice seed length. Plant Cell Physiol 8:1315–1329
Kepiro J, McClung A, Chen M, Yeater K, Fjellstrom R (2008) Mapping QTLs for milling yield and grain characteristics in a tropical japonica long grain cross. J Cearal Sci 48:477–485
Koutroubas S, Mazzini F, Pons B, Ntanos D (2004) Grain quality variation and relationships with morpho-physiological traits in rice (Oryza sativa L.) genetic resources in Europe. Field Crop Res 86:115–130
Li YB, Fan CC, Xing YZ, Jiang YH, Luo LJ, Sun L et al (2011) Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet 243:1266–1270
Li YB, Fan CC, Xing YZ, Yun P, Luo LJ, Yan B et al (2014) Chalk5 encodes a vacuolar H + -translocating pyrophosphatase influencing grain. Nat Genet 46:398–404
Luo LL, Zhang YX, Chen DB, Zhan XD, Shen XH, Cheng SH et al (2014) Mapping QTL of rice hull thickness and related traits in hybrid rice Xieyou 9308. Rice Sci 21:29–38
Luo Y, Yang J (1998) Elite Germplasm Evaluation in China., p 3
Ma L, Bao J, Guo L, Zeng D, Li X, Ji Z et al (2009a) Quantitative trait Loci for panicle layer uniformity identified in doubled haploid lines of rice in two environments. J Integr Plant Biol 51:818–824
Ma L, Yang C, Zeng D, Cai J, Li X, Ji Z et al (2009b) Mapping QTLs for heading synchrony in a doubled haploid population of rice in two environments. J Genet Genomics 36:297–304
McCouch SR CY, Yano M, Paul E, Blinstrub M, Morishima H, T K (1997) Report on QTL nomenclature. Rice Genet Newsl 14: 11-13.
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8:4321–4326
Ren DY, Li YF, Zhao FM, Sang XC, Shi JQ, Wang N et al (2013) MULTI-FLORET SPIKELET1, which encodes an AP2/ERF protein, determines spikelet meristem fate and sterile lemma identity in rice. Plant Physiol 162:872–884
Ren DY, Rao YC, Wu LW, Xun QK, Li ZZ, Yu HP et al (2015) The pleiotropic ABNORMAL FLOWER AND DWARF1 affects plant height, floral development and grain yield in rice. J Integr Plant Biol doi: 10.1111/jipb.12441.
Rosegrant M, Cline S (2003) Global food security: challenges and policies. Science 302:1917–1919
Sogawa K, Qian Q, Zeng DL, Hu J, Zeng LJ (2005) Differential expression of whitebacked planthopper resistance in the japonica/indica doubled haploid rice population under field evaluation and seedbox screening test. Rice Sci 12:63–67
Sogawa K, Sun ZX, Qian Q, Zeng DL (2004) Phenotypic expression of whitebacked planthopper resistance in the newly established japonica/indica doubled haploid rice population. Rice Sci 11:155–160
Song X, Huang W, Shi M, Zhu M, Lin H (2007) A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39:623–630
Song XJ, Kuroha T, Ayano M, Furuta T, Nagai K, Komeda N et al (2015) Rare allele of a previously unidentified histone H4 acetyltransferase enhances grain weight, yield, and plant biomass in rice. Proc Natl Acad Sci U S A 112:76–81
Su Y, Rao YC, Hu SK, Yang YL, Gao ZY, Zhang GH et al (2011) Map-based cloning proves qGC-6, a major QTL for gel consistency of japonica/indica cross, responds by Waxyin rice (Oryza sativa L.). Theor Appl Genet 123:859–867
Tan Y, Xing Y, Li J, Yu S, Xu C, Zhang Q (2000) Genetic bases of appearance quality of rice grains in Shanyou 63, an elite rice hybrid. Theor Appl Genet 101:823–829
Qi P, Lin YS, Song XJ, Shen JB, Huang W, Shan JX et al (2012) The novel quantitative trait locus GL3.1 controls rice grain size and yield by regulating Cyclin-T1;3. Cell Res 22:1666–1680
Wan X, Wan J, Jiang L, Wang J, Zhai H, Weng J et al (2006) QTL analysis for rice grain length and fine mapping of an identified QTL with stable and major effects. Theor Appl Genet 112:1258–1270
Wang E, Wang JJ, Zhu XD, Hao W, Wang LY, Li Q et al (2008) Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat Genet 40:1370–1374
Wang S, Li S, Liu Q, Wu K, Zhang J, Wang S, et al (2015a) The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat Genet 47: 949954.
Wang SK, Wu K, Yuan QB, Liu XY, Liu ZB, Lin XY et al (2012) Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet 44:950–955
Wang Y, Xiong G, Hu J, Jiang L, Yu H, Xu J, et al (2015b) Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat Genet doi:10.1038/ng.3346.
Xing Y, Zhang Q (2010) Genetic and molecular basis of rice yield. Annu Rev Plant Biol 61:421–442
Yang HY (2005) Rice Reproductive Biology. Zhejiang University Press, Hangzhou, pp 19–25
Zeng DL, Hu J, Dong GJ, Liu J, Zeng LJ, Zhang GH et al (2009) Quantitative trait loci mapping of flag-leaf ligule length in rice and alignment with ZmLG1 gene. J Integr Plant Biol 51:360–366
Zhang M, Peng Z (1997) Gnetic analysis of milling quality for color rice. Crop Varieties 77:1–4
Zhang X, Wang J, Huang J, Lan H, Wang C, Yin C et al (2012) Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proc Natl Acad Sci U S A 109:21534–21539
Zhang Y, Li J, Zhou JW, Xu P, Deng XN, Yang F et al (2014) Seed length controlled by same locus in four different AA genome species of Genus Oryza. Rice Sci 21:20–28
Zuo JR,Li JY (2014) Molecular dissection of complex agronomic traits of rice: a team effort by Chinese scientists in recent years. Natl Sci Rev doi: 10.1093/nsr/nwt004.
Acknowledgments
This work was supported by the National Science and Technology Major Project (2016ZX08009003-003), National Natural Science Foundation of China (Grant Nos. 91435105, 31171531, 31401464 and 31201183), Zhejiang Provincial Natural Science Foundation of China (Grant No. Y3110194, LY16C130001), China Postdoctoral Science Foundation (Grant No, 2014M561108), the Open Foundation from Zhejiang Provincial Top Key Discipline of Biology.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DL Z and Q Q designed this research. DY R, YC R, LC H, YJ L, M L, J H and GH Z performed this research. DY R, YC R, L Z, ZY G, GJ D and LB G analyzed data. DY R and DL Z wrote the paper. All authors approved the final manuscript.
Additional file
Additional file 1: Figure S1.
The distribution of other seed traits in the DH population. Figure S2. The thickness of hull and brown rice in CJ06, TN1 and CSSL1-2. Table S1. QTL identified for brown rice rate in the DH population. Table S2. QTL identified for other related traits in the DH population. Table S3. Primers used in the study. Table S4. SSR markers selected to identify the CSSLs. (DOC 171 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ren, D., Rao, Y., Huang, L. et al. Fine Mapping Identifies a New QTL for Brown Rice Rate in Rice (Oryza Sativa L.). Rice 9, 4 (2016). https://doi.org/10.1186/s12284-016-0076-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-016-0076-7