
Abstract
Cyclic adenosine 3’, 5’ monophosphate (cAMP)-dependent Protein Kinase A (PKA) is a multi-functional serine/threonine kinase that regulates a wide variety of physiological processes including gene transcription, metabolism, and synaptic plasticity. Genomic sequencing studies have identified both germline and somatic variants of the catalytic and regulatory subunits of PKA in patients with metabolic and neurodevelopmental disorders. In this review we discuss the classical cAMP/PKA signaling pathway and the disease phenotypes that result from PKA variants. This review highlights distinct isoform-specific cognitive deficits that occur in both PKA catalytic and regulatory subunits, and how tissue-specific distribution of these isoforms may contribute to neurodevelopmental disorders in comparison to more generalized endocrine dysfunction.
Similar content being viewed by others


Background
Regulation of signal transduction by phosphorylation is one of the most well studied post-translational modifications to date. Seminal work from many laboratories identified phosphorylation to be a critical form of regulation in neuronal signaling in response to Ca2+ transients [1,2,3,4]. The dependence of this phosphorylation on cyclic adenosine 3’,5’ monophosphate (cAMP) would help differentiate Protein Kinase A (PKA) from Ca2+/Calmodulin-dependent protein kinase II (CaMKII) as critical Ca2+-sensitive messengers in the brain [5, 6]. It is now well established that PKA plays a central role in the induction of both functional and structural changes in neurons such as long-term potentiation (LTP), and long-term depression (LTD) [7,8,9] (For reviews see Abel and Nguyen 2008 and Christensen and Nairn 2021 [10, 11].
In addition to signal transduction in the adult brain, there is emerging evidence that suggests PKA plays a critical role in neurodevelopment, and pathogenic variants of PKA can produce neurodevelopmental disorders that impair learning and memory, cognition, motor function and coordination, pain sensing, and social behavior. In this review, we will discuss the known genetic risk factors and functional outputs of the cAMP signaling pathway and their contributions to neurodevelopmental disorders.
PKA structure/function
PKA is a basophilic serine/threonine kinase with broad substrate specificity, regulating diverse signaling pathways from glucose homeostasis to gene transcription. In the native state, the PKA holoenzyme exists as a hetero-tetrameric complex of two catalytic subunits (C subunits) autoinhibited by two regulatory subunits (R subunits). Binding of cAMP to the R subunits promotes a large conformational change that unleashes the C subunits from their dimeric holoenzyme and promotes phosphorylation of downstream targets. Due to its broad substrate specificity, cells take advantage of targeted PKA phosphorylation through subcellular localization and anchoring of the R subunits to discrete compartments by association with A-Kinase Associated Proteins (AKAP’s). Localized phosphodiesterase (PDE) activity further controls cAMP signaling and activation of PKA in discrete compartments within the cell [12,13,14].
The catalytic subunit
Eukaryotic protein kinases have a small N-terminal lobe with 5 β strands and 3 α helices, and a larger, mostly helical, C-terminal lobe, connected through a disordered hinge region that binds Mg2+/adenosine 5’-triphosphate (ATP) between them (Fig. 1A). The majority of PKA C subunits expressed in the human body are encoded by two genes – PRKACA and PRKACB, which gives rise to the Cα and Cβ catalytic isozymes [12,13,14,15]. Two other putative PKA C subunit related genes, PRKACG and PRKX which encode the Cγ and Cχ isozymes, have also been identified, but their biological relevance is currently unclear [16,17,18]. The gene that encodes the predominant isoform, Cα, is located on the reverse strand of chromosome 19 at p13.1 and is ubiquitously expressed in all tissues [19]. Alternative splicing produces three splice-site isoforms of the Cα subunit, creating Cα1, Cα2, and Cα3. Cα1 is the predominant PKA C subunit and is widely expressed in most tissues. This isoform has a longer N-terminal transcript compared to the Cα2 isoform, which is specifically expressed in the testes [20]. Cα3 is the longest Cα isozyme but has not been widely studied [13, 21, 22] (Fig. 1B).

The Catalytic Subunit: A Representative AlphaFold Structure of the Cα1 catalytic subunit of PKA. The protein N-terminus (MGNAAAAKGSEQES) is highlighted in hot pink, and the more conserved regions of the catalytic subunit are colored in green. B Multiple sequence alignment of the N-terminus for the PKA catalytic subunit isoforms Cα1, Cα2, Cα3, Cβ1, Cγ, and Cχ
In contrast, PRKACB has four alternative 5’ exons that can produce up to 16 different, catalytically active splice isoforms [12, 23,24,25,26]. PRKACB shares about 93% sequence homology with PRKACA and encode for a structurally and functionally similar catalytic domain core [27]. Like Cα1, Cβ1 is ubiquitously expressed in most tissues [25]. Expression of other PRKACB variants are more distinct. Cβ2 is enriched in brain and lymphoid tissues [25, 28], while Cβ3 and Cβ4 are brain specific isozymes [23, 25]. In situ hybridization studies have indicated high levels of expression for Cβ1 in the thalamic areas, dentate gyrus, and pyramidal cells of the hippocampus, while Cβ2 hybridization was more specific to the prelimbic cortex, bed nucleus of the stria terminalis, amygdala, and ventral medial hypothalamic nucleus [23].
The regulatory subunit
The R subunits are encoded by four genes: PRKAR1A, PRKAR1B, PRKAR2A, and PRKAR2B. When PKA holoenzymes were first being purified, two major peaks were observed upon elution from strong anion exchange columns and led to the naming of Type I and Type II PKA holoenzymes [29,30,31,32]. The α and β isoforms of each Type of R subunit were discovered later through molecular cloning experiments [33,34,35,36], resulting in the expression of the RIα, RIβ, RIIα, and RIIβ isoforms. These isoforms all encode for the same general structural features including a Dimerization/Docking (D/D) domain, a small inhibitory segment, and two cyclic nucleotide binding (CNB) domains, typically referred to as CNB-A and CNB-B (Fig. 2A, B). The ratio of Type I and Type II PKA holoenzymes varies among tissues, but many studies suggest that actively proliferating or differentiating tissues express higher ratios of Type I holoenzymes, while terminally differentiated or mature tissues have higher concentrations of Type II holoenzymes (for a review, see Cho-Chung Y. 1995 [37]).

The Regulatory Subunit: A Schematic representation of the structural domains encoded by the PKA regulatory subunits. B Two representations of the predicted AlphaFold structure of a PRKAR1B dimer. The DD domain is colored in magenta, the inhibitory segment is colored in grey, CNB-A in blue, and CNB-B in orange. The linker region between the two CNB’s is colored in black
While they all have the same structural features, tissue distribution of the R subunits varies. Both the RIα and RIIα subunits are ubiquitously expressed in most tissues [33, 35]. In contrast, the RIβ subunit is primarily expressed in the brain [38] and the RIIβ subunit is expressed in the brain, endocrine tissues, fat, and reproductive organs [39,40,41].
The docking/dimerization domain
As the name suggests, the D/D domain serves two important roles for the PKA holoenzyme: dimerization of two regulatory subunits and “docking” of the PKA holoenzyme into specific subcellular compartments by binding to an amphipathic α-helix in AKAPs [42, 43]. AKAP specificity to R subunits is mediated by amino acids that occupy the groove formed within the D/D domain [42], which allows for PKA C subunits to be compartmentalized in a cell/tissue dependent manner for highly specific cAMP signaling to occur.
While the α and β isoforms of Type I and Type II R subunits are known to form isoform-specific heterodimers, there appears to be no significant evidence for heterodimerization between Type I and Type II R subunits. Additionally, holoenzymes prefer to form homodimers of α and β R subunits of the same isoform [44,45,46]. The bias toward homodimerization of α and β subunits within each type may provide an additional layer of PKA signaling regulation in a tissue-dependent manner based on relative expression levels of these isoforms.
The cyclic nucleotide binding domains
For PKA, each R subunit contains two CNB’s, often denoted as the A-site for the more N-terminal lobe and the B-site for the more C-terminal lobe. These two sites have drastically different affinities for cAMP, with the A-site having faster on and off rates of cAMP compared to the B-site. The two CNB’s form an allosteric network that provides a basis for cooperative activation of PKA by cAMP. Upon binding cAMP to the A-site, a large conformational change between the two CNB’s occurs that allosterically modulates binding of cAMP to the B-site. Phosphorylation of downstream PKA targets can only occur after cAMP binds to both the A- and B-sites to induce dissociation of the C subunits from the R subunits. The general conformation of the A and B-sites are drastically different in crystal structures of Type I and Type II R subunits, which may provide a structural basis for differences in Type I and Type II holoenzyme cooperativity and sensitivity to activation by cAMP [47,48,49].
Activation of PKA by GPCR’s
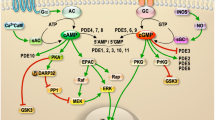
Neuronal function primarily responds to two main mechanisms – fast synaptic transmission through ionotropic receptors and slow synaptic transmission through metabotropic or G protein-coupled receptors (GPCR’s). GPCR’s make up one of the largest families of transmembrane proteins with > 800 GPCR’s identified to date, more than 90% of which are expressed in the brain [50]. These receptors help cells respond to changes in their extracellular environment and induce biochemical signaling pathways upon activation by their specific ligands, as is the case in synaptic transmission. GPCR’s are composed of a heterotrimeric G protein complex coupled to a transmembrane receptor where the G protein complex contains Gα, Gβ, and Gγ subunits [51,52,53]. The Gβ and Gγ subunits constitutively dimerize and form a trimeric complex with Gα subunits that are bound to guanosine 5’-diphosphate (GDP). Binding of the appropriate ligand to the GPCR’s transmembrane receptor promotes a conformational change in the G proteins that catalyzes the exchange of GDP for guanosine 5’-triphosphate (GTP) within the Gα subunit, and in turn leads to the dissociation or rearrangement of the Gα subunit and the Gβγ dimer [54,55,56]. Both the Gα subunit and the Gβγ dimer can then bind to downstream targets and initiate their unique biochemical pathways. Signaling through Gαs initiates the conversion of ATP to cAMP by Adenylyl Cyclases (AC’s), which in turn activates proteins with CNB’s such as PKA (Fig. 3). cAMP binding to the R subunit unleashes the C subunit for downstream phosphorylation to occur. Once AC’s become activated by GPCR’s, intrinsic GTPase activity of the Gα subunit hydrolyzes GTP to GDP and dissociates the Gα subunit from the AC, thus ending the signal induced by the initial ligand binding event.

Common PKA signaling pathways: Diagram of the MAPK and PKA pathways. Signaling events leading to Elk1-mediated transcription are shown on the left, while those leading to CREB-mediated transcription are shown on the right. Depending on cell context, PKA either positively or negatively regulates Ras-MAPK signaling. Moreover, MAPK indirectly stimulates CREB-mediated transcription via activation of ribosomal protein S6 kinases (RSKs)
PKA’s role in learning and memory
Some of the first studies implicating a role for PKA in learning and memory were performed in Aplysia. Aplysia have a gill and siphon withdrawal reflex that can be sensitized following repeated stimulation [57]. Given Aplysia’s relatively simple nervous system, this allows for a comprehensive dissection of the mechanisms underlying this response [58]. Specifically, this sensitization requires long-term facilitation (LTF), the growth of new synaptic connections for neurons involved in Aplysia’s withdrawal reflex. Induction of LTF in cultured Aplysia sensory and motor neurons could be stimulated with the application of serotonin (5-HT) [59, 60], which increases cAMP levels in cells to activate PKA [61].
Early studies using this system discovered that LTF induction requires gene transcription and translation, as inhibitors of both RNA and protein synthesis prevented 5-HT-induced increases in excitatory post-synaptic potentials (EPSPs) [59]. Once activated by cAMP, the PKA C subunit can translocate to the nucleus, where it phosphorylates cAMP response element-binding protein (CREB) [62]. In neurons, CREB is constitutively bound to the cAMP responsive element (CRE) promoter [63, 64]. Following phosphorylation, CREB initiates transcription of genes associated with the promoters it is bound to [58]. In Aplysia sensory neurons, Dash and colleagues found that injection of CRE oligonucleotide sequences prevented induction of LTF following 5-HT stimulation [65]. A greater understanding of the mechanism underlying LTF in Aplysia was uncovered by introducing mutations into CREB. Specifically, CREB Ser119, the residue required for PKA- or CaMKII-dependent initiation of transcription, was mutated to an alanine to prevent CREB phosphorylation. 5-HT treatment of Aplysia neurons expressing the CREB Ser119Ala mutant showed no induction of LTF [66]. To determine which protein phosphorylated CREB, Kaang and colleagues generated a CREB mutant that lacked the PKA phosphorylation motif but still had the CaMKII phosphorylation motif. Expression of this mutant CREB also prevented LTF induction, suggesting that PKA, not CaMKII, phosphorylated CREB at Ser119 [66]. Finally, Bacskai and colleagues showed that the PKA C subunit moved into the nucleus of neurons following elevation of cAMP levels resulting from forskolin, IBMX, and 5-HT stimulation [67]. These results support the idea that the cAMP pathway plays a role in LTF.
While they showed that PKA and CREB were important in memory, the experiments in Aplysia did not provide information about where PKA and CREB act to regulate LTF/long-term memory (LTM). Location-specific information about where LTM is regulated was only acquired with electrophysiology and behavioral data collected from transgenic mice expressing an inhibitory form of PKA RIα (R(AB)) [68]. Electrophysiological recordings of hippocampal area CA1 slices from R(AB) mice revealed deficits in the late phase of long-term potentiation (L-LTP) relative to slices from wild-type (WT) mice [68]. These mice also performed worse than their WT counterparts in the Morris water maze, indicating that they had deficits in spatial memory [68]. Altogether, the experiments in Aplysia and mice reveal that PKA plays a significant role in learning and memory and that these processes appear to be evolutionarily conserved across species.
MAPK signaling
Previous work has shown that PKA regulates the mitogen-activated protein kinase (MAPK) /extracellular signal-regulated kinase (ERK) signaling pathway. Specifically, Li and colleagues showed that PKA phosphorylation of B-Raf Ser365 inhibits ERK signaling by blocking binding of B-Raf to Ras [69]. Other mechanisms of negative regulation of Ras-MAPK signaling have also been reported [70, 71]. However, while this phosphorylation event disrupts B-Raf binding to Ras, PKA can stimulate ERK signaling by phosphorylating different proteins in the ERK pathway in a cell context dependent manner [72,73,74,75,76]. ERK signaling can be stimulated through Rap1 binding to B-Raf, but this interaction is blocked by PKA phosphorylation [77]. However, if PKA also phosphorylates Rap1, B-Raf can indirectly bind to Rap1 by binding to kinase suppressor of Ras (KSR), which itself binds to phosphorylated Rap1 [77].
The aforementioned PKA phosphorylation events in the ERK signaling pathway are important because of Elk1 activation by MAPK/ERK. Elk1 is a transcription factor expressed in neurons that plays a role in differentiation, cancer, inflammation, and cell growth [78]. Once phosphorylated by ERK, Elk1 translocates from the cytoplasm into the nucleus, where it binds serum response factor (SRF) at the serum response element (SRE) on DNA [79]. Elk1 can also recruit CREB binding protein (CBP) to DNA, which can acetylate histones on specific regions of DNA to promote gene transcription [78]. Elk1 activation also promotes NMDA-dependent LTD in vivo [80] and is associated with taste memory [81] and one-trial avoidance learning [82].
PKA variants in neurodevelopmental disorders
Pathogenic variants that contribute to some forms of a neurodevelopmental disorder (NDD) have been identified in both PKA catalytic and regulatory subunits. Some of these variants cause disorders that are not primarily associated with neurodevelopment, such as acrodysostosis, but can result in intellectual disability (ID) as a secondary phenotype in a small portion of affected patients. Other pathogenic variants have been shown to directly cause ID with few, if any, secondary phenotypes. An overview of these protein variants and the disorders they cause is described below.
PRKACA and PRKACB
Pathogenic somatic variants of PRKACA often manifest as endocrine and metabolic disorders such as Cushing’s syndrome, cortisol-producing adenomas (CPA’s), hypothalamic hamartomas, and cardiac myxomas [83,84,85,86,87,88]. Between 23 and 67% of CPA’s can be attributed to a L205R somatic variant in PKA Cα, resulting in constitutive activation of PKA Cα and altering subcellular localization by decreasing interaction with AKAPs [87,88,89,90,91]. Several residues proximal to L205 on PKA Cα localize to a catalytic domain “hot spot” commonly mutated in patients with CPA’s [86,87,88, 90, 91]. This hot spot alters the structural determinants of the catalytic domain’s specificity for both autoinhibition by R subunits and substrate specificity. Phosphoproteomic analysis of common PRKACA and PRKACB variants associated with CPA’s identified a profound shift in PKA’s phosphorylation profile, with consistent hyperphosphorylation of citron rho-interacting kinase (CIT), the mitochondrial import receptor subunit TOM34, histone H1.2, and histone H1.4 by three activating PRKACA somatic variants [92].
Both pathogenic somatic and germline variants in either PRKACA or PRKACB were discovered in patients with several congenital abnormalities such as atrioventricular septal defect (AVSD) and polydactyly [93]. Of the seven patients described, two patients with de novo PKA Cβ H88R or H88N mutations had mild-to-severe ID, anxiety, autistic features, and medically refractory focal epilepsy [93]. Using bioluminescence resonance energy transfer (BRET), Palencia-Campos et al. demonstrated the H88 mutants rapidly dissociate from the R subunits upon stimulation with Forskolin/IBMX and have slower reassociation kinetics to all R subunit isoforms compared to WT. Molecular Dynamic simulations suggest the H88 mutations likely disrupt the glycine-rich loop of the catalytic domain N-terminus, which packs onto the gamma-phosphate of ATP during phosphotransferase activity [94]. These models were corroborated by Fluorescence Polarization experiments that demonstrated a significant decrease in binding affinity for a FAM-labeled PKA inhibitor IP20 when varying H88R PKA Cβ at 1 mM ATP, concomitant with a decrease in ATP binding affinity at 5 nM H88R PKA Cβ, but independently of any changes in cAMP affinity [93]. These results suggest that perturbations in the catalytic domain that coordinate ATP binding destabilize substrate binding affinity and autoinhibition.
PRKAR1A
Pathogenic germline nonsense insertion/deletion variants of PRKAR1A are the most common genetic causes of Carney complex, a hereditary condition associated with spotted skin pigmentation, cardiac myxomas, and endocrine tumors [95, 96]. Nonsense-mediated decay of mRNA decreases total expression of the RIα subunit, resulting in constitutive activation of PKA C subunits throughout the body [97].
Similarly, researchers identified several individuals with de novo pathogenic germline variants in PRKAR1A that had acrodysostosis, an autosomal dominant disorder characterized by hormone resistance, brachydactyly, craniofacial abnormalities, and short stature [98,99,100,101]. Unlike Carney complex, many of the pathogenic variants in PRKAR1A that are found in patients with acrodysostosis may lead to decreased sensitivity to cAMP, resulting in a dominant negative effect on PKA activity (reviewed in Michot 2018 [98]). Several patients with acrodysostosis have been reported with mild-to-severe intellectual disability or mental retardation as a comorbidity [102, 103], but the cooccurrence of ID with PRKAR1A variants is relatively low. In a recent report, only 1/9 acrodysostosis patients with PRKAR1A variants were comorbid with ID [98]. Thus, clinical manifestation of ID is more closely associated with decreased PKA activity through decreased sensitivity of RIα to cAMP. It should be mentioned that while many PRKAR1A variants have been identified in Carney Complex, so too have variants in the phosphodiesterases PDE11A and PDE8 which more commonly result in ID, suggesting cAMP concentrations in distinct compartments or cell types may contribute to ID more strongly than total regulation of PKA activity by the RIα subunit [98].
PRKAR1B
Although RIβ is relatively understudied in comparison to the other R subunits, it has been associated with numerous neurological phenotypes. In 2014, a novel rare pathogenic germline variant in PRKAR1B (L50R) was identified by Wong et al. in a three generational family [104]. Twelve members of the family had a neurodegenerative disorder with dementia, frontotemporal dementia (FTD)-like and Parkinsonism symptoms, and the L50R germline variant segregated within the family in affected members. Additional evidence from this study supporting an association between PRKAR1B and neurodegeneration was the accumulation of PRKAR1B in neuronal inclusions in the affected cases. The age of onset was between 45 and 64 years old and the most common symptoms noted were behavioral changes including self-neglect and delusions, anxiety, memory problems, and motor deficits. Wong and colleagues predicted that the L50R variant altered PRKAR1B binding, and therefore the PKA interactome, due to its location in the D/D domain. Notably, no pathogenic PRKAR1B variants were found in a cohort of Parkinson’s Disease, neuronal intermediate filament inclusion disease (NIFID) or FTD [104, 105], suggesting this may be a novel and rare variant. A subsequent genetic study of the PRKAR1B gene identified 7 variants in FTD and Alzheimer’s disease (AD) patients but these were not predicted to be pathogenic [106]. In contrast to some of the other studies mentioned, common variants located just downstream of PRKAR1B have been associated with late-onset AD and present in a punctate formation [107]. Importantly, the Agora database has ranked PRKAR1B with a score of 4.43, which suggests a high relevance between PRKAR1B and AD based on published evidence including a significant reduction in RNA expression in the temporal cortex and a significant reduction in protein expression in the dorsolateral prefrontal cortex ( https://agora.adknowledgeportal.org/genes ). Overall, these findings suggest an important role for PRKAR1B in neurodegeneration and AD.
Recent exome sequencing studies have identified 13 individuals in 2 cohorts with novel pathogenic variants of PRKAR1B that leads to global developmental delay, ID, and autism spectrum disorder (ASD) known as Marbach-Schaaf Neurodevelopmental Syndrome (MASNS) [108, 109]. Interestingly, 11/13 individuals identified with MASNS had a c.1003C > T missense variant of PRKAR1B, which resulted in an R335W RIβ subunit of PKA. The two other patients in the cohort were identified with c.586G > A (E196K) and c.500_501 inv (Q167L) variants. These variants can be found in the R subunit CNB’s, and R335 can be seen making electrostatic contacts with the phosphate group of cAMP in crystal structures, and thus may perturb cAMP binding to the R1β subunit [110].
Many neurologic anomalies are associated with MASNS, including delayed development of fine motor skills, dyspraxia, clumsiness, tremor, dystonia, and congenital hypotonia [108, 109]. Behavioral abnormalities in MASNS patients usually observed in the context of ASD include self-harm, hand/arm flapping, and repetitive, sensory-seeking behavior. Many of these patients also experienced hyperactivity or restlessness, some of whom were formally diagnosed with attention-deficit/hyperactivity disorder (ADHD).
The authors identified PRKAR1B to be highly expressed in the pituitary, diencephalon, mesencephalon, and hypothalamus in human embryos and Carnegie stage 22, suggesting a critical role for the RIβ subunit during the development of the embryonic brain. However, no significant abnormalities in brain structures were detected in children with MASNS using magnetic resonance imaging (MRI) or electroencephalography (EEG).
Interestingly, a phenotype unique to the R335W variant, but not the Q167L or E196K variants, was an inherent insensitivity to pain. Parents of children with R335W MASNS have informally reported an intrinsic lack of pain sensitivity during self-injuring behaviors or when holding hot beverages but had normal temperature perception.
Transient expression of R335W RIβ in HEK293 cells has shown both low basal and stimulated cAMP stimulated PKA activity compared to WT (Glebov-McCloud et al., unpublished), suggesting a lack of cAMP sensitivity for PKA activation in mutant RIβ. Thus, PKA signaling in both developing neurons and primary sensory neurons may be impaired by R335W RIβ PKA holoenzymes.
Conclusion
Taken together, these data provide valuable insights into the role of cAMP/PKA signaling during neurodevelopment. Many of the variants in the cAMP/PKA signaling pathway that are more widely expressed throughout the body to contribute to broader morphological, metabolic, and endocrine dysfunction. This is highlighted by the prevalence of Cushing’s Syndrome and acrodysostosis for patients with PRKACA or PRKAR1A variants, respectively. However, variants of genes that are enriched or specifically expressed in brain tissues, such as PRKACB or PRKAR1B, appear to produce more cognitive deficits, as is the case with MASNS. Furthermore, inactivating variants of either C or R subunits, producing a dominant negative phenotype on catalytic PKA activity, appear to manifest as cognitive or behavioral deficits more so than activating variants. A PDE4 inhibitor Zatomilast® is currently in Phase III Clinical Trials for the treatment of cognitive deficits produced by Alzheimer’s disease and Fragile-X Syndrome, further demonstrating the importance of activating the cAMP/PKA pathway.
Due to PKA’s broad substrate specificity, it’s hard to say exactly which downstream targets are more significantly impacted by variants in the cAMP/PKA pathway. The neurodegenerative-linked L50R variant of PRKAR1B lies within the D/D domain which is responsible for PKA localization with AKAP’s in distinct subcellular compartments. Recent work from the Strack Lab indicate this variant impairs PKA C subunit compartmentalization (Glebov-McCloud et al., unpublished) and may prevent specific substrate phosphorylation through this intracellular targeting. However, there is a clear link between regulating transcription/translation of CRE promoted genes and neurodevelopmental disorders.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Abbreviations
- 5-HT:
-
5-Hydroxytryptamine/serotonin
- AC’s:
-
Adenylyl cyclases
- AD:
-
Alzheimer’s disease
- ADHD:
-
Attention-deficit/hyperactivity disorder
- AKAP’s:
-
A-Kinase Associated Proteins
- ASD:
-
Autism spectrum disorder
- ATP:
-
Adenosine triphosphate
- AVSD:
-
Atrioventricular septal defect
- BRET:
-
Bioluminescence resonance energy transfer
- C subunits:
-
Catalytic subunits
- CaMKII:
-
Ca2+/Calmodulin-dependent protein kinase II
- cAMP:
-
Cyclic adenosine 3’,5’ monophosphate
- CBP:
-
CREB binding protein
- CIT:
-
Citron rho-interacting kinase
- CNB domains:
-
Cyclic nucleotide binding domains
- CPA’s:
-
Cortisol-producing adenomas
- CRE:
-
CAMP responsive element
- CREB:
-
CAMP response element-binding protein
- D/D domain:
-
Dimerization/Docking domain
- EEG:
-
Electroencephalography
- EPSPs:
-
Excitatory post-synaptic potentials
- ERK:
-
Extracellular signal-regulated kinase
- FTD:
-
Frontotemporal dementia
- GDP:
-
Guanosine diphosphate
- GPCR’s:
-
G protein-coupled receptors
- GTP:
-
Guanosine-5’-triphosphate
- ID:
-
Intellectual disability
- KSR:
-
Kinase suppressor of Ras
- L-LTP:
-
The late phase of long-term potentiation
- LTD:
-
Long-term depression
- LTF:
-
Long-term facilitation
- LTM:
-
Long-term memory
- LTP:
-
Long-term potentiation
- MAPK:
-
Mitogen-activated protein kinase
- MASNS:
-
Marbach-Schaaf Neurodevelopmental Syndrome
- MRI:
-
Magnetic resonance imaging
- NDD:
-
Neurodevelopmental disorder
- NIFID:
-
Neuronal intermediate filament inclusion disease
- PDE:
-
Phosphodiesterase
- PKA:
-
Protein Kinase A
- R subunits:
-
Regulatory subunits
- R(AB):
-
An inhibitory form of PKA RIα
- SRE:
-
Serum response element
- SRF:
-
Serum response factor
- WT:
-
Wild-type
References
Schulman H, Greengard P. Stimulation of brain membrane protein phosphorylation by calcium and an endogenous heat-stable protein. Nature. 1978;271:478–9. https://doi.org/10.1038/271478a0.
Schulman H, Greengard P. Ca2+-dependent protein phosphorylation system in membranes from various tissues, and its activation by “calcium-dependent regulator.” Proc Natl Acad Sci USA. 1978;75:5432–6.
Walsh D, Perkins JP, Krebs EG. An adenosine 3’,5’-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;10:3763–5.
Reimann E, Walsh DA, Krebs EG. Purification and properties of rabbit skeletal muscle adenosine 3’5’-monophosphate-dependent protein kinases. J Biol Chem. 1971;246:1986–95.
Kennedy MB, McGuinness T, Greengard P. A calcium/calmodulin-dependent protein kinase from mammalian brain that phosphorylates Synapsin I: partial purification and characterization. J Neurosci. 1983;3:818–31. https://doi.org/10.1523/JNEUROSCI.03-04-00818.1983.
Walter U, Lohmann SM, Sieghart W, Greengard P. Identification of the cyclic AMP-dependent protein kinase responsible for endogenous phosphorylation of substrate proteins in synaptic membrane fraction from rat brain. J Biol Chem. 1979;254:12235–9.
Hell JW. How Ca2+-permeable AMPA receptors, the kinase PKA, and the phosphatase PP2B are intertwined in synaptic LTP and LTD. Sci Signal. 2016;9:e2. https://doi.org/10.1126/scisignal.aaf7067.
Sanderson JL, Dell’Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–36. https://doi.org/10.1177/1073858410384740.
Wang H, Peng RY. Basic roles of key molecules connected with NMDAR signaling pathway on regulating learning and memory and synaptic plasticity. Mil Med Res. 2016;3:26. https://doi.org/10.1186/s40779-016-0095-0.
Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008;169:97–115. https://doi.org/10.1016/S0079-6123(07)00006-4.
Christensen KR, Nairn AC. cAMP-regulated phosphoproteins DARPP-32, ARPP16/19, and RCS modulate striatal signal transduction through protein kinases and phosphatases. Adv Pharmacol. 2021;90:39–65. https://doi.org/10.1016/bs.apha.2020.09.005.
Uhler MD, Chrivia JC, McKnight GS. Evidence for a second isoform of the catalytic subunit of cAMP-dependent protein kinase”. J Biol Chem. 1986;261:15360–3.
Uhler MD, McKnight GS. Expression of cDNAs for two isoforms of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 1987;262:15202–7.
Jahnsen T, Hedin L, Kidd VJ, Schulz T, Richards JS. Molecular cloning of cDNA for a hormone-regulated isoform of the regulatory subunit of type II cAMP-dependent protein kinase from rat ovaries. Methods Enzymol. 1988;159:318–24. https://doi.org/10.1016/0076-6879(88)59032-8.
Uhler MD, et al. Isolation of cDNA clones coding for the catalytic subunit of mouse cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1986;83:1300–4. https://doi.org/10.1073/pnas.83.5.1300.
Zimmermann B, Chiorini JA, Ma Y, Kotin RM, Herberg FW. PrKX is a novel catalytic subunit of the cAMP-dependent protein kinase regulated by the regulatory subunit type I. J Biol Chem. 1999;274:5370–8. https://doi.org/10.1074/jbc.274.9.5370.
Beebe, S. J. S., P.; Jahnsen, T.; Li, Y. The C gamma subunit is a unique isozyme of the cAMP-dependent protein kinase. J Biol Chem 1992;267:25505–25512.
Schiebel K, et al. Abnormal XY interchange between a novel isolated protein kinase gene, PRKY, and its homologue, PRKX, accounts for one third of all (Y+)XX males and (Y-)XY females. Hum Mol Genet. 1997;6:1985–9. https://doi.org/10.1093/hmg/6.11.1985.
Taskén K, et al. The gene encoding the catalytic subunit C alpha of cAMP-dependent protein kinase (locus PRKACA) localizes to human chromosome region 19p13.1. Genomics. 1996;36:535–8. https://doi.org/10.1006/geno.1996.0501.
San Agustin JT, Witman GB. Differential expression of the C(s) and Calpha1 isoforms of the catalytic subunit of cyclic 3’,5’-adenosine monophosphate-dependent protein kinase testicular cells. Biol Reprod. 2001;65:151–64. https://doi.org/10.1095/biolreprod65.1.151.
Showers MO, Maurer RA. Cloning of cDNA for the catalytic subunit of cAMP-dependent protein kinase. Methods Enzymol. 1988;159:311–8. https://doi.org/10.1016/0076-6879(88)59031-6.
Showers MO, Maurer RA. A cloned bovine cDNA encodes an alternate form of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 1986;261:16288–91.
Guthrie CR, Skålhegg BS, McKnight GS. Two novel brain-specific splice variants of the murine Cbeta gene of cAMP-dependent protein kinase. J Biol Chem. 1997;272:29560–5. https://doi.org/10.1074/jbc.272.47.29560.
Wiemann, S. K., V.; Pyerin, W. Isoform C beta 2, an unusual form of the bovine catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 1991;266:5140–5146.
Ørstavik S, et al. Identification of novel splice variants of the human catalytic subunit Cbeta of cAMP-dependent protein kinase. Eur J Biochem. 2001;268:5066–73. https://doi.org/10.1046/j.0014-2956.2001.02429.x.
Kvissel AK, et al. Induction of Cbeta splice variants and formation of novel forms of protein kinase A type II holoenzymes during retinoic acid-induced differentiation of human NT2 cells. Cell Signal. 2004;16:577–87. https://doi.org/10.1016/j.cellsig.2003.08.014.
Soberg K, Moen LV, Skalhegg BS, Laerdahl JK. Evolution of the cAMP-dependent protein kinase (PKA) catalytic subunit isoforms. PLoS One. 2017;12:e0181091. https://doi.org/10.1371/journal.pone.0181091.
Funderud A, et al. Identification, cloning and characterization of a novel 47 kDa murine PKA C subunit homologous to human and bovine Cbeta2. BMC Biochem. 2006;7:20. https://doi.org/10.1186/1471-2091-7-20.
Beavo JA, Bechtel PJ, Krebs EG. Preparation of homogeneous cyclic AMP-dependent protein kinase(s) and its subunits from rabbit skeletal muscle. Methods Enzymol. 1974;38:299–308. https://doi.org/10.1016/0076-6879(74)38046-9.
Hofmann FB, Beavo JA, Bechtel PJ, Krebs EG. Comparison of adenosine 3’:5’-monophosphate-dependent protein kinases from rabbit skeletal and bovine heart muscle. J Biol Chem. 1975;250:7795–801.
Rosen OM, Rubin CS, Erlighman J. Properties of the cyclid AMP-dependent protein kinase from bovine and porcine heart. Adv Enzyme Regul. 1975;13:173–85. https://doi.org/10.1016/0065-2571(75)90014-x.
Taylor SS, Lee CY, Swain L, Stafford PH. Cyclic AMP-dependent protein kinase: purification of the holoenzyme by affinity chromatography. Anal Biochem. 1976;76:45–52. https://doi.org/10.1016/0003-2697(76)90262-1.
Scott JD, et al. The molecular cloning of a type II regulatory subunit of the cAMP-dependent protein kinase from rat skeletal muscle and mouse brain. Proc Natl Acad Sci U S A. 1987;84:5192–6. https://doi.org/10.1073/pnas.84.15.5192.
Sandberg M, Tasken K, Oyen O, Hansson V, Jahnsen T. Molecular cloning, cDNA structure and deduced amino acid sequence for a type I regulatory subunit of cAMP-dependent protein kinase from human testis. Biochem Biophys Res Commun. 1987;149:939–45. https://doi.org/10.1016/0006-291x(87)90499-2.
Lee DC, Carmichael DF, Krebs EG, McKnight GS. Isolation of a cDNA clone for the type I regulatory subunit of bovine cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1983;80:3608–12. https://doi.org/10.1073/pnas.80.12.3608.
Øyen O, Myklebust F, Scott JD, Hansson V, Jahnsen T. Human testis cDNA for the regulatory subunit RIIα of CAMP-dependent protein kinase encodes an alternate amino-terminal region. FEBS Letters. 2001;246:57–64. https://doi.org/10.1016/0014-5793(89)80253-4.
Cho-Chung YS, Pepe S, Clair T, Budillon A, Nesterova M. cAMP-dependent protein kinase: role in normal and malignant growth. Crit Rev Oncol Hematol. 1995;21:33–61. https://doi.org/10.1016/1040-8428(94)00166-9.
Cadd G, McKnight GS. Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron. 1989;3:71–9. https://doi.org/10.1016/0896-6273(89)90116-5.
Zhang P, et al. An Isoform-Specific Myristylation Switch Targets Type II PKA Holoenzymes to Membranes. Structure. 2015;23:1563–72. https://doi.org/10.1016/j.str.2015.07.007.
Taskén K, et al. Structure, function, and regulation of human cAMP-dependent protein kinases. Adv Second Messenger Phosphoprotein Res. 1997;31:191–204. https://doi.org/10.1016/s1040-7952(97)80019-5.
Jahnsen T, et al. Molecular cloning, cDNA structure, and regulation of the regulatory subunit of type II cAMP-dependent protein kinase from rat ovarian granulosa cells. J Biol Chem. 1986;261:12352–61.
Gold MG, et al. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell. 2006;24:383–95. https://doi.org/10.1016/j.molcel.2006.09.006.
Sarma GN, et al. Structure of D-AKAP2PKA RI complex insights into AKAP specificity and selectivity. Structure. 2010;18:155–66. https://doi.org/10.1016/j.str.2009.12.012.
Ogreid D, et al. Activation of protein kinase isozymes by cyclic nucleotide analogs used singly or in combination Principles for optimizing the isozyme specificity of analog combinations. Eur J Biochem. 1985;150:219–27. https://doi.org/10.1111/j.1432-1033.1985.tb09010.x.
Carlson CR, Ruppelt A, Taskén K. A kinase anchoring protein (AKAP) interaction and dimerization of the RIalpha and RIbeta regulatory subunits of protein kinase a in vivo by the yeast two hybrid system. J Mol Biol. 2003;327:609–18. https://doi.org/10.1016/s0022-2836(03)00093-7.
Tasken K, Skalhegg BS, Solberg R, Andersson KB, Taylor SS, Lea T, Blomhoff HK, Jahnsen T. Novel isozymes of cAMP-dependent protein kinase exist in human cells due to formation of RI alpha-RI beta heterodimeric complexes. J Biol Chem. 1993;268:21276–83.
Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971–1005. https://doi.org/10.1146/annurev.bi.59.070190.004543.
Gibbs CS, Knighton DR, Sowadski JM, Taylor SS, Zoller MJ. Systematic mutational analysis of cAMP-dependent protein kinase identifies unregulated catalytic subunits and defines regions important for the recognition of the regulatory subunit. J Biol Chem. 1992;267:4806–14.
Anand GS, et al. R-subunit isoform specificity in protein kinase A: distinct features of protein interfaces in PKA types I and II by amide H/2H exchange mass spectrometry. J Mol Biol. 2007;374:487–99. https://doi.org/10.1016/j.jmb.2007.09.035.
Vassilatis DK, et al. The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci U S A. 2003;100:4903–8. https://doi.org/10.1073/pnas.0230374100.
Levitzki A. From epinephrine to cyclic AMP. Science. 1988;241:800–6. https://doi.org/10.1126/science.2841758.
Cassel D, Selinger Z. Mechanism of adenylate cyclase activation through the beta-adrenergic receptor: catecholamine-induced displacement of bound GDP by GTP. Proc Natl Acad Sci U S A. 1978;75:4155–9. https://doi.org/10.1073/pnas.75.9.4155.
Hamm HE, Gilchrist A. Heterotrimeric G proteins. Curr Opin Cell Biol. 1996;8:189–96. https://doi.org/10.1016/s0955-0674(96)80065-2.
Neer EJ, Clapham DE. Roles of G protein subunits in transmembrane signalling. Nature. 1988;333:129–34. https://doi.org/10.1038/333129a0.
Janetopoulos C, Jin T, Devreotes P. Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science. 2001;291:2408–11. https://doi.org/10.1126/science.1055835.
Bunemann M, Frank M, Lohse MJ. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci U S A. 2003;100:16077–82. https://doi.org/10.1073/pnas.2536719100.
Byrne JH. Cellular analysis of associative learning. Physiol Rev. 1987;67:329–439. https://doi.org/10.1152/physrev.1987.67.2.329.
Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–48. https://doi.org/10.1146/annurev.neuro.21.1.127.
Montarolo PG, et al. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–54. https://doi.org/10.1126/science.3775383.
Meneses A, Liy-Salmeron G. Serotonin and emotion, learning and memory. Rev Neurosci. 2012;23:543–53. https://doi.org/10.1515/revneuro-2012-0060.
Kandel ER, Schwartz JH. Molecular biology of learning: modulation of transmitter release. Science. 1982;218:433–43. https://doi.org/10.1126/science.6289442.
Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–80. https://doi.org/10.1016/0092-8674(89)90013-5.
Cha-Molstad H, Keller DM, Yochum GS, Impey S, Goodman RH. Cell-type-specific binding of the transcription factor CREB to the cAMP-response element. Proc Natl Acad Sci U S A. 2004;101:13572–7. https://doi.org/10.1073/pnas.0405587101.
Riccio A, et al. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol Cell. 2006;21:283–94. https://doi.org/10.1016/j.molcel.2005.12.006.
Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–21. https://doi.org/10.1038/345718a0.
Kaang BK, Kandel ER, Grant SG. Activation of cAMP-responsive genes by stimuli that produce long-term facilitation in Aplysia sensory neurons. Neuron. 1993;10:427–35. https://doi.org/10.1016/0896-6273(93)90331-k.
Bacskai BJ, et al. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 1993;260:222–6. https://doi.org/10.1126/science.7682336.
Abel T, et al. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–26. https://doi.org/10.1016/s0092-8674(00)81904-2.
Li Y, Takahashi M, Stork PJS. Ras-mutant cancer cells display B-Raf binding to Ras that activates extracellular signal-regulated kinase and is inhibited by protein kinase A phosphorylation. J Biol Chem. 2013;288:27646–57. https://doi.org/10.1074/jbc.M113.463067.
Pursiheimo JP, Kieksi A, Jalkanen M, Salmivirta M. Protein kinase A balances the growth factor-induced Ras/ERK signaling. FEBS Letters. 2002;521(1–3):157–64. https://doi.org/10.1016/S0014-5793(02)02864-8.
Bengis-Garber C, Gruener N. Protein Kinase A Downregulates the Phosphorylation of p47 Phox in Human Neutrophils: A Possible Pathway for Inhibition of the Respiratory Burst. Cell Signal. 1996;8(4):291–6. https://doi.org/10.1016/0898-6568(96)00052-6.
Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross Talk between ERK and PKA Is Required for Ca2+ Stimulation of CREB-Dependent Transcription and ERK Nuclear Translocation. Neuron. 1998;21(4):869–83. https://doi.org/10.1016/S0896-6273(00)80602-9.
Law NC, Donaubauer EM, Zeleznik AJ, Hunzicker-Dunn M. How Protein Kinase A Activates Canonical Tyrosine Kinase Signaling Pathways To Promote Granulosa Cell Differentiation. Endocrinology. 2017;158(7):2043–51. https://doi.org/10.1210/en.2017-00163.
Paul S, Snyder GL, Yokakura H, Picciotto MR, Nairn AC, Lombroso PJ. The Dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. J Neurosci. 2000;20(15):5630–8. https://doi.org/10.1523/JNEUROSCI.20-15-05630.2000.
Schmitt JM, Stork PJ. beta 2-adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein rap1 and the serine/threonine kinase B-Raf. J Biol Chem. 2000;275(33):25342–50. https://doi.org/10.1074/jbc.M003213200.
Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89(1):73–82. https://doi.org/10.1016/s0092-8674(00)80184-1.
Takahashi M, Li Y, Dillon TJ, Stork PJ. Phosphorylation of Rap1 by cAMP-dependent Protein Kinase (PKA) Creates a Binding Site for KSR to Sustain ERK Activation by cAMP. J Biol Chem. 2017;292:1449–61. https://doi.org/10.1074/jbc.M116.768986.
Besnard A, Galan-Rodriguez B, Vanhoutte P, Caboche J. Elk-1 a transcription factor with multiple facets in the brain. Front Neurosci. 2011;5:35. https://doi.org/10.3389/fnins.2011.00035.
Herrera RE, Shaw PE, Nordheim A. Occupation of the c-fos serum response element in vivo by a multi-protein complex is unaltered by growth factor induction. Nature. 1989;340:68–70. https://doi.org/10.1038/340068a0.
Thiels E, Kanterewicz BI, Norman ED, Trzaskos JM, Klann E. Long-term depression in the adult hippocampus in vivo involves activation of extracellular signal-regulated kinase and phosphorylation of Elk-1. J Neurosci. 2002;22:2054–62. https://doi.org/10.1523/JNEUROSCI.22-06-02054.2002.
Berman DE, Hazvi S, Rosenblum K, Seger R, Dudai Y. Specific and differential activation of mitogen-activated protein kinase cascades by unfamiliar taste in the insular cortex of the behaving rat. J Neurosci. 1998;18:10037–44. https://doi.org/10.1523/JNEUROSCI.18-23-10037.1998.
Cammarota M, et al. Learning-associated activation of nuclear MAPK, CREB and Elk-1, along with Fos production, in the rat hippocampus after a one-trial avoidance learning: abolition by NMDA receptor blockade. Brain Res Mol Brain Res. 2000;76:36–46. https://doi.org/10.1016/s0169-328x(99)00329-0.
Tseng IC, et al. Microinsertions in PRKACA cause activation of the protein kinase A pathway in cardiac myxoma. J Pathol. 2017;242:134–9. https://doi.org/10.1002/path.4899.
Hildebrand MS, et al. Mutations of the Sonic Hedgehog Pathway Underlie Hypothalamic Hamartoma with Gelastic Epilepsy. Am J Hum Genet. 2016;99:423–9. https://doi.org/10.1016/j.ajhg.2016.05.031.
Calebiro D, et al. PKA catalytic subunit mutations in adrenocortical Cushing’s adenoma impair association with the regulatory subunit. Nat Commun. 2014;5:5680. https://doi.org/10.1038/ncomms6680.
Ronchi CL, et al. Genetic Landscape of Sporadic Unilateral Adrenocortical Adenomas Without PRKACA p.Leu206Arg Mutation. J Clin Endocrinol Metab. 2016;101:3526–38. https://doi.org/10.1210/jc.2016-1586.
Di Dalmazi G, et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab. 2014;99:E2093-2100. https://doi.org/10.1210/jc.2014-2152.
Beuschlein F, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med. 2014;370:1019–28. https://doi.org/10.1056/NEJMoa1310359.
Sato Y, et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science. 2014;344:917–20. https://doi.org/10.1126/science.1252328.
Cao Y, et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science. 2014;344:913–7. https://doi.org/10.1126/science.1249480.
Goh G, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet. 2014;46:613–7. https://doi.org/10.1038/ng.2956.
Bathon K, et al. Alterations in Protein Kinase A Substrate Specificity as a Potential Cause of Cushing Syndrome. Endocrinology. 2019;160:447–59. https://doi.org/10.1210/en.2018-00775.
Palencia-Campos A, et al. Germline and Mosaic Variants in PRKACA and PRKACB Cause a Multiple Congenital Malformation Syndrome. Am J Hum Genet. 2020;107:977–88. https://doi.org/10.1016/j.ajhg.2020.09.005.
Hemmer W, McGlone M, Tsigelny I, Taylor SS. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J Biol Chem. 1997;272:16946–54. https://doi.org/10.1074/jbc.272.27.16946.
Rhayem Y, et al. Functional Characterization of PRKAR1A Mutations Reveals a Unique Molecular Mechanism Causing Acrodysostosis but Multiple Mechanisms Causing Carney Complex. J Biol Chem. 2015;290:27816–28. https://doi.org/10.1074/jbc.M115.656553.
Bertherat J, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. 2009;94:2085–91. https://doi.org/10.1210/jc.2008-2333.
Kirschner LS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. https://doi.org/10.1038/79238.
Michot C, et al. Expanding the phenotypic spectrum of variants in PDE4D/PRKAR1A: from acrodysostosis to acroscyphodysplasia. Eur J Hum Genet. 2018;26:1611–22. https://doi.org/10.1038/s41431-018-0135-1.
Linglart A, et al. PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J Clin Endocrinol Metab. 2012;97:E2328-2338. https://doi.org/10.1210/jc.2012-2326.
Muhn F, et al. Novel mutations of the PRKAR1A gene in patients with acrodysostosis. Clin Genet. 2013;84:531–8. https://doi.org/10.1111/cge.12106.
Michot C, et al. Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. Am J Hum Genet. 2012;90:740–5. https://doi.org/10.1016/j.ajhg.2012.03.003.
Robinow M, Pfeiffer RA, Gorlin RJ, McKusick VA, Renuart AW, Johnson GF, Sumit RL. Acrodysostosis A syndrome of peripheral dysostosis, nasal hypoplasia, and mental retardation. Am J Dis Child. 1971;121:195–203.
Butler MG, Rames LJ, Wadlington WB. Acrodysostosis: report of a 13-year-old boy with review of literature and metacarpophalangeal pattern profile analysis. Am J Med Genet. 1988;30:971–80. https://doi.org/10.1002/ajmg.1320300416.
Wong TH, et al. PRKAR1B mutation associated with a new neurodegenerative disorder with unique pathology. Brain. 2014;137:1361–73. https://doi.org/10.1093/brain/awu067.
Pottier C, Baker M, Dickson DW, Rademakers R. PRKAR1B mutations are a rare cause of FUS negative neuronal intermediate filament inclusion disease. Brain. 2015;138:e357. https://doi.org/10.1093/brain/awu332.
Cohn-Hokke PE, et al. Mutation frequency of PRKAR1B and the major familial dementia genes in a Dutch early onset dementia cohort. J Neurol. 2014;261:2085–92. https://doi.org/10.1007/s00415-014-7456-y.
Wang X, et al. A Bayesian Framework for Generalized Linear Mixed Modeling Identifies New Candidate Loci for Late-Onset Alzheimer’s Disease. Genetics. 2018;209:51–64. https://doi.org/10.1534/genetics.117.300673.
Marbach F, et al. Variants in PRKAR1B cause a neurodevelopmental disorder with autism spectrum disorder, apraxia, and insensitivity to pain. Genet Med. 2021;23:1465–73. https://doi.org/10.1038/s41436-021-01152-7.
Marbach F, et al. Phenotypic characterization of seven individuals with Marbach-Schaaf neurodevelopmental syndrome. Am J Med Genet A. 2022;188:2627–36. https://doi.org/10.1002/ajmg.a.62884.
Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307:690–6. https://doi.org/10.1126/science.1104607.
Acknowledgements
Not applicable.
Funding
This work was supported by R21 AG080472-01 (M.E.G., S.S.), the Simons Foundation Autism Research Initiative (SFARI, S.S.), the Eagles Autism Foundation (M.E.G., S.S.), and training grants T32GM144636 and T32NS045549 (to A.G.M.).
Author information
Authors and Affiliations
Contributions
A.G.P.G.M and W.S.S prepared the manuscript and figures, with contributions from M.E.G, and review and editing by M.E.G and S.S. All authors read and approved the final manuscript before submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Glebov-McCloud, A.G.P., Saide, W.S., Gaine, M.E. et al. Protein Kinase A in neurological disorders. J Neurodevelop Disord 16, 9 (2024). https://doi.org/10.1186/s11689-024-09525-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11689-024-09525-0