
Abstract
Alternative splicing of pre-mRNAs is a fundamental step in RNA processing required for gene expression in most metazoans. Serine and arginine-rich proteins (SR proteins) comprise a family of multifunctional proteins that contain an RNA recognition motif (RRM) and the ultra-conserved arginine/serine-rich (RS) domain, and play an important role in precise alternative splicing. Increasing research supports SR proteins as also functioning in other RNA-processing-related mechanisms, such as polyadenylation, degradation, and translation. In addition, SR proteins interact with N6-methyladenosine (m6A) regulators to modulate the methylation of ncRNA and mRNA. Dysregulation of SR proteins causes the disruption of cell differentiation and contributes to cancer progression. Here, we review the distinct biological characteristics of SR proteins and their known functional mechanisms during carcinogenesis. We also summarize the current inhibitors that directly target SR proteins and could ultimately turn SR proteins into actionable therapeutic targets in cancer therapy.
Similar content being viewed by others
Introduction
Serine and arginine-rich proteins (SR proteins) are conserved RNA-binding proteins present in all plants and metazoans [1]. SR proteins are involved in constitutive and alternative splicing (AS) of pre-mRNA, which is one of the most important steps during RNA processing, and contributes to the diversity of the transcriptome and proteome [2, 3]. Apart from this canonical role, SR proteins have noncanonical roles in RNA regulation by participating in alternative cleavage and polyadenylation (APA), nonsense-mediated decay (NMD), mRNA export, mRNA translation, and interaction with ncRNA [4, 5]. In particular, SR proteins also participate posttranscriptionally in RNA N6-methyladenosine (m6A) modification of various RNAs by directly or indirectly interacting with methyltransferases [5] (Fig. 1).

The multifaceted role of SR proteins and their direct targeted inhibitors for cancer treatment. a SR proteins regulate the alternative splicing of pre-mRNA and contribute to the diversity of the transcriptome and proteome. b SR proteins regulate alternative cleavage and polyadenylation to generate distinct 3′ ends of mRNAs and ncRNA. c SR proteins regulate nonsense-mediated decay to prevent the translation of potentially deleterious truncated proteins. d SR proteins regulate mRNA translation. e SR proteins interact with noncoding RNA to regulate biological processes. f SR proteins participate in N6-methyladenosine modification. g Potential medicines directly target SR proteins, including chemicals, RNA oligonucleotides, and SR protein regulators
SR proteins are critical to metazoan development. Inactivation of serine and arginine-rich splicing factor 1 (SRSF1) and SRSF6 causes embryonic lethality in chickens and Drosophilae [6]. In addition, SRSF1, SRSF2, SRSF3, SRSF5, and transformer 2 beta homolog (TRA2B) have been reported to play a pivotal role in heart, skeletal muscle, liver, and central nervous system development in the corresponding gene knockout mice [7,8,9,10,11]. On the other hand, dysregulation of SR proteins also contributes to tumorigenesis and metastasis. For example, the generation of oncogenic isoforms of SRSF1, SRSF3, SRSF6, and TRA2B have been associated with lung cancer, breast cancer, and colon cancer [12]. These cancer-related isoforms can enhance cell escape from apoptosis and confer drug resistance [13]. Therefore, SR proteins act as pivotal regulators to affect RNA metabolism and gene expression in tumor cells. In the present review, we summarize the diverse functions of SR proteins and their characteristics, as well as the therapeutic compounds and RNA oligonucleotides that directly target SR proteins in the treatment of cancer (Fig. 1).
Biochemical characteristics of SR proteins
SRSF1 was first identified in the early 1990s, and was the first SR protein found to be involved in preventing exon skipping and ensuring splicing accuracy in AS [14, 15]. Intensive studies on RNA splicing have subsequently shown that other SR proteins (i.e., SRSF2-12) take part in splicing complementation [16,17,18,19,20,21,22]. These classical SR proteins are characterized by one or two RNA recognition motifs (RRM) at the N-terminus, and an RS domain, rich in serine/arginine dipeptide repeats, at the C-terminus [1]. Remarkably, two TRA family members, TRA2A and TRA2B, are now regarded as SR-like proteins. They have only one RRM domain but two RS domains, and function as sequence-specific splicing activators [23]. TRA2A is functionally conserved because human TRA2A can replace its Drosophila homolog to affect both female sexual differentiation and AS of dsx pre-mRNA [24]. TRA2B has been reported to stimulate full-length survival motor neuron 2 (SMN2) expression through its ability to regulate AS [25].
Among the 14 known SR proteins, SRSF1, SRSF4, SRSF5, SRSF6, and SRSF9 have one canonical RRM and one pseudo-RRM [26]. The main role of RRMs is to recognize specific pre-mRNA binding sites and dictate the position of SR proteins on RNA sequences [1]. A recent study has reported that the pseudo-RRM of SRSF1 frequently competes with splicing repressors, such as hnRNPA1, rather than recruiting spliceosomal components to regulate splicing [27]. Structural NMR studies revealed that the conserved residues located in α-helix 1 of the pseudo-RRM contribute to recognition of the specific GGA motif in pre-mRNA, and this unusual mode of RNA recognition is conserved in all pseudo-RRMs [27]. In addition, the RS domain of SR proteins is conserved across vertebrates and invertebrates, and coordinates protein–protein or protein–RNA interactions by phosphorylating serine residues [28]. SR protein kinases, including SR protein kinases (SRPKs), Cdc2-like kinases (CLKs), and dual-specificity tyrosine-regulated kinases (DYRKs), have been shown to be specifically responsible for catalyzing the numerous serine residue phosphorylations on the RS domain [29,30,31]. The RS domain phosphorylation/dephosphorylation state is important for two nuclear import receptors, transportin-SR1 (TRN-SR1) and transportin-SR2 (TRN-SR2) to target SR proteins to the nucleus, thereby affecting their cellular distribution and functional flexibility [32,33,34]. It has been reported that TRN-SR2 imports TRA2B only when it is phosphorylated, but its splice variant TRN-SR1 can import both unphosphorylated and phosphorylated TRA2B [35]. Moreover, phosphorylation of the RS domain is necessary for SR proteins to leave nuclear speckles and bind to RNA, whereas a hypophosphorylated RS domain is required for SR protein splicing activity and the transport of RNA from the nucleus to cytoplasm with the help of the TAP/NFX1 nuclear export receptor [36, 37]. Dephosphorylation of the RS domain also promotes cytoplasmic mRNA binding to SR proteins and enhances SR proteins in translational activity [38]. In addition to its RRM and RS domains, SRSF7 has a unique CCHC-type zinc finger domain, which is thought, together with the RRM domain, to confer RNA-binding specificity [23, 39].
Functional mechanisms of SR proteins in cancer
Essential roles of SR proteins have been reported for sex determination [40, 41], cell differentiation [42, 43], development of the brain [44, 45] and heart [10, 46, 47], the immune system [48, 49], and many types of cancer [12, 50, 51]. Here, we focus on the known functional mechanisms of SR proteins, and discuss the importance of these functions in various cancers.
Regulation of constitutive and alternative splicing of pre-mRNAs by SR proteins
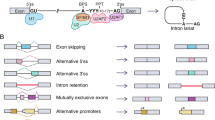
Constitutive and alternative splicing of pre-mRNA is a crucial part of eukaryotic gene expression in metazoans, and contributes to proteomic by enabling a single gene to encode multiple different transcripts with distinct functions [52, 53]. Nuclear pre-mRNA splicing is catalyzed by a macromolecular ribonucleoprotein (RNP) complex termed the spliceosome [54]. Spliceosomes recognize the four major regulatory sequences in pre-mRNA introns, including the 5′ splice site (5′SS), the intron branch point site (BP), the 3′ splice site (3′SS), and the polypyrimidine tract [55]. SR proteins serve as molecular adapters to assemble spliceosomes and produce diverse pre-mRNAs in the splicing of transcripts [1]. Two major models have been proposed to explain the mechanism by which SR proteins affect cis-regulatory elements, called exonic splicing enhancers (ESEs), on pre-mRNAs to regulate exon inclusion (Fig. 2a). The “U2AF-recruitment” model involves SR proteins binding to an ESE to interact with U2AF35 at the 3’SS to stabilize the binding of U2AF65 at the polypyrimidine tract. Simultaneously, SR proteins also recruit and stabilize U1 snRNP at the 5′SS to activate splicing in a process known as exon definition [3, 56]. The “coactivator” model proposes that ESE-bound SR proteins can interact with basal components of the spliceosome through a bridging factor named the SRm160/300 splicing coactivator, or communicate with U1 snRNP and U2 snRNP [56, 57]. Abnormal expression of SR proteins are found in almost all tumor types, and results in dysregulated RNA splicing and tumor progression [12].

Mechanisms of SR proteins in human cancers. a Two models of SR protein function in splicing. U2AF-recruitment model: ESE-bound SR proteins interact with U2AF35 at the 3′SS to stabilize the binding of U2AF65 at the polypyrimidine tract and recruit U1 snRNP at the 5′SS to active splicing. Coactivator model: ESE-bound SR proteins interact with splicing coactivator SRm160/300 and communicate with U1 snRNP and U2 snRNP. b Model of SRSF3- and SRSF7-mediated regulation of APA by interacting with the APA component on PASs in an antagonistic manner. c Model of SR proteins recognizing NMD factor UPF1 downstream of the PTC to activate the NMD pathway, thus inducing protein degradation. d Example of SR protein function in translation. SRSF1 recruits mammalian target of rapamycin complex 1 (mTORC1) to mRNA to promote phosphorylation of 4E-BP, leading to release of eIF4E and initiation of translation. e SR proteins can interact with lncRNA, circRNA and microRNA to regulate tumorigenesis. f SR proteins can directly or indirectly interact with m6A methyltransferases, or colocalize with m6A readers to modulate m6A modification
SRSF2 is involved in the switch in AS of various pre-mRNAs for apoptotic genes, such as caspase-8, caspase-9 and Bcl-x, to favor proapoptotic splice variants in lung carcinoma [58]. Similarly, downregulation of SRSF2 in renal tumors results in concomitant changes of splicing profiles of apoptosis-associated genes by decreasing the expression of proapoptotic isoforms caspase-9a, Smac3, Surv-2B, BimS, Bimα3, and MCL-1S, while increasing the expression of anti-apoptotic isoforms caspases-9b and caspase-8L, therefore leading to cell proliferation [59]. Furthermore, the low level of SRSF3 in relapsed B-cell acute lymphoblastic leukemias inhibits the retention of exon 2 in CD19, causing failure to trigger killing by chimeric antigen receptor-armed T cells (CART-19), which leads to resistance to CART-19 immunotherapy [60]. These findings show anti-tumor roles of SR proteins by AS. In contrast, in breast cancer, overexpressed SRSF1, through its function in constitutive and alternative splicing, increases BIN1 isoforms that lack pro-apoptotic functions, thereby causing BIN1 failed interaction with MYC, and leading to MYC-induced epithelial cell transformation [61]. In addition, upregulated TRA2A in triple-negative breast cancer (TNBC) promotes the generation of biologically inactive RSRC2, which retains exon 4 in its transcript, thus contributing to the migration, invasion, and paclitaxel resistance of TNBC cells [62]. Overall, the above studies indicate that SR proteins play a dual role in different types of cancers by regulating pre-mRNA constitutive and alternative splicing.
Regulation of alternative cleavage and polyadenylation by SR proteins
Alternative cleavage and polyadenylation is a widespread RNA-processing mechanism across all eukaryotic species and refers to the regulated selection of polyadenylation sites (PASs) to generate distinct 3′ ends on RNA polymerase II transcribed RNAs, including mRNAs and ncRNA [63]. APA frequently occurs in the 3′ untranslated region (3′UTR) of mRNAs, thus producing multiple mRNA and protein isoforms derived from a single gene to regulate RNA stability and facilitate RNA nuclear export [63]. APA is initiated by the APA machinery assembling from four elements at each PAS, comprising cleavage stimulatory factor (CSTF), cleavage and polyadenylation specificity factor (CPSF), cleavage factor Im (CFIm), and cleavage factor IIm (CFIIm) [64]. SRSF3 and SRSF7 have been reported to regulate APA processing by interacting with APA components on PASs in an antagonistic manner in an embryonic carcinoma cell line [65] (Fig. 2b). Specifically, SRSF7 interacts with FIP1, a subunit of CPSF, independently of RNA via its hypophosphorylated RS domain to activate proximal PAS (pPAS) usage directly in a splicing-independent manner, thus promoting short 3′UTRs. Conversely, SRSF3 controls the levels of active CFIm, which enhances cleavage at distal PASs (dPASs) and directly counteracts SRSF7 levels to promote long 3′UTRs by inhibiting pPAS usage [65]. The distinct function between SRSF3 and SRSF7 is owing to the unique domain present in SRSF7, which containing a stretch of 27 amino acids enriched in hydrophobic residues and a Zn knuckle, which are absent in SRSF3 [65]. It has been widely confirmed that mRNA isoforms with shorter 3′UTRs lose microRNA-mediated repression, therefore increasing oncogene RNA stability and translation to promote tumorigenesis [66,67,68]. In addition, SRSF3 and SRSF7 are both reported to connect polyadenylation with NXF1-mediated mRNA export, therefore regulating the transcripts with alternative 3′ end export from the nucleus to the cytoplasm [69].
Regulation of nonsense-mediated decay by SR proteins
Nonsense-mediated decay is regarded as a cellular surveillance mechanism to promote degradation of erroneous transcripts containing premature termination codons (PTCs), thus preventing the translation of potentially deleterious truncated proteins [70]. One major molecular mechanism involved in the NMD pathway depends on the formation of a key regulatory player called the exon junction complex (EJC) [71]. An EJC is deposited ~20–24 nucleotides upstream of most exon–exon junctions and is removed from the mRNA by ribosomes during translation. In certain transcripts, ribosomes stop at the PTC, resulting in EJCs remaining downstream of the PTC, allowing recognition of EJCs by NMD factors to activate NMD [72] (Fig. 2c). NMD is an important biological mechanism of RNA processing, partially regulated by SR proteins in tumor development. In HeLa cells, SRSF1 promotes the recruitment of EJC factors and UPF1 to downstream of a PTC by its RS domain to elicit NMD [73]. In addition, an SRSF2 Pro95 mutation enhances the deposition of EJCs downstream of PTCs, associating with key NMD factors to enhance mRNA decay in acute myeloid leukemia [74].
Interestingly, owing to a highly ultraconserved PTC known as a poison exon (PE), SR proteins not only regulate other genes, they usually participate in their own NMD regulation. For example, binding of TRA2B to TRA2B-PE enhances TRA2B-PE inclusion, thus generating a transcript that cannot be translated [75]. Conversely, knockdown of SRSF3 has been shown to contribute to SRSF3-PE inclusion [76]. In addition, it has been reported that SRSF1 negatively regulates SRSF4- and SRSF11-PE inclusion, and positively regulates SRSF2-, SRSF3-, SRSF6-, SRSF7-, and TRA2B-PE inclusion [76]. SRSF4 and SRSF5 both regulate SRSF6-PE inclusion positively [76]. These cross-regulations and/or autoregulations among SR proteins via the NMD pathway precisely control the balance of their different spliced isoforms. Thus, the role of the SR proteins in RNA processing not only depends on their phosphorylated state, but also relies on their total protein level, distinct isoform levels and protein ratio between the cytoplasm and nucleus. In this respect, systematic dissection of the specific roles of each SR protein and the precise mechanism to control its expression in different cells may help us clarify the importance of each SR protein in cancer. Since SR proteins cross-regulate shared target genes, including the transcripts of each other, and form a dense coordinated network [76], a systems biology approach can be applied to globally identify the most dysregulated SR protein–RNA circuits under cancer conditions. This complementary approach enables the researcher to simultaneously characterize all alterations in SR proteins and to balance their multifunctionality without being trapped into negligible details [77, 78].
Regulation of translation by SR proteins
The continuous shuttling of SR proteins between the nucleus and cytoplasm not only plays a role in RNA transcription, but also influences the regulation of mRNA translation. SRSF1 has been reported to take part in cap-dependent translation. Specifically, SRSF1 can bind to specific mRNAs that contain SRSF1-binding sites and serve as an adapter protein to recruit mTORC1 to the target mRNA by its second RRM domain and promote eIF4E-binding protein (4E-BP) phosphorylation, leading to the release of cytoplasmic cap-binding protein eIF4E and eliciting the activation of translation [79] (Fig. 2d). Additionally, SRSF1 was found to cosediment with the 80S ribosome and polysomes to promote translation in an RS domain-dependent manner [80]. Other cytoplasmic SR proteins also function in mRNA translation. For instance, SRSF1 and SRSF9 can bind to β-catenin mRNA and enhance its protein synthesis in a mTOR-dependent manner to promote β-catenin accumulation, thus mediating tumorigenesis [81]. SRSF10 interacts with the peptidyl transferase center of 28S rRNA to regulate ribosome biogenesis and translation [82]. On the contrary, hypoxia-induced SRSF3 promotes specific retention of intron 12 in EIF2B5, resulting in EIF2B5 protein decrease by the NMD pathway; therefore, inhibiting overall initiation of translation to protect head and neck cancer cells from extreme hypoxia, ensuring cell survival [83]. In addition, SRSF3 interacts with the 5′UTR of programmed cell death 4 (PDCD4) mRNA to repress translation and recruit PDCD4 mRNA to P-bodies for mRNA silencing [84]. Furthermore, SRSF7 can bind to its own pre-mRNA to promote inclusion of a PE and cause NMD-mediated transcript degradation to autoregulate itself at the translational level, thus modulating protein homeostasis during carcinogenesis [85].
SR proteins interact with noncoding RNA
SR proteins also interact with a variety of ncRNA, including lncRNA, circRNA, and microRNA, to regulate biological processes (Fig. 2e). Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is a typical lncRNA that has been widely reported to associate with SR proteins to modulate downstream pathways. MALAT1 localizes to nuclear speckles and interacts with the RRM domain of SR proteins. MALAT1 acts as a regulator of the phosphorylation/dephosphorylation status of SR proteins, contributing to alter SR protein-associated AS modulation, NMD pathway, and translation [36]. In breast cancer cells, SRSF1 interacts with mutant p53 and ID4 in a MALAT1-dependent manner. Mutant p53 and ID4 proteins promote the stabilization of SRSF1 binding of to MALAT1, thus inducing an increase of proangiogenic VEGFA isoforms while inhibiting the production of anti-angiogenic VEGFA isoforms [86]. Additionally, Pushkar et al. have reported that overexpression of MALAT1 in hepatocellular carcinoma activates the transcription of SRSF1, inducing the shorter spliced variant of S6K1, called Iso-2, an oncogene that can activate mTORC1 and induce increased 4E-BP phosphorylation [87]. SR proteins also interact with circRNA. For instance, SRSF10 can bind to the back-splice junction of cTTN1, a class of RBM20-dependent circRNAs that collectively regulate targets downstream of SRSF10 [88]. In colorectal cancer, the tumor promoter circPLCE1 directly binds to SRSF2, resulting in the repression of SRSF2-dependent PLCE1 pre-RNA splicing, which leads to tumor progression [89]. Furthermore, CLIP-seq analysis has shown that microRNAs associate with SR proteins [90], though the precise mechanism is still unknown. One possibility is that circRNAs work as a sponge for microRNA and protein, offering an opportunity for microRNAs to compete with SR proteins for binding to circRNA, thus modulating downstream molecular events, such as RNA splicing or RNA export. Overall, the study of SR protein–ncRNA interaction is still in the early stage and needs to be further investigated.
SR proteins involvement in N6-methyladenosine modification
N6-methyladenosine (m6A) is one of the most common reversible modifications in eukaryotic RNAs [91]. Regulation of m6A modification plays an essential role in RNA metabolism, including mRNA decay, pre-mRNA splicing, mRNA export, translation regulation, and ncRNA processing [92]. Several SR proteins have been reported to participate in m6A modification, thereby controlling RNA fate (Fig. 2f). For example, Zu et al. have shown that the upregulation of SRSF3 in pancreatic cancer cells contributes to increased m6A modification on lncRNA ANRIL [93]. The m6A decorations on ANRIL are responsible for SRSF3 binding to ANRIL and generating the ANRIL-L isoform via AS. ANRIL-L associates with another two proteins, Ring1B and EZH2, to form a complex to enhance drug resistance and DNA homologous recombination repair [93]. Furthermore, SRSF3, through both its RRM and RS domain, was also found to directly bind to the m6A reader YTHDC1 and the mRNA export receptor NXF1 to facilitate the export of m6A methylated mRNAs [94, 95]. In addition, SRSF7 colocalizes with the methyltransferase complex containing METTL3, METTL14, and WTAP in the nucleus; knockdown of SRSF7 decreases the m6A modification near the SRSF7 m6A modification binding sites on mRNA. SRSF7 modulates mRNA m6A methylation through recruiting METTL3, independent of its canonical role in AS and APA, to promote proliferation and migration of glioblastoma cells [96]. Recently, we have demonstrated that TRA2A induces esophageal cancer progression via MALAT1 [97]. Mechanistically, TRA2A directly interacts with core methyltransferase METTL3 and the m6A reader RBMX to regulate the methylation and stability of MALAT1 [98].
As we describe above, SR proteins participate in AS, APA, NMD, mRNA translation, ncRNA interaction, and m6A modification, so the mystery is how SR proteins accurately engage their function in a specific process, or how they precisely switch between their canonical and noncanonical roles during RNA processing. In eukaryotes, m6A methylation has been shown to be involved in almost the entire RNA life cycle, including splicing, translation, degradation, and transportation [99, 100]. Thus, we postulate that SR proteins may be guided by the m6A marks in RNA to dynamically execute their function to comprehensively regulate RNA metabolism. Indeed, Zhao et al. have reported that m6A sites are enriched in exonic regions flanking 5′- and 3′-splice sites, that overlap with the ESE binding regions, suggesting that m6A signals may be related to the activity of SR proteins [101]. Further supporting this point, we and others recently found that several SR proteins, including TRA2A, SRSF3, and SRSF7, can interact with METTL3 and other proteins in RNA methylation, and the m6A signal in pre-mRNA is essential for SR proteins to participate in RNA processing [94, 96, 98, 102].
Designing drugs to target SR–RNA interactions for clinical use
As mentioned above, phosphorylation of the SR protein RS domain controls the localization and activity of SR proteins. Much effort has been spent in the development of small molecular inhibitors to target SR protein-associated kinases. Dozens of compounds have been discovered and have demonstrated promising antitumor activity in vivo. Interested readers are referred to more extensive reviews for available inhibitors currently being assessed in preclinical/clinical studies [103,104,105]. Here, we mainly focus on inhibitors directly targeting the SR proteins themselves by means of chemicals and RNA oligomers (Table 1).
Repurposing chemicals from approved drugs
Since the early findings of inhibitors that alter HIV-1 splicing via SR proteins, attempts have been focused on screening for new antiviral compounds [106]. These results are surely valuable for viral-induced cancer treatment. For cancers independent of viral infection, therapeutic strategies targeting SR proteins have also been actively developed in recent years. SRSF6 is frequently amplified and upregulated in several cancers, including colon, lung, and breast cancer [107]. On the basis of the predicted three-dimensional (3D) structure from homology modeling, Wan et al. virtually screened compounds in the DrugBank database and identified indacaterol as a potent inhibitor targeting the RRM2 domain of SRSF6. Indacaterol is known as a β2-adrenergic receptor agonist approved for chronic obstructive pulmonary disease treatment. Functional experiments showed that indacaterol can inhibit cancer progression in colorectal tumor cells in a murine xenograft model [108]. Inspired by this preliminary study, we utilized indacaterol as a structural template to select nebivolol, a β1-adrenergic receptor antagonist, as an RRM domain antagonist of TRA2A. Follow-up experiments confirmed that nebivolol can compete with RNA targets of TRA2A to interfere with its downstream signaling in esophageal cancer cells [98]. Similarly, potent compounds have also been repurposed for SRSF3 inhibition [109].
However, SR proteins harbor similar structural domains and often cross-regulate each other or autoregulate itself [76, 110]. For example, the expression levels of SRSF10 expression correlate with other SR proteins, and depletion of SRSF10 autoregulation affects the expression of all SR proteins [110]. Thus, it needs to be investigated whether there are profound effects on other SR proteins when testing the compounds. Indeed, there is a report that GPS167/192, which impacts the activity of SRSF10, demonstrated a modest but statistically significant effect on other SR proteins [111].
Designer RNA-based therapeutic agents
Effective compound optimization needs the exact protein crystal structure, which is usually limited for SR family proteins. As an alternative approach, RNA oligonucleotides, which solely depends on the sequence information and can be easily synthesized, have shown promising results on the modulation of splicing via SR proteins. We categorize these kinds of inhibitors in the following three classes: small interfering RNAs (siRNAs), antisense oligonucleotides (ASOs), and decoy RNAs.
(A) Small interfering RNAs (siRNAs) are synthetic double-stranded RNAs and can induce target RNA cleavage via AGO2. They have long been used in research to downregulate endogenous SRSFs, such as SRSF3, SRSF6, SRSF7, and TRA2A [93, 96, 108]. However, the clinical application of siRNA to SR proteins is still not possible owing to off-target effects and lack of efficiency in in vivo delivery methods.
(B) Antisense oligonucleotides (ASOs) are short, single-stranded oligonucleotides that trigger different mechanisms, such as RNA degradation, altered splicing, and translational arrest, to modulate target transcript expression [112]. As previously described, inclusion of a PE triggers auto downregulation of SR protein expression via a NMD pathway. Members of the SR family can bind to the PE-splicing regulatory sequences, therefore allowing SR proteins to compete or cooperate to regulate the AS of PE. Based on this principle, ASOs targeting the regulatory sequence to promote or reduce PE inclusion have achieved successful control of TRA2B and SRSF3 expression, suggesting that such splice switching oligomers can be potential anticancer drugs [76, 113].
(C) Denichenko et al. synthesized three tandem motif repeats against the RRM domain of SRSF1 but not resemble the known ESE [114]. Experiments have shown these RNA decoys directly bind SRSF1 and affect splicing. The main advantage of this approach is that decoy oligonucleotides affect SR protein-RNA binding activities without disturbing the interaction between SR protein and other proteins, thus providing target selectivity and less toxicity than complete knockdown of SR protein.
Although RNA oligonucleotides can target specific SR proteins, there is still the possibility that siRNAs, ASOs, or decoy RNAs may cause dysregulation of AS events in multiple downstream molecules owing to the ability of one SR protein to regulate multiple AS events. To further enhance selectivity and to reduce toxicity, it is expected that treatment to control gene-specific splicing events without globally affecting cellular splicing will be developed. This would be achieved by carefully disentangling the specific interactions between splicing sites and the domains in SR proteins under disease conditions, and by utilizing a targeted approach, i.e., CRISPR/Cas9-mediated deletion, to modify the regulatory binding sequence of SR proteins. Moreover, it is necessary to assess the inhibitory effects by setting proper normal controls to ensure that the cancer cells are sensitive to subtle AS changes induced by the compounds while the normal cells can tolerate such alteration.
Developing treatments based on novel mechanisms
Detailed mechanistic studies have found additional clues for SR protein regulation. For instance, Zhou et al. found a significant increase of SRSF transcripts, including SRSF6, in T-cell acute lymphoblastic leukemia (T-ALL) compared with normal T cells [115]. Furthermore, they found that USP7 controls SRSF6 degradation via deubiquitination. Thus, proteasome inhibitors could be exploited for therapeutic use to control SR protein expression and inhibit T-ALL growth. Similar ubiquitylation-mediated control of protein degradation has been found for SRSF3, SRSF6, and TRA2A [115,116,117]. Furthermore, TRA2A is transcriptionally induced by hypoxia-inducible factor 1 subunit alpha (HIF1α) in pancreatic cancer [118]. Thus, these regulatory mechanisms can be developed into novel treatments in the future.
Conclusions
In this review, we summarize the canonical and noncanonical functions of SR proteins during carcinogenesis. In the future, more efforts are expected to reveal the functional significance of SR proteins and the relevant pathways during cell transformation. To translate the mechanistic findings of SR proteins into the clinic, novel drug developmental strategies are also needed to improve the specificity, safety, and efficiency in modulating the SR proteins and gene splicing, which will ultimately turn SR proteins into actionable therapeutic targets in cancer.
Availability of data and materials
Not applicable.
Abbreviations
- SR protein:
-
Serine and arginine-rich protein
- RRM:
-
RNA recognition motif
- ncRNA:
-
Non-coding RNA
- AS:
-
Alternative splicing
- APA:
-
Alternative cleavage and polyadenylation
- NMD:
-
Nonsense-mediated decay
- SRPK:
-
SR protein kinase
- CLK:
-
Cdc2-like kinase
- DYRK:
-
Dual-specificity tyrosine phosphorylation regulated kinase
- U2AF:
-
U2 auxiliary factor
- 5′SS:
-
5′ Splice sites
- 3′SS:
-
3′ Splice sites
- ESE:
-
Exonic splicing enhancer
- ESS:
-
Exonic splicing silencer
- siRNA:
-
Small interfering RNA
References
Kumar K, Sinha SK, Maity U, Kirti PB, Kumar KRR. Insights into established and emerging roles of SR protein family in plants and animals. Wiley Interdiscip Rev RNA. 2023;14(3): e1763.
Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009;23(4):482–95.
Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010;11(5):345–55.
Zhu Y, Wang X, Forouzmand E, Jeong J, Qiao F, Sowd GA, et al. Molecular mechanisms for CFIm-mediated regulation of mRNA alternative polyadenylation. Mol Cell. 2018;69(1):62–74.
Sliskovic I, Eich H, Muller-McNicoll M. Exploring the multifunctionality of SR proteins. Biochem Soc Trans. 2022;50(1):187–98.
Jumaa H, Wei G, Nielsen PJ. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Curr Biol. 1999;9(16):899–902.
Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell. 2005;120(1):59–72.
Guo R, You X, Meng K, Sha R, Wang Z, Yuan N, et al. Single-cell RNA sequencing reveals heterogeneity of Myf5-derived cells and altered myogenic fate in the absence of SRSF2. Adv Sci (Weinh). 2022;9(18): e2105775.
Sen S, Jumaa H, Webster NJ. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun. 2013;4:1336.
Zhang X, Wang Z, Xu Q, Chen Y, Liu W, Zhong T, et al. Splicing factor Srsf5 deletion disrupts alternative splicing and causes noncompaction of ventricular myocardium. iScience. 2021;24(10): 103097.
Storbeck M, Hupperich K, Gaspar JA, Meganathan K, Martinez Carrera L, Wirth R, et al. Neuronal-specific deficiency of the splicing factor Tra2b causes apoptosis in neurogenic areas of the developing mouse brain. PLoS ONE. 2014;9(2): e89020.
Bradley RK, Anczukow O. RNA splicing dysregulation and the hallmarks of cancer. Nat Rev Cancer. 2023;23(3):135–55.
Kedzierska H, Piekielko-Witkowska A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017;396:53–65.
Krainer AR, Conway GC, Kozak D. The essential pre-mRNA splicing factor SF2 influences 5’ splice site selection by activating proximal sites. Cell. 1990;62(1):35–42.
Ge H, Manley JL. A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell. 1990;62(1):25–34.
Zahler AM, Lane WS, Stolk JA, Roth MB. SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 1992;6(5):837–47.
Cavaloc Y, Popielarz M, Fuchs JP, Gattoni R, Stevenin J. Characterization and cloning of the human splicing factor 9G8: a novel 35 kDa factor of the serine/arginine protein family. EMBO J. 1994;13(11):2639–49.
Soret J, Gattoni R, Guyon C, Sureau A, Popielarz M, Le Rouzic E, et al. Characterization of SRp46, a novel human SR splicing factor encoded by a PR264/SC35 retropseudogene. Mol Cell Biol. 1998;18(8):4924–34.
Screaton GR, Caceres JF, Mayeda A, Bell MV, Plebanski M, Jackson DG, et al. Identification and characterization of three members of the human SR family of pre-mRNA splicing factors. EMBO J. 1995;14(17):4336–49.
Matsuo N, Ogawa S, Imai Y, Takagi T, Tohyama M, Stern D, et al. Cloning of a novel RNA binding polypeptide (RA301) induced by hypoxia/reoxygenation. J Biol Chem. 1995;270(47):28216–22.
Zhang WJ, Wu JY. Functional properties of p54, a novel SR protein active in constitutive and alternative splicing. Mol Cell Biol. 1996;16(10):5400–8.
Barnard DC, Patton JG. Identification and characterization of a novel serine-arginine-rich splicing regulatory protein. Mol Cell Biol. 2000;20(9):3049–57.
Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417(1):15–27.
Dauwalder B, Amaya-Manzanares F, Mattox W. A human homologue of the Drosophila sex determination factor transformer-2 has conserved splicing regulatory functions. Proc Natl Acad Sci U S A. 1996;93(17):9004–9.
Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci USA. 2000;97(17):9618–23.
Wagner RE, Frye M. Noncanonical functions of the serine-arginine-rich splicing factor (SR) family of proteins in development and disease. BioEssays. 2021;43(4): e2000242.
Clery A, Sinha R, Anczukow O, Corrionero A, Moursy A, Daubner GM, et al. Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc Natl Acad Sci USA. 2013;110(30):E2802-2811.
Xiang S, Gapsys V, Kim HY, Bessonov S, Hsiao HH, Mohlmann S, et al. Phosphorylation drives a dynamic switch in serine/arginine-rich proteins. Structure. 2013;21(12):2162–74.
Colwill K, Feng LL, Yeakley JM, Gish GD, Caceres JF, Pawson T, et al. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J Biol Chem. 1996;271(40):24569–75.
Song M, Pang L, Zhang M, Qu Y, Laster KV, Dong Z. Cdc2-like kinases: structure, biological function, and therapeutic targets for diseases. Signal Transduct Target Ther. 2023;8(1):148.
Arbones ML, Thomazeau A, Nakano-Kobayashi A, Hagiwara M, Delabar JM. DYRK1A and cognition: a lifelong relationship. Pharmacol Ther. 2019;194:199–221.
Allemand E, Dokudovskaya S, Bordonne R, Tazi J. A conserved Drosophila transportin-serine/arginine-rich (SR) protein permits nuclear import of Drosophila SR protein splicing factors and their antagonist repressor splicing factor 1. Mol Biol Cell. 2002;13(7):2436–47.
Lai MC, Lin RI, Tarn WY. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc Natl Acad Sci USA. 2001;98(18):10154–9.
Kataoka N, Bachorik JL, Dreyfuss G. Transportin-SR, a nuclear import receptor for SR proteins. J Cell Biol. 1999;145(6):1145–52.
Yun CY, Velazquez-Dones AL, Lyman SK, Fu XD. Phosphorylation-dependent and -independent nuclear import of RS domain-containing splicing factors and regulators. J Biol Chem. 2003;278(20):18050–5.
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–38.
Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proc Natl Acad Sci U S A. 2004;101(26):9666–70.
Sanford JR, Ellis JD, Cazalla D, Caceres JF. Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc Natl Acad Sci USA. 2005;102(42):15042–7.
Cavaloc Y, Bourgeois CF, Kister L, Stevenin J. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA. 1999;5(3):468–83.
Amrein H, Gorman M, Nothiger R. The sex-determining gene tra-2 of Drosophila encodes a putative RNA binding protein. Cell. 1988;55(6):1025–35.
Okkema PG, Kimble J. Molecular analysis of tra-2, a sex determining gene in C. elegans. EMBO J. 1991;10(1):171–6.
Liu J, You M, Yao Y, Ji C, Wang Z, Wang F, et al. SRSF1 plays a critical role in invariant natural killer T cell development and function. Cell Mol Immunol. 2021;18(11):2502–15.
Wei N, Cheng Y, Wang Z, Liu Y, Luo C, Liu L, et al. SRSF10 plays a role in myoblast differentiation and glucose production via regulation of alternative splicing. Cell Rep. 2015;13(8):1647–57.
Ramond F, Dalgliesh C, Grimmel M, Wechsberg O, Vetro A, Guerrini R, et al. Clustered variants in the 5’ coding region of TRA2B cause a distinctive neurodevelopmental syndrome. Genet Med. 2023;25(4): 100003.
Hsu SY, Chen CH, Mukda S, Leu S. Neuronal Pnn deficiency increases oxidative stress and exacerbates cerebral ischemia/reperfusion injury in mice. Antioxidants. 2022;11(3):466.
Ortiz-Sanchez P, Villalba-Orero M, Lopez-Olaneta MM, Larrasa-Alonso J, Sanchez-Cabo F, Marti-Gomez C, et al. Loss of SRSF3 in cardiomyocytes leads to decapping of contraction-related mRNAs and severe systolic dysfunction. Circ Res. 2019;125(2):170–83.
de Bruin RG, Rabelink TJ, van Zonneveld AJ, van der Veer EP. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur Heart J. 2017;38(18):1380–8.
Paz S, Ritchie A, Mauer C, Caputi M. The RNA binding protein SRSF1 is a master switch of gene expression and regulation in the immune system. Cytokine Growth Factor Rev. 2021;57:19–26.
Wu P, Geng B, Chen Q, Zhao E, Liu J, Sun C, et al. Tumor cell-derived TGFbeta1 attenuates antitumor immune activity of T cells via regulation of PD-1 mRNA. Cancer Immunol Res. 2020;8(12):1470–84.
Sciarrillo R, Wojtuszkiewicz A, Assaraf YG, Jansen G, Kaspers GJL, Giovannetti E, et al. The role of alternative splicing in cancer: from oncogenesis to drug resistance. Drug Resist Updat. 2020;53: 100728.
Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22(9):976–86.
Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–63.
Maniatis T, Tasic B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature. 2002;418(6894):236–43.
Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–18.
Wilkinson ME, Charenton C, Nagai K. RNA splicing by the spliceosome. Annu Rev Biochem. 2020;89:359–88.
Blencowe BJ. Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci. 2000;25(3):106–10.
Blencowe BJ, Issner R, Nickerson JA, Sharp PA. A coactivator of pre-mRNA splicing. Genes Dev. 1998;12(7):996–1009.
Merdzhanova G, Edmond V, De Seranno S, Van den Broeck A, Corcos L, Brambilla C, et al. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008;15(12):1815–23.
Kedzierska H, Poplawski P, Hoser G, Rybicka B, Rodzik K, Sokol E, et al. Decreased expression of SRSF2 Splicing factor inhibits apoptotic pathways in renal cancer. Int J Mol Sci. 2016;17(10):1598.
Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–95.
Anczukow O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19(2):220–8.
Liu T, Sun H, Zhu D, Dong X, Liu F, Liang X, et al. TRA2A promoted paclitaxel resistance and tumor progression in triple-negative breast cancers via regulating alternative splicing. Mol Cancer Ther. 2017;16(7):1377–88.
Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18(1):18–30.
Kumar A, Clerici M, Muckenfuss LM, Passmore LA, Jinek M. Mechanistic insights into mRNA 3’-end processing. Curr Opin Struct Biol. 2019;59:143–50.
Schwich OD, Blumel N, Keller M, Wegener M, Setty ST, Brunstein ME, et al. SRSF3 and SRSF7 modulate 3′UTR length through suppression or activation of proximal polyadenylation sites and regulation of CFIm levels. Genome Biol. 2021;22(1):82.
Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673–84.
Zhang Y, Liu L, Qiu Q, Zhou Q, Ding J, Lu Y, et al. Alternative polyadenylation: methods, mechanism, function, and role in cancer. J Exp Clin Cancer Res. 2021;40(1):51.
Frankiw L, Baltimore D, Li G. Alternative mRNA splicing in cancer immunotherapy. Nat Rev Immunol. 2019;19(11):675–87.
Muller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, et al. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 2016;30(5):553–66.
Sato H, Singer RH. Cellular variability of nonsense-mediated mRNA decay. Nat Commun. 2021;12(1):7203.
Nagar P, Islam MR, Rahman MA. Nonsense-mediated mRNA decay as a mediator of tumorigenesis. Genes (Basel). 2023;14(2):357.
Wan L, Deng M, Zhang H. SR splicing factors promote cancer via multiple regulatory mechanisms. Genes. 2022;13(9):1659.
Aznarez I, Nomakuchi TT, Tetenbaum-Novatt J, Rahman MA, Fregoso O, Rees H, et al. Mechanism of nonsense-mediated mRNA decay stimulation by splicing factor SRSF1. Cell Rep. 2018;23(7):2186–98.
Rahman MA, Lin KT, Bradley RK, Abdel-Wahab O, Krainer AR. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev. 2020;34(5–6):413–27.
Stoilov P, Daoud R, Nayler O, Stamm S. Human tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum Mol Genet. 2004;13(5):509–24.
Leclair NK, Brugiolo M, Urbanski L, Lawson SC, Thakar K, Yurieva M, et al. Poison exon splicing regulates a coordinated network of SR protein expression during differentiation and tumorigenesis. Mol Cell. 2020;80(4):648–65.
Jiang L, Chen Q, Bei M, Shao M, Xu J. Characterizing the tumor RBP-ncRNA circuits by integrating transcriptomics, interactomics and clinical data. Comput Struct Biotechnol J. 2021;19:5235–45.
Jiang L, Hao S, Lin L, Gao X, Xu J. fRNC: uncovering the dynamic and condition-specific RBP-ncRNA circuits from multi-omics data. Comput Struct Biotechnol J. 2023;21:2276–85.
Michlewski G, Sanford JR, Caceres JF. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol Cell. 2008;30(2):179–89.
Sanford JR, Gray NK, Beckmann K, Caceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18(7):755–68.
Fu Y, Huang B, Shi Z, Han J, Wang Y, Huangfu J, et al. SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing beta-catenin biosynthesis. EMBO Mol Med. 2013;5(5):737–50.
Liu KJ, Harland RM. Inhibition of neurogenesis by SRp38, a neuroD-regulated RNA-binding protein. Development. 2005;132(7):1511–23.
Brady LK, Wang H, Radens CM, Bi Y, Radovich M, Maity A, et al. Transcriptome analysis of hypoxic cancer cells uncovers intron retention in EIF2B5 as a mechanism to inhibit translation. PLoS Biol. 2017;15(9): e2002623.
Kim J, Park RY, Chen JK, Kim J, Jeong S, Ohn T. Splicing factor SRSF3 represses the translation of programmed cell death 4 mRNA by associating with the 5’-UTR region. Cell Death Differ. 2014;21(3):481–90.
Konigs V, de OliveiraFreitasMachado C, Arnold B, Blumel N, Solovyeva A, Lobbert S, et al. SRSF7 maintains its homeostasis through the expression of Split-ORFs and nuclear body assembly. Nat Struct Mol Biol. 2020;27(3):260–73.
Pruszko M, Milano E, Forcato M, Donzelli S, Ganci F, Di Agostino S, et al. The mutant p53-ID4 complex controls VEGFA isoforms by recruiting lncRNA MALAT1. EMBO Rep. 2017;18(8):1331–51.
Malakar P, Shilo A, Mogilevsky A, Stein I, Pikarsky E, Nevo Y, et al. Long noncoding RNA MALAT1 promotes hepatocellular carcinoma development by SRSF1 upregulation and mTOR activation. Cancer Res. 2017;77(5):1155–67.
Tijsen AJ, Cocera Ortega L, Reckman YJ, Zhang X, van der Made I, Aufiero S, et al. Titin circular RNAs Create a back-splice motif essential for SRSF10 splicing. Circulation. 2021;143(15):1502–12.
Chen Z, Chen H, Yang L, Li X, Wang Z. CircPLCE1 facilitates the malignant progression of colorectal cancer by repressing the SRSF2-dependent PLCE1 pre-RNA splicing. J Cell Mol Med. 2021;25(15):7244–56.
Sanford JR, Wang X, Mort M, Vanduyn N, Cooper DN, Mooney SD, et al. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19(3):381–94.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42.
Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112: 108613.
Wang ZW, Pan JJ, Hu JF, Zhang JQ, Huang L, Huang Y, et al. SRSF3-mediated regulation of N6-methyladenosine modification-related lncRNA ANRIL splicing promotes resistance of pancreatic cancer to gemcitabine. Cell Rep. 2022;39(6): 110813.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017;6: e31311.
Tatsuno T, Ishigaki Y. Multiple phosphorylations of SR protein SRSF3 and its binding to m(6)A reader YTHDC1 in human cells. Cells. 2022;11(9):1461.
Cun Y, An S, Zheng H, Lan J, Chen W, Luo W, et al. Specific regulation of m(6)A by SRSF7 promotes the progression of glioblastoma. Genomics Proteomics Bioinform. 2023;21(4):707–28.
Zhao X, Chen Q, Cai Y, Chen D, Bei M, Dong H, et al. TRA2A binds with LncRNA MALAT1 to promote esophageal cancer progression by regulating EZH2/beta-catenin pathway. J Cancer. 2021;12(16):4883–90.
Bei M, Hao S, Lin K, Chen Q, Cai Y, Zhao X, et al. Splicing factor TRA2A contributes to esophageal cancer progression via a noncanonical role in lncRNA m(6) A methylation. Cancer Sci. 2023;114(8):3216–29.
An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21(1):14.
Fang Z, Mei W, Qu C, Lu J, Shang L, Cao F, et al. Role of m6A writers, erasers and readers in cancer. Exp Hematol Oncol. 2022;11(1):45.
Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24(12):1403–19.
An S, Huang W, Huang X, Cun Y, Cheng W, Sun X, et al. Integrative network analysis identifies cell-specific trans regulators of m6A. Nucleic Acids Res. 2020;48(4):1715–29.
Tang J, Xie Y, Huang J, Zhang L, Jiang W, Li Z, et al. A critical update on the strategies towards small molecule inhibitors targeting Serine/arginine-rich (SR) proteins and Serine/arginine-rich proteins related kinases in alternative splicing. Bioorg Med Chem. 2022;70: 116921.
Naro C, Bielli P, Sette C. Oncogenic dysregulation of pre-mRNA processing by protein kinases: challenges and therapeutic opportunities. FEBS J. 2021;288(21):6250–72.
Martin Moyano P, Nemec V, Paruch K. Cdc-Like Kinases (CLKs): biology, chemical probes, and therapeutic potential. Int J Mol Sci. 2020;21(20):7549.
Soret J, Bakkour N, Maire S, Durand S, Zekri L, Gabut M, et al. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. Proc Natl Acad Sci USA. 2005;102(24):8764–9.
She W, Shao J, Jia R. Targeting splicing factor SRSF6 for cancer therapy. Front Cell Dev Biol. 2021;9: 780023.
Wan L, Yu W, Shen E, Sun W, Liu Y, Kong J, et al. SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut. 2019;68(1):118–29.
Zhang Y, Wang M, Meng F, Yang M, Chen Y, Guo X, et al. A novel SRSF3 inhibitor, SFI003, exerts anticancer activity against colorectal cancer by modulating the SRSF3/DHCR24/ROS axis. Cell Death Discov. 2022;8(1):238.
Meinke S, Goldammer G, Weber AI, Tarabykin V, Neumann A, Preussner M, et al. Srsf10 and the minor spliceosome control tissue-specific and dynamic SR protein expression. Elife. 2020;9: e56075.
Sohail M, Shkreta L, Toutant J, Rabea S, Babeu JP, Huard C, et al. A novel class of inhibitors that target SRSF10 and promote p53-mediated cytotoxicity on human colorectal cancer cells. NAR Cancer. 2021;3(2): zcab019.
Crooke ST, Baker BF, Crooke RM, Liang XH. Antisense technology: an overview and prospectus. Nat Rev Drug Discov. 2021;20(6):427–53.
Guo J, Che X, Wang X, Jia R. Inhibition of the expression of oncogene SRSF3 by blocking an exonic splicing suppressor with antisense oligonucleotides. RSC Adv. 2018;8(13):7159–63.
Denichenko P, Mogilevsky M, Clery A, Welte T, Biran J, Shimshon O, et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat Commun. 2019;10(1):1590.
Zhou Y, Han C, Wang E, Lorch AH, Serafin V, Cho BK, et al. Posttranslational regulation of the exon skipping machinery controls aberrant splicing in leukemia. Cancer Discov. 2020;10(9):1388–409.
Kumar D, Das M, Sauceda C, Ellies LG, Kuo K, Parwal P, et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J Clin Invest. 2019;129(10):4477–91.
Lu Y, Jiang B, Peng K, Li S, Liu X, Wang B, et al. Differential degradation of TRA2A and PYCR2 mediated by ubiquitin E3 Ligase E4B. Front Cell Dev Biol. 2022;10: 833396.
Cao W, Lei S, Zeng Z, Xiao C, Sun B, Xie P, et al. Transformer 2 alpha homolog is a downstream gene of hypoxia-inducible factor 1 subunit alpha and is involved in the progression of pancreatic cancer. Bioengineered. 2022;13(5):13238–51.
Acknowledgements
Not applicable.
Funding
This study was supported by the Guangdong Basic and Applied Basic Research Foundation, China (no. 2024A1515011328 to JZX).
Author information
Authors and Affiliations
Contributions
M.B. and J.X. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bei, M., Xu, J. SR proteins in cancer: function, regulation, and small inhibitor. Cell Mol Biol Lett 29, 78 (2024). https://doi.org/10.1186/s11658-024-00594-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-024-00594-6