
Abstract
Background
The etiology and most risk factors for a sporadic first primary neoplasm in childhood or subsequent second primary neoplasms are still unknown. One established causal factor for therapy-associated second primary neoplasms is the exposure to ionizing radiation during radiation therapy as a mainstay of cancer treatment. Second primary neoplasms occur in 8% of all cancer survivors within 30 years after the first diagnosis in Germany, but the underlying factors for intrinsic susceptibilities have not yet been clarified. Thus, the purpose of this nested case–control study was the investigation and comparison of gene expression and affected pathways in primary fibroblasts of childhood cancer survivors with a first primary neoplasm only or with at least one subsequent second primary neoplasm, and controls without neoplasms after exposure to a low and a high dose of ionizing radiation.
Methods
Primary fibroblasts were obtained from skin biopsies from 52 adult donors with a first primary neoplasm in childhood (N1), 52 with at least one additional primary neoplasm (N2+), as well as 52 without cancer (N0) from the KiKme study. Cultured fibroblasts were exposed to a high [2 Gray (Gy)] and a low dose (0.05 Gy) of X-rays. Messenger ribonucleic acid was extracted 4 h after exposure and Illumina-sequenced. Differentially expressed genes (DEGs) were computed using limma for R, selected at a false discovery rate level of 0.05, and further analyzed for pathway enrichment (right-tailed Fisher’s Exact Test) and (in-) activation (z ≥|2|) using Ingenuity Pathway Analysis.
Results
After 0.05 Gy, least DEGs were found in N0 (n = 236), compared to N1 (n = 653) and N2+ (n = 694). The top DEGs with regard to the adjusted p-value were upregulated in fibroblasts across all donor groups (SESN1, MDM2, CDKN1A, TIGAR, BTG2, BLOC1S2, PPM1D, PHLDB3, FBXO22, AEN, TRIAP1, and POLH). Here, we observed activation of p53 Signaling in N0 and to a lesser extent in N1, but not in N2+. Only in N0, DNA (excision-) repair (involved genes: CDKN1A, PPM1D, and DDB2) was predicted to be a downstream function, while molecular networks in N2+ were associated with cancer, as well as injury and abnormalities (among others, downregulation of MSH6, CCNE2, and CHUK). After 2 Gy, the number of DEGs was similar in fibroblasts of all donor groups and genes with the highest absolute log2 fold-change were upregulated throughout (CDKN1A, TIGAR, HSPA4L, MDM2, BLOC1SD2, PPM1D, SESN1, BTG2, FBXO22, PCNA, and TRIAP1). Here, the p53 Signaling-Pathway was activated in fibroblasts of all donor groups. The Mitotic Roles of Polo Like Kinase-Pathway was inactivated in N1 and N2+. Molecular Mechanisms of Cancer were affected in fibroblasts of all donor groups. P53 was predicted to be an upstream regulator in fibroblasts of all donor groups and E2F1 in N1 and N2+. Results of the downstream analysis were senescence in N0 and N2+, transformation of cells in N0, and no significant effects in N1. Seven genes were differentially expressed in reaction to 2 Gy dependent on the donor group (LINC00601, COBLL1, SESN2, BIN3, TNFRSF10A, EEF1AKNMT, and BTG2).
Conclusion
Our results show dose-dependent differences in the radiation response between N1/N2+ and N0. While mechanisms against genotoxic stress were activated to the same extent after a high dose in all groups, the radiation response was impaired after a low dose in N1/N2+, suggesting an increased risk for adverse effects including carcinogenesis, particularly in N2+.
Similar content being viewed by others

Introduction
Malignant neoplasms or any neoplasm in the central nervous system occurring in children and adolescents before the age of 20 years are defined as childhood cancer (IARC 2016). Despite extensive research on this topic, risk factors for sporadic childhood cancer remain largely unknown (Thun et al. 2017). Inherited genetic predispositions only account for 10% of childhood cancer cases (Saletta et al. 2015). Such predispositions can affect pathways that are needed for radiation-response in the course of treatment for the first primary neoplasm (Saletta et al. 2015; Vogelstein et al. 2004). Genotoxic cancer therapies using cytostatic drugs and ionizing radiation (IR) in high doses [HDIR, ≥ 2 Gray (Gy)] during radiation therapy are established risk factors for the development of second primary neoplasms later in life (Spector et al. 2015; Tukenova et al. 2011; Scholz-Kreisel et al. 2018; Travis et al. 2012; Ron et al. 1988; Inskip et al. 2016; Meadows et al. 1980; Tucker et al. 1991). Young age at exposure is an additional and important risk factor for iatrogenic second primary neoplasms (Hodgson et al. 2017). Of all survivors of a first primary neoplasm occurring before the age of 15 years in Germany, 8% develop a second primary neoplasm within 30 years of the first diagnosis (Scholz-Kreisel et al. 2018). Since the incidence and survival rates of childhood cancer are increasing worldwide, the number of second primary neoplasms is expected to increase as well (Scholz-Kreisel et al. 2018; Kutanzi et al. 2016; Kaatsch et al. 2017). Contrary to the established cancer risk associated with exposure to HDIR, the carcinogenic potential of low doses of IR (LDIR, < 0.1 Gy) is still being discussed and controversial (Inskip et al. 2016; Spycher et al. 2015; Cardarelli et al. 2018; Sutou 2018; Ji et al. 2019; Luckey 1982; Graupner et al. 2017; Ma et al. 2013; Velegzhaninov et al. 2015). In children, studies suggest elevated leukemia risk in association with diagnostic applications of LDIR, such as computed tomography (Berrington de Gonzalez et al. 2016; Little et al. 2018). Despite the potential risk not being fully understood, LDIR are increasingly used in medical diagnostics (Smith-Bindman et al. 2008; Linet et al. 2012). In Germany, diagnostic applications account for about 41% of total radiation deposited in organs per person and year (1.6 of 3.9 millisievert) (Claudia Hachenberger et al. 2016). Also, in radiation therapy, the unwanted but inevitable exposure to LDIR outside the primary radiation beam is regarded as an additional risk factor for second primary neoplasms besides the impact of HDIR deposited within the tumor volume (Diallo et al. 2009).
Irrespective its dosage, exposure to IR causes a variety of cellular responses primarily based on the potent induction of deoxyribonucleic acid (DNA) damage (Nikitaki et al. 2016). On the cellular level, radiation-induced DNA damage activates complex signaling cascades of DNA repair, cell cycle regulation, apoptosis, senescence, and immunogenic responses (Mavragani et al. 2016). The induction of these signaling pathways is dose-dependent (Graupner et al. 2017; Velegzhaninov et al. 2015; Ghandhi et al. 2015; Tilton et al. 2016; Helm et al. 2020), whereby signaling pathways for the inactivation of cells by apoptosis or premature senescence predominate after HDIR (Sokolov et al. 2015; Ding et al. 2005). Such mechanisms of radiation-response can be measured by changes in gene expression (Wahba et al. 2016) and ribonucleic acid (RNA)-sequencing has shown to be a potent and precise method for transcriptome analysis, replacing probe-based tools such as microarrays (Kukurba et al. 2015). Studies that examined the molecular response to IR via transcriptome analysis have been conducted using vastly different cell types, irradiation doses, time points of extraction of messenger RNA (mRNA), and sequencing-methods depending on their research intent (Sokolov et al. 2015; Ding et al. 2005; Ray et al. 2012; Yunis et al. 2012; Warters et al. 2009; Stecca et al. 1998). In a previous work, we have reported the downregulation of Cell cycle checkpoint control protein RAD9A (RAD9A and Cyclin Dependent Kinase Inhibitor 1A (CDKN1A) in non-irradiated (Victor et al. 2013a) and irradiated [1 Gy, (Weis et al. 2011)] fibroblasts of former childhood cancer patients with at least one second primary neoplasm (N2+), compared to former childhood cancer patients with a first primary neoplasm only (N1). Both of these genes are associated with the cellular response and repair of radiation-induced DNA damages. These findings indicate a compromised radiation-response in N2+ that might be associated with an increased risk to their second primary malignancies. Additionally, we hypothesize that former childhood cancer patients show differences in the transcriptional radiation-response in their skin fibroblasts compared to those of cancer-free controls (N0). To date, changes in gene expression induced by IR have not been examined and results have not been compared between N1 and N2+ using next-generation sequencing and subsequent pathway analysis.
The present study aimed to unravel molecular mechanisms of susceptibilities to sporadic cancer in early childhood and therapy-associated second primary neoplasms related to the genotoxic radiation-response. For this purpose, we explored gene expression profiles using Illumina-based next-generation sequencing and Ingenuity Pathway Analysis (IPA) in N1 (n = 52), N2+ (n = 52), and N0 (n = 52) after exposure to a low and a high dose of IR.
Design, participants, and methods
Study design and participants
The KiKme study is a nested case–control study with emphasis on the interplay between hereditary dispositions, cellular reaction to IR, risk for sporadic childhood cancer, as well as therapy-related second primary neoplasms. From 2013 to 2019, 591 participants were included in the study. The study population, data collection, and strategies for recruitment were described elsewhere (Marron et al. 2021). For the experiments in this project, primary skin fibroblasts, obtained from skin biopsies of 156 participants from the KiKme-study, were used (Table 1). Former childhood cancer patients registered in the German Childhood Cancer Registry (Scholz-Kreisel et al. 2018) provided the basis for the recruitment of cancer patients (52 with at least one second primary neoplasm and 52 with first primary neoplasms only). Participants had to be at least 18 years old and diagnosed with one first primary neoplasm in childhood. The first primary neoplasm had to be one of the three most common childhood cancers, including leukemia, lymphoma, or a tumor of the central nervous system. The second primary neoplasm had to occur at an anatomic site potentially exposed during radiation therapy, such as thyroid carcinoma, breast cancer, skin carcinoma, malignant melanoma, leukemia, or ependymomas and choroid plexus tumors (Dracham et al. 2018). In addition, 52 cancer-free controls were recruited from patients undergoing an elective surgery unrelated to cancer at the Department of Orthopedic Surgery at the University Medical Center Mainz. One long-term childhood cancer survivor with at least one second primary neoplasm, one without a second primary neoplasm, and one cancer-free control were matched according to age at sampling, as well as sex, and then processed as triplets. The long-term cancer survivors of a triplet were also matched according to the tumor entity as well as the age at and year of diagnosis of the first primary neoplasm.
Samples and experiments
The acquisition of biosamples, the establishment of primary fibroblasts, radiation experiments, RNA extraction and processing, as well as the analysis for differentially expressed genes (DEGs) have been described in depth elsewhere (Marron et al. 2021; Brackmann et al. 2020). In summary, fibroblasts were obtained via skin biopsies from former childhood cancer patients as a 3 mm punch biopsy from the inside of the cubital region. Samples from cancer-free controls were taken from the scar region of the surgery. Primary fibroblasts were expanded from biopsies for 14–15 days and cryopreserved. For radiation experiments, cells were thawed, cultured, and synchronized in G1 by confluency. An overview of the experimental workflow is provided as Fig. 1. Cells were exposed at room temperature to a dose of 2 Gy with 140 kilovolt X-rays at a dose rate of 3.62 Gy per minute or to 0.05 Gy with 50 kilovolt X-rays at a dose rate of 0.34 Gy per minute using the D3150 X-ray Therapy System (Gulmay Medical Ltd, Byfleet, UK). This was achieved by reducing voltage to 50 kilovolt and a source-to-target distance of 30 cm. Thus, exposure times were 0.55 and 0.15 min for HDIR and LDIR, respectively. Sham-irradiated cells were kept at the same conditions in the radiation device control room. Cells from matched triplets were cultivated and treated simultaneously to prevent batch effects within groups. mRNA was extracted four hours after radiation exposure. In previous work, we showed the highest number of DEGs in fibroblasts at this time point during an overall observational period of 15 min to 24 h postexposure to IR (Brackmann et al. 2020). For mRNA-sequencing (RNA-Seq), libraries were processed on a HiSeq2500 instrument (Illumina, San Diego, California, USA) set to high-output mode. Single-end reads with a length of 50 base pairs were generated using TruSeq Single Read Cluster Kit v3 (Illumina, San Diego, California, USA) and TruSeq SBS Kit v3 (Illumina, San Diego, California, USA). Base calling was performed by Real-Time Analysis (Version 1.8.4) and the resulting data were converted into FASTQ format using bcl2fastq (Version 1.8.4, Illumina, San Diego, California, USA).

Experimental workflow. Primary fibroblasts of all donors were processed as matched triplets consisting of a donor with a first primary neoplasm only, a donor with a second primary neoplasm and a cancer-free control. The triplets were exposed to a low dose of ionizing radiation (LDIR, 0.05 Gray), a high dose of ionizing radiation (HDIR, 2 Gray), or were sham-irradiated (0 Gray). mRNA was extracted 4 h after exposure, Illumina-sequenced, and processed for the investigation of differentially expressed genes for each donor with regard to the regulation/level in sham-irradiated cells (dashed arrows). Differentially expressed genes as a result of groupwise-expression models were further subjected to Ingenuity Pathway Analysis and resulting gene-sets of the intra-group comparisons were analyzed for over-representation using the Gene Ontology via ConsensusPathDB
Processing of RNA-Seq data
To detect DEGs and conduct pathway analysis, RNA-Seq data were processed (Marron et al. 2021). First, Trimmomatic was employed to clean adapter sequences from the raw reads (Bolger et al. 2014). Here, a quality of less than 3 was defined as the threshold to remove bases. The reads with an average quality less than 15 over 4 bases were trimmed. Processed reads were aligned to the human reference genome (GRCh38) using STAR [STAR_2.6.0c, Dobin et al. 2013]. The number of aligned reads per gene (seen as equal to the expression per gene), was quantified using FeatureCounts (Liao et al. 2014). Data were then normalized using the voom method (Law et al. 2014).
Analysis of differential gene expression
DEGs dependent on radiation dose were detected using linear models implemented in the limma package (Ritchie et al. 2015), accounting for the individual donor as block variable, as well as the factors donor group and radiation dose. The differential gene expression after irradiation was computed by comparing measurements of fibroblasts of each participant with measurements after sham-irradiation (e.g., counts of transcripts in cells of each individual after 0 Gy versus counts after 2 Gy; Additional files 1, 2). Differential expression was detected using three different models: (1) crude model, (2) considering age and sex (model 1), and (3) considering age, sex, age at and year of diagnosis of the first neoplasm, and tumor type (model 2). The latter was used in analysis with N1 and N2+ data only. P-values were computed for the interaction between the effect of radiation and donor group, as well as the effect of radiation alone. Genes with a p-value adjusted at a false discovery rate [FDR, Benjamini–Hochberg procedure (Benjamini et al. 1995)] below 0.05 were flagged as significant for further analyses. Since multiple pair-wise comparisons were calculated, p-values from the separate analysis for each donor group have to be regarded as explorative.
Quantitative real-time reverse-transcriptase polymerase chain reaction (qRT-PCR)
Total RNAs were prepared from treated and untreated fibroblast cultures using the Nucleo Spin RNA Plus Kit from Macherey–Nagel (Düren, Germany). 2 µg of the RNA samples were reversely transcribed into complementary DNA using the SuperScript IV First-Strand random hexamer Synthesis System (Invitrogen, Waltham, United States). Exon-spanning forward and reverse primers were designed with Primer-BLAST (Ye et al. 2012) or purchased from Qiagen (Hilden, Germany), depending on the availability of primers at Qiagen (Additional file 3a). Transcripts of the TATA-Box Binding Protein gene were used as endogenous reference gene control. Each 10 µl reaction volume contained 25 ng complementary DNA or DNA template in 5 µl Sybr-Green Master Mix (Biozym), 2 µl ribonuclease-free PCR graded water (MN), and 1 µl each of forward and reverse primer (10 µM). All reactions were performed in triplicate and two stages, with one cycle of 95 °C for 10 min (first stage) and 45 cycles of 94 °C for 10 s, 10 s at the primer-specific melting temperature, and 72 °C for 10 s (second stage) using the LightCycler 480II Roche. Amplification qualities were assayed using melting curves and agarose gel analysis. The qRT-PCR amplification efficiency was calculated using the LinReg program (Ruijter et al. 2009) and the cycle threshold values were corrected using the mean amplification efficiency. Relative quantification was carried out with the comparative cycle threshold method using the endogenous control gene and the control at 0 Gy for calibration.
Ingenuity pathway and gene ontology analysis
Pathway analyses were conducted via IPA [Content Version 57662101, QIAGEN Inc., 2020, (Krämer et al. 2014)]. As input, lists of DEGs and the corresponding gene-wise p-values (adjusted with the FDR procedure), as well as the log2 fold-change (LFC) retrieved from the analysis of differential expression were used. The cutoff for the FDR was set to 0.05. The settings for IPA were selected as experimentally observed data in human fibroblasts and commercial fibroblast cell lines (Additional file 4). In IPA, Fisher’s Exact Test is employed to compute the significance of the overlap of a list of DEGs and a gene list, representing a pathway. Negative logarithms (-log) of the resulting p-values are reported by IPA and a value of at least 1.3 (≙ p-value = 0.05) after Benjamini–Hochberg correction was regarded as significant here. Such pathways were marked as affected. (In-) activation z-score threshold was set to z ≥|2| (Krämer et al. 2014). The z-score indicates pathway activation or inactivation by comparing given expressional directions of pathway components with information from the data set entered for analysis. If z ≥|2|, a pathway was marked as modulated. Moreover, pathways with a negative z-score ≤ −2 were marked as inactivated, pathways with a z-score ≥ 2 were marked as activated. The ratio, calculated as the number of DEGs (k) per number of postulated genes in the pathway in IPA database (K), was documented as well. For some pathways, z-score calculation was not possible [z = Not a number (NaN)]. This was either because of insufficient data in the IPA database (lack of information on “expected direction of differential expression”) or the information provided by our data was not sufficient. For these pathways, either activity prediction is not useful due to its circular or multi-cellular nature, or data are not sufficient for proper calculation of the z-score. The complete output from the IPA analyses can be found in Additional file 5. For all experiments, molecular networks from IPA were extracted. For the sake of legibility, only networks with a network score > 10 were included into the main body of this work, as all data sets showed only 1–2 networks with a score of this magnitude, with the next networks per donor group and dose combination reaching scores in the range of 1–6. The full list can be found in Additional file 5d. IPA was also employed to predict upstream regulators. These are transcription factors that are likely to have induced the DEGs and the direction of differential expression. Again, Fisher’s Exact Test was used to calculate a p-value for the significance of overlap of DEGs and genes, known to be regulated by a specific transcription factor. As described above, a z-score for the prediction of the activation state was calculated where applicable by IPA. This method was also applied for the prediction of downstream functions and diseases (Additional file 5c). As with pathway analyses, results from upstream and downstream analyses were adjusted using the Benjamini–Hochberg procedure to adjust for FDR. All results of the pathway analyses for model 1 are displayed in heat maps in Additional file 6. A comparison of differential expression values of genes per donor group and treatment in the analyzed pathways was also plotted as heat maps (Additional file 7). In all analyzed pathways, the molecule activity prediction of IPA was used (Additional file 8). Moreover, genes found in the IPA network analysis were displayed in a heat map, as well (Additional file 9). To account for even small effects in the interaction of radiation and donor group, results of the interaction analysis were filtered for the top 50 genes per donor group comparison with regard to the p-value irrespective of the significance threshold. These gene sets were then analyzed for over-representation using the ConsensusPathDB [Additional file 5e, (Kamburov et al. 2012)] against the gene background provided in Additional file 5f. The resulting Gene Ontology (GO) terms were then summarized as well as displayed in tree maps using REVIGO (Supek et al. 2011) with an allowed semantic similarity of 0.9 and organism set to Homo sapiens (Additional file 10a and b). Moreover, we filtered the genes per comparison for those within the 0.01% lowest p-values of each respective data set, a p-value < 0.15, or LFC >|0.75|. For additional sensitivity analyses, DEGs and subsequent IPA analyses were also computed for data (1) stratified by sex and adjusted for age (Additional file 11) and (2) after exclusion of self-reported non-Caucasian participants (n = 1) prior to computing DEGs using model 1 (Additional file 12). In addition to that, we repeated the IPA analyses using only genes showing absolute changes larger than 20% compared to the expression levels after 0 Gy (Additional file 13).
Results
Study participants
Primary fibroblasts of 156 donors were analyzed (52 N0, 52 N1, and 52 N2+; Table 1). The median age of participants was 32.0 and 32.5 years at sampling for long-term survivors of childhood cancer with at least one second primary neoplasm and without, respectively (interquartile range (IQR): 28.0–38.2 years) and 33.0 years (IQR: 27.8–38.0 years) for cancer-free controls. In each donor group, 27 (51.92%) of the participants were female. For long-term childhood cancer survivors, the median age at diagnosis for the first primary neoplasm was 8.0 years (IQRsecond primary neoplasm: 4.0–11.2 years, IQRfirst primary neoplasm: 3.8–12.0 years). The median age at diagnosis for the second primary neoplasm was 23.0 years (IQR: 17.0–30.0 years).
Differential gene expression after exposure to LDIR
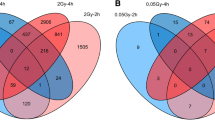
In fibroblasts of all donor groups and for all adjustment models (crude model/model 1/model 2), more genes were down- than upregulated after exposure to LDIR (Fig. 2a). The total number of DEGs was about three times higher in both, N2+ (model 1: 694) and N1 (model 1: 653), compared to N0 (model 1: 236). A detailed list of DEGs after LDIR is provided in Additional file 1a. The amount of DEGs was similar across adjusted models for each group of donors. Less than half of all DEGs were upregulated across all three groups in all adjustment models in reaction to LDIR (Fig. 2a). N0 showed the highest fraction of upregulated genes (44.07%, model 1), albeit the lowest amount (n = 104, model 1) of total upregulated genes, followed by N2+ (40.63%, model 1), and N1 (37.67%, model 1) after LDIR. Compared to female N1 and N2+ and male N0, male N1 and N2+ showed a larger number of DEGs after LDIR (Additional file 11a). These were mostly centered at the p-value threshold of significance. The top genes with regard to the p-value in the main analysis were differentially expressed in all groups nevertheless (Additional file 11b). The removal of self-reported non-Caucasian participants did not change the results of the differential expression analysis after LDIR (Additional file 12a). Moreover, upregulated genes after LDIR showed higher peaks concerning the -log(p-value) in the volcano plots (Fig. 2b). Only upregulated genes showed a p-value < 10–10 across donor groups after LDIR. The top 10 genes with regard to p-value after LDIR are shown stratified by donor group and LFC-direction in Fig. 2c. All of the highest-ranking upregulated genes were present in all groups after LDIR. Across these top-ranking genes that were downregulated, mutS homolog 6 (MSH6), Disabled homolog 2 (DAB2), Peroxisome proliferator-activated receptor gamma coactivator-related protein 1 (PPRC1), Nucleoporin 153 (NUP153), Serum response factor (SRF), 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), and TBC1 Domain Family Member 10A (TBC1D10A) were observed to be only downregulated in N1 and N2+. Cyclin E2 (CCNE2) was only downregulated in N2+. Nevertheless, qPCR conducted on MSH6 for a subsample (n = 6 per group) did not confirm differential expression after LDIR (Additional file 3b). In a previous work within the collective of the KiKme study [n = 2 per group, (Brackmann et al. 2020)], we showed that across groups Mouse double minute 2 (MDM2) is upregulated after LDIR (Additional file 3c).

Summarized results on differential gene expression after 0.05 Gray. Differentially expressed genes in irradiated compared to sham-irradiated fibroblasts from donors with a first primary neoplasm only (N1), donors with at least one second primary neoplasm (N2+), and cancer-free controls (N0) 4 h after exposure to 0.05 Gray (false discovery rate adjusted p-value < 0.05). The data are presented for the crude model, model 1 (considering age at sampling and sex), and model 2 [considering age at sampling, sex, age at and year of diagnosis of the first neoplasm, and tumor type (not applicable for N0 data)]. In total, 14,756 genes were detected in the samples. Shown are A the proportion of up- and downregulated genes stratified by dose, group, and model, B volcano plots for the results of model 1, and C top 10 genes with regard to p-value, stratified by direction of log2 fold-change
Analysis for interactions of the effect of radiation dose and donor group was used to identify genes showing a differential reaction to radiation exposure between donor groups. Only one gene showed a differential reaction after exposure to LDIR: Comparing N2+ and N0, Calmodulin binding transcription activator 2 (CAMTA2) was upregulated with a borderline significant p-value [LFC: 0.101, p-value = 0.056 (crude model)] after LDIR (Table 2). All genes that were differentially expressed after LDIR in more than one donor group showed the same direction of differential expression (Additional files 1b and 2b). Comparing fibroblasts of cancer survivors with N0, member RAS oncogene family (RAB41), CAMTA2, POZ/BTB and AT hook containing zinc finger 1 (PATZ1), receptor interacting serine/threonine kinase 1 (RIPK1), speedy/RINGO cell cycle regulator family member E3 (SPDYE3), and zinc finger protein 226 (ZNF226) were of interest based on the extended criteria after LDIR (Table 2, Fig. 3a). Comparing N1 with N2+, nine genes of interest were observed after LDIR: ADNP antisense RNA 1 (ADNP-AS1), ALG9 alpha-1,2-mannosyltransferase (ALG9), clustered mitochondria homolog pseudogene 3 (CLUHP3), deleted in lymphocytic leukemia 2 (DLEU2), uncharacterized LOC101927630, myosin VIIB (MYO7B), transcription factor CP2 (TFCP2), Transforming growth factor beta-induced anti-apoptotic factor 1 (TIAF1), and long intergenic non-protein coding RNA 2257 (LINC02257). Due to multiple occurrences of identical p-values, filtering for the top 50 genes with regard to the p-value per donor group comparison for the use in GO enrichment analysis resulted in more than 50 genes in two instances (n = 82 for N1/N2+ vs. N0 and n = 55 for N1 vs. N0). All of these were used in the GO analyses.

Summarized results on interaction effects. Results of analysis for differentially expressed genes in irradiated compared to sham-irradiated fibroblasts from donors with a first primary neoplasm only (N1), donors with at least one second primary neoplasm (N2+), and cancer-free controls (N0) 4 h after exposure to A 0.05 Gray and B 2 Gray including the interaction of group status and radiation dose. The data are presented for model 1 (considering age at sampling and sex). The highlighted genes were selected based on the data structure of each comparison, to highlight top-ranking genes: if their p-value was among the top 0.01% of that data set, p-value < 0.15, and/or log2 fold-change> |0.75|. Dashed horizontal lines indicate the significance threshold of the adjusted p-value < 0.05
Functional analysis of DEGs after exposure to LDIR
The over-representation analysis resulted in distinct GO terms for all comparisons of interactions between radiation dose and donor group and can be found in Additional files 5e and 10b. Noteworthy, N2+ compared to N0 showed a large cluster of seven GO terms subsumed under the term tRNA methylation. Comparing N1/N2+ with N0, stem cell differentiation and xenobiotic transport (including ion transport) were the largest clusters of GO terms (three terms each).
Data of DEGs per donor group, radiation dose, and adjustment model were used for upstream analyses in IPA (Fig. 4, Additional file 5b). We observed Tumor protein p53 (p53) as a common predicted upstream regulator in fibroblasts of all donor groups after LDIR (Additional file 5a). N2+ and N0 showed E2F Transcription Factor 1 (E2F1) to be an additional upstream regulator after LDIR, which was not significant after correction for false discovery.

Results of Ingenuity Pathway Analysis. Overview of affected (false discovery rate adjusted at p-value < 0.05) and (in-) activated pathways (|z|≥ 2), predicted upstream effectors, downstream biofunctions and diseases, and observed molecular networks after irradiation with a low (0.05 Gray) or a high dose (2 Gray) ordered by p-value. For molecular networks, the network score instead of a p-value and no z-score was calculated by Ingenuity Pathway Analysis and is given inside the tiles. For each combination of dose, model, and donor group, the top functions of the network with a score > 10 is presented, except for N2+ after 2 Gray, where two networks had a score above 10 in all models. On the right-hand side, the functions associated with the networks are shown. *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001. Model 1: considering age at sampling and sex, model 2: considering age at sampling, sex, age at and year of diagnosis of the first neoplasm, and tumor type (not used with data from N0), N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm
Correspondingly, pathway analysis showed that p53 Signaling was affected and activated after LDIR, however only in N0 (crude model and model 1) and N1 (crude model and model 2), but not in N2+ (Fig. 4, Additional file 5b). Here, Proliferating cell nuclear antigen (PCNA) and Tumor Protein P53 Inducible Nuclear Protein 1 (TP53INP1) were not differentially expressed in N2+, P53-Induced Death Domain Protein 1 (PIDD1), and B-cell lymphoma 2 (BCL2) only in N0 (Additional file 7b). Using only DEGs with an LFC higher than |0.25|, p53 Signaling was not activated in any donor group and only affected in all donor groups and models but N1, model 1 (Additional file 13). In this analysis, Cyclins and Cell Cycle Regulation and Estrogen-mediated S-phase Entry were inactivated in all models of N2+. In the sensitivity analysis Molecular Mechanisms of Cancer were only affected in N2+ after LDIR. Downstream functions in reaction to LDIR for N0 were predicted by IPA as well (Fig. 4, Additional file 5c). These were Cell viability of fibroblast cell lines [CDKN1A, DNA Polymerase Eta (POLH), and Breast Cancer Gene 2, DNA repair associated (BRCA2)], Nucleation of fibroblast cell lines (CDKN1A and BRCA2), Excision repair of DNA [CDKN1A and DNA damage-binding protein 2 (DDB2)], and Repair of DNA (CDKN1A, PPM1D, and DDB2). Except the latter, all of these were only present in data of the crude model, whereas Repair of DNA was also found in model 1.
In each donor group and adjustment model, one network with a score over 10 was identified by IPA in the data from the LDIR treatment (Fig. 4, Additional files 5d and 9). The annotated functions of the network in N0 were Cell Cycle, Cell Death and Survival, and DNA Replication, Recombination, and Repair in the crude model and model 1. For N1, annotations in the crude model and model 2 were Cell Cycle, Cell Death and Survival, and Gene Expression after LDIR. In model 1, the network included Cell Cycle, Cellular Development, and DNA Replication, Recombination, and Repair after LDIR. For N2+, top diseases and functions were Cancer, Cell Death and Survival, and Organismal Injury and Abnormalities for all models. Together, we observed a trend towards the activation of cell cycle control and DNA repair in N0 and cell death- and cancer-associated signaling pathways in N2+ following LDIR.
Differential gene expression after exposure to HDIR
As in treatment with LDIR, there were more downregulated than upregulated DEGs in fibroblasts of all donor groups after exposure to HDIR (Fig. 5a, Additional file 1a). The total number of DEGs was slightly higher in N2+ and N1 compared to N0 in all adjustment models and the number of DEGs per group differed only slightly between adjustment models after HDIR. As with reaction to LDIR, upregulated genes after HDIR showed lower p-values compared to downregulated genes (Fig. 5b, model 1 only). In detail, only upregulated genes showed a p-value in the range of 10–75 to 10–275 in all donor groups. All of the highest-ranking up- and downregulated genes were found in all groups after HDIR (Fig. 5c, model 1 only). Here, qRT-PCR conducted for PCNA for a subsample (n = 6 per group) did confirm differential expression after HDIR for N1 (Additional file 3b). Also, the upregulation of CDKN1A and MDM2 across all groups in the present study has been previously confirmed by us using qRT-PCR [(Brackmann et al. 2020), Additional file 3c]. Neither stratifying for sex, nor the prior removal of self-reported non-Caucasian participants did change the results of the differential expression main analysis for exposure to HDIR. Contrary to the reaction to LDIR, several genes were differentially expressed when comparing the response to HDIR between donor groups (Table 2, Fig. 3b, Additional file 1b). Bridging integrator 3 (BIN3), cordon-bleu WH2 repeat protein like 1 (COBLL1), eukaryotic translation elongation factor 1 alpha lysine and N-terminal methyltransferase (EEF1AKNMT), long intergenic non-protein coding RNA 601 (LINC00601), sestrin 2 (SESN2), and Tumor necrosis factor receptor superfamily member 10a (TNFRSF10A) were differentially expressed comparing fibroblasts of both cancer groups to N0 after HDIR (model 1). Additionally, BTG Anti-Proliferation Factor 2 (BTG2) was differentially expressed comparing N1 to N0 (model 1). As with the gene expression after LDIR, the general direction of the gene expression after HDIR in genes that were commonly differentially expressed in more than one donor group, was identical (Additional files 1b and 2b). The additional filtering for non-significant genes with a p-value smaller than 0.15, the 0.01% lowest p-values of the respective data, or with an LFC larger than |0.75|, found five genes of interest for the comparison of cancer survivor groups after HDIR: ADAM metallopeptidase with thrombospondin type 1 motif 17 (ADAMTS17), dual oxidase maturation factor 1 (DUOXA1), hyaluronan synthase 13 (HAS3), the uncharacterized locus LOC100505622, and zinc finger protein 2 (ZNF2) (Table 2, Fig. 3b).

Summarized results on differential gene expression after 2 Gray. Differentially expressed genes in irradiated compared to sham-irradiated fibroblasts from donors with a first primary neoplasm only (N1), donors with at least one second primary neoplasm (N2+), and cancer-free controls (N0) 4 h after exposure to 2 Gray (false discovery rate adjusted p-value < 0.05). The data are presented for the crude model, model 1 (considering age at sampling and sex), and model 2 [considering age at sampling, sex, age at and year of diagnosis of the first neoplasm, and tumor type (not applicable for N0 data)]. In total, 14,756 genes were detected in the samples. Shown are A the proportion of up- and downregulated genes stratified by dose, group, and model, B volcano plots for the results of model 1, and C top 10 genes with regard to p-value, stratified by direction of log2 fold-change
Functional analysis of DEGs after exposure to HDIR
Comparing reaction to HDIR between donor groups, analysis for GO term enrichment resulted in several group-specific differences (Additional files 5e and 10b). Comparing the reaction of N0 to N1 and N2+ , the largest GO term clusters were regulation of signal transduction and DNA damage response signal transduction by p53 class mediator after HDIR. Comparing N0 and N1, the largest cluster (8 terms) was signal transduction in response to DNA damage and comparing N0 with N2+ the largest cluster (2 terms) was DNA methylation. The comparison between the cancer groups (N1 and N2+) resulted in two GO terms (transporter complex and cation channel complex).
Fibroblasts of all donor groups had p53 as a predicted upstream regulator in reaction to HDIR (Fig. 4, Additional file 5a) in the main analysis. Moreover, E2F1 was predicted to be an upstream regulator in all adjustment models of N1 and N2+ after HDIR. In the sensitivity analysis using an LFC threshold of |0.25|, p53 was predicted to be an active upstream regulator with a z-score > 2 for all models of N0 and N1, but not N2+ (Additional file 13). Additionally, MYB Proto-Oncogene Like 2 (MYBL2), Lin-9 homolog (LIN9), Chromobox homolog 7 (CBX7), and H2.0 Like Homeobox (HLX) were inactivated, Bromodomain Containing 7 (BRD7) was an activated upstream regulator for all donor groups and all models after HDIR, based on the IPA prediction. Only in all models of N2+ Jun Proto-Oncogene (JUN) was an active upstream regulator.
In the pathway analysis of the main analysis, p53 Signaling was found to be affected after HDIR. Contrary to LDIR, it was affected in all groups and models. Moreover, it was activated in all groups and models except N2+ (crude model) after HDIR. The Mitotic Role of Polo-Like Kinase pathway was inactivated in all donor groups, but not affected in any model for N0. Here, STE20-like serine/threonine-protein kinase (SLK) and Checkpoint kinase 2 (CHEK2) were only downregulated in all models of N1 and N2+ (Additional file 7a). In the sensitivity analysis, p53 Signaling was activated in both models for N0 and affected in all models of N1 and N2+ (Additional file 13). Here, the Mitotic Role of Polo-Like Kinase pathway was inactivated in all models of all groups. Moreover, only in the sensitivity analysis, Cyclins and Cell Cycle Regulation, Kinetochore Metaphase Signaling Pathway, and Estrogen-mediated S-phase entry were inactivated in all models of all donor groups after HDIR. In the main analysis, there were five pathways where z-score calculation was not possible by IPA that were affected in at least one group. The Molecular Mechanisms of Cancer pathway was affected in all donor groups and models. Chronic Myeloid Leukemia Signaling was affected in N2+ (crude model and model 1), Hereditary Breast Cancer Signaling in all models of N0 and N2+ , Prostate Cancer Signaling in all models for N1, and DNA Double-Strand Break Repair by Homologous Recombination in N2+ (crude model and model 1) after HDIR.
In the analysis for downstream diseases and bio functions (Fig. 4, Additional file 5c), senescence (senescence of cells and senescence of fibroblast cell lines) showed a trend toward activation, interphase [(N0: interphase and interphase of fibroblasts (both, crude model and model 1), N2+ : interphase (all models) and interphase of fibroblasts (crude model)] toward inactivation after HDIR. In N0, transformation (cell transformation and transformation of fibroblast cell lines) showed a trend toward inactivation. Synthesis of DNA was found in N2+ (all models) with a trend toward inactivation in N0 and N2+ after HDIR. Repair of DNA was found to be a downstream function in N0 and N1, though not significant.
Following HDIR, associated functions for the network in N0 across all adjustments were Cellular Development, Cellular Growth and Proliferation, and Connective Tissue Development and Function (Fig. 4, Additional files 5d and 9). In N1, networks for both adjustment models showed associated functions to be Cell Cycle, Cell Death and Survival and Cellular Development after HDIR. In N2+, we observed two networks for model 1 and model 2. For both adjustment models, associated functions for the first respective network were Cellular Development, Embryonic Development, and Organismal Development in N2+. The second network in N2+ differed and was Cell Cycle, Cell Death and Survival, and Cellular Development for model 1 and Cell Cycle, Cellular Development, and DNA Replication, Recombination, and Repair for model 2.
Discussion
In the present nested case–control study we examined and compared the gene expression and associated pathways in primary skin fibroblasts of childhood cancer survivors without or with a subsequent second primary neoplasm and of cancer-free controls after exposure to different doses of IR. Our results revealed alterations in the response to a low and a high dose of IR in fibroblasts of former childhood cancer patients compared to cancer-free controls. The IPA-analysis of upstream regulators, molecular networks, and predicted downstream-effects supported these findings and indicate impaired protection and higher vulnerability to the genotoxic impact of IR in former childhood cancer patients. In particular, compromised DNA repair and cell cycle-regulation may foster the development of therapy-associated second primary neoplasm already after exposure to low doses of radiation.
Exposure to LDIR
P53 Signaling is the most prominent pathway activated by genotoxic stress, e.g., shown after irradiation of whole blood with a variety of doses and dose rates (Ghandhi et al. 2015). Furthermore, the predicted downstream activities are in line with previous findings on the response to LDIR (Ding et al. 2005). In our data, activity prediction in N0 and N1 was similar: cell cycle progression, apoptosis, and DNA repair were increased with cell survival being inactivated. This pathway not being affected in any model for N2+ and being only weakly affected in N1 is an important finding for the personalized risk assessment of the application of LDIR in diagnostics.
Results from pathway analysis were further supported by the corresponding molecular networks and their attributed functions and diseases, where N0 and N1 had DNA Repair as associated function, while only N2+ included Cancer. After LDIR, a key molecule among others (PGR, PCNA, TP53INP1 IL15, HSPA8, SOD1, CHUK, IRF1, CDC6, CCNE2) in the network of N0 was DDB2, with important functions for (excision-) repair and cell fate decision (Sugasawa et al. 2019; Stoyanova et al. 2009). Additionally, the downstream functions Excision Repair and Repair of DNA were only found for N0. This implies, that if an insufficient or no apoptotic response occurs, induced genomic mutations may be transmitted to following generations carrying a potential carcinogenic risk by inducing resistance and increased DNA damage tolerance. Multiple exposures to LDIR could then amplify this effect (Martin et al. 2010).
We additionally hypothesize that the high difference in number of DEGs following LDIR between N0 and N1/N2+ may implicate an undirected reaction in N1 and N2+, including expression of genes without necessity for radiation-response, while N0 react in a more precise manner. Interestingly, after stratifying by sex, this was found only in male N1 and N2+, whereas the number of DEGs in female N1 and N2+ was similar to those in male and female N0 after LDIR. Sex-specific differences in radiation-response have been proposed before (Alsbeih et al. 2016). Nevertheless, further research is needed to elaborate on such differences in fibroblasts of childhood cancer survivors after LDIR.
Exposure to HDIR
After HDIR, our most important finding was that p53 Signaling was affected and activated in all models of N1 and N0 but not activated in the crude model or N2+. When we applied a 20% LFC-threshold, only N0 showed activation of this pathway. The inactivation of the Mitotic Roles of Polo-Like Kinase pathway after HDIR in N1 and N2+ was in line with a radiation-induced G1 arrest as Polo-like kinases are, amongst others, responsible for G1/S-transition (Zitouni et al. 2014). A notable difference to N0 was the upregulation of Heat shock protein 90-α (Hsp90α) after HDIR in N2+ and N1 donors that has been previously described in various pathological conditions (Zuehlke et al. 2015) and stabilization of the Polo-like kinase 1. Future efforts are needed to investigate this signal in detail in order to validate our findings. Molecular Mechanisms of Cancer was affected in all donor groups after HDIR and cell survival was inactivated via the radiation-triggered activation of the nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-kB), a well-documented mechanism (Pordanjani et al. 2016). In N1 and N0, cell cycle progression was inactivated and apoptosis was activated after HDIR. Remarkably, we observed an inverse response to HDIR in N2+ that was mediated by the up-regulation of RAC-alpha serine/threonine-protein kinase 1 (AKT1). The different isoforms of the proto-oncogene AKT promote radiation resistance by fostering DNA repair, survival, and proliferation (Sahlberg et al. 2014; Hollander et al. 2011). Here, activation of cell cycle progression and inactivation of apoptosis may lead to accumulation of DNA-damage that is subsequently carried on in further cellular generations.
Comparison of the reactions to LDIR and HDIR
In reaction to LDIR, a limited subset of genes has been found to be consistently upregulated by other studies, like CDKN1A (Sokolov et al. 2015), which is activated in a p53-dependent manner and plays a pivotal role in cell cycle arrest (Sokolov et al. 2015; Maier et al. 2016). Kabacik et al. (2011) proposed a set of genes to be positively associated with the radiation response after 2, 4, and 24 h, following HDIR (2 and 4 Gy) in cultured lymphocytes and peripheral whole blood, among them PCNA, Sestrin 1 (SESN1), Growth arrest and DNA-damage-inducible protein GADD45 alpha (GADD45A), CDKN1A, Cyclin G1 (CCNG1), Ferredoxin Reductase (FDXR), BCL2 Binding Component 3 (BBC3), and MDM2. The results for the upregulation of these genes after LDIR or HDIR were confirmed in the present work for all donor groups. Comparing the prediction of pathway-specific downstream activity of p53 Signaling, results were similar between LDIR and HDIR: After both treatments, activation of apoptosis and downregulation of cell survival was inferred. However, Cell Cycle Progression was activated after LDIR and inactivated after HDIR, which may be rooted in the cellular effort to rather repair than induce apoptosis after the lower genotoxic insult that LDIR entails compared to HDIR.
Interactions between radiation dose and donor groups
CAMTA2, the only gene with a borderline significant p-value when comparing group-specific differential expression in reaction to LDIR (comparing N2+ and N0, crude model) has been shown to be involved in cancer (Luan et al. 2019), stress reaction across species (Mollet et al. 2016), and cardiac hypertrophy (Song et al. 2006). The genes differentially expressed after HDIR (N2+ /N1 vs. N0 and N1 vs. N0) were associated to regulation of apoptosis [TNFRSF10A, (Pan et al. 1997)], survival in chronic lymphocytic leukemia [COBLL1, (Plešingerová et al. 2018; Plesingerova et al. 2017)], prevention of accumulation of reactive oxygen species [SESN2, (Wang, et al. 2019)], cell growth/proliferation, heat shock response, and tumorigenesis [EEF1A1 (also known as METTL13), which is methylated by EEF1AKNMT (Vera et al. 2014; Liu et al. 2019)].
Strengths and limitations
To the best of our knowledge, this is the first study that examined the genome-wide transcriptome response to IR in primary fibroblasts of a large group of former childhood cancer patients compared to cancer-free controls. Moreover, the participants were carefully matched for sex, age at sampling, as well as age at and type of first cancer diagnosis to account for the largest sources of variation. In addition, cases were matched by year of diagnosis to account for differences in treatment. Following our previous work, we used fibroblasts from skin biopsies, that were collected from a large, well-defined pool of cell donors [n = 156, (Marron et al. 2021)]. Unlike other studies, we chose to not use commercial cell lines (Velegzhaninov et al. 2015; Ding et al. 2005; Mezentsev et al. 2011; Hou et al. 2015; Ghandhi et al. 2008; Jen et al. 2005). Although commercial cell lines may be well researched, they do not adequately depict intrinsic individual biological variability. Therein also lies a limitation, as this unknown variation might have influenced our results. Our study controlled for this with the aforementioned careful matching of subjects, accounting for variation in the R software package limma, and adjustment of differential expression for factors that introduce most variation, such as age and sex. Despite other cell types being more accessible, such as lymphocytes via venipuncture, we chose fibroblasts to perform our radiation experiments on, because survival and cultivation of lymphocytes without immortalization by Epstein-Barr virus transformation is very limited (Neitzel 1986). Additionally, untransformed fibroblasts keep an intact cell cycle that may provide a more complete analysis of gene expression, even in confluency. Moreover, as some of our participating long-term childhood cancer survivors have received bone marrow transplants, blood samples would have contained foreign blood cells of the bone marrow donors (Victor et al. 2013b). This would have precluded analysis of germline mutations, which was our main aim. Finally, pathways were analyzed using IPA. This steadily updated and curated database allows the analysis of complex RNA-sequencing data. It gives insight beyond expression of single genomic features by incorporating measurements and inferring further information from the transcriptional patterns provided. Moreover, we conducted sensitivity analyses, such as the exclusion of self-reported non-Caucasian participants, which did not change the results of this study after LDIR or HDIR. This came unsurprisingly, as to date there is no data that reports on disparities in radiation-response based on ethnicity. Despite the strengths mentioned, important issues for radiation carcinogenesis such as systemic or non-targeted radiation effects like the bystander effect or genomic instability (Mavragani et al. 2016) were not addressed in our work. We aimed to provide initial results for future studies by using primary cells and performing radiation experiments in vitro. It was essential to examine basic cellular events before accounting for highly complex intercellular or systemic effects. As this is an important factor for radiation-related adverse effects and carcinogenesis, we aim to address these important issues in future works for tissues, cellular networks, and different cell types, with particular regard for an involvement of the immune response/system (Mavragani et al. 2016; El-Saghire et al. 2013). As the design of the study allowed only the inclusion of long-term cancer survivors, aggressive tumor entities with high mortality rates could not be taken into account. However, most cancers share constitutive characteristics, such as autonomous and unlimited proliferation, evasion of cell death, or invasion of normal tissues (Spector et al. 2015). This work investigated the impact of two different radiation doses at a single time point post-exposure. A low and a high dose were chosen to simulate clinically relevant exposure during radiologic examinations or radiation therapy, respectively (Pearce et al. 2012; Seidlitz et al. 2017). Even though gene expression patterns after LDIR show a dynamic transient response within 24 h post-radiation (Albrecht et al. 2012; Berglund et al. 2008), we (Brackmann et al. 2020) and others (Yunis et al. 2012; Chaudhry et al. 2012) observed the highest amount of DEGSs at 3–4 h after irradiation compared to sham-irradiated cells and we hence chose this time point of analysis for our investigations. Although dose rate effects at the transcriptome level are known for radiation exposures, such studies usually compare acute and very protracted irradiations. The latter is usually performed at dose rates of a few mGy per minute for several hours until the desired cumulative dose is reached. In the present study, however, both exposure scenarios with very short irradiation times represent acute exposures. Thus, the difference in absolute dose is expected to dominate the impact on the transcriptome, but the influence of varying dose rates between HDIR and LDIR cannot be ruled out. In irradiation experiments with human lymphocytes, different dose rates (1.03 Gy/min vs. 3.1 Gy/min) had no effect on activation of p53 signaling at the same cumulative doses (Ghandhi et al. 2015).
Conclusion
In this study, we report alterations in the radiation-response in primary fibroblasts of former childhood cancer patients compared to cancer-free donors that were most pronounced in reaction to a low dose of IR in long-term survivors which developed a subsequent second primary neoplasm. Mechanisms directed against genotoxic stress were activated to the same extent after a high dose in all donor groups. However, the radiation response was impaired after a low dose in fibroblast of childhood cancer survivors. This suggests an increased risk for adverse effects by accumulation of DNA-damage following genotoxic insult that may subsequently lead to carcinogenesis, particularly in fibroblasts of donors with at least one second primary neoplasm. This work provides the foundation for further projects and facilitates guided validatory works on the proposed differences in molecular mechanisms coping with IR.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional information files.
Abbreviations
- ADAMTS17:
-
ADAM metallopeptidase with thrombospondin type 1 motif 17
- ADNP-AS1:
-
ADNP antisense ribonucleic acid 1
- AKT1:
-
RAC-alpha serine/threonine-protein kinase 1
- ALG9:
-
ALG9 alpha-1,2-mannosyltransferase
- BBC3:
-
BCL2 binding component 3
- BCL2:
-
B-cell lymphoma 2
- BIN3:
-
Bridging integrator 3
- BRCA2:
-
Breast cancer gene 2, deoxyribonucleic acid repair associated
- BRD7:
-
Bromodomain containing 7
- BTG2:
-
BTG anti-proliferation factor 2
- CAMTA2:
-
Calmodulin binding transcription activator 2
- CBX7:
-
Chromobox homolog 7
- CCNE2:
-
Cyclin E2
- CCNG1:
-
Cyclin G1
- CDC6:
-
Cell division cycle 6
- CDKN1A:
-
Cyclin Dependent Kinase Inhibitor 1A
- CLUHP3:
-
Clustered mitochondria homolog pseudogene 3
- CHEK2:
-
Checkpoint kinase 2
- CHUK:
-
Inhibitor of nuclear factor kappa-B kinase subunit alpha
- COBLL1:
-
Cordon-bleu WH2 repeat protein-like 1
- DAB2:
-
Disabled homolog 2
- DDB2:
-
Deoxyribonucleic acid damage-binding protein 2
- DEG:
-
Differentially expressed genes
- DLEU2:
-
Deleted in lymphocytic leukemia 2
- DNA:
-
Deoxyribonucleic acid
- DUOXA1:
-
Dual oxidase maturation factor 1
- E2F1:
-
E2F transcription factor 1
- EEF1AKNMT:
-
Eukaryotic translation elongation factor 1 alpha lysine and N-terminal methyltransferase
- FDR:
-
False discovery rate
- FDXR:
-
Ferredoxin reductase
- GADD45A:
-
Growth arrest and deoxyribonucleic acid-damage-inducible protein GADD45 alpha
- GO:
-
Gene ontology
- Gy:
-
Gray
- HAS3:
-
Hyaluronan synthase 3
- HDIR:
-
High dose of ionizing radiation
- HLX:
-
H2.0 like homeobox
- HMGCR:
-
3-Hydroxy-3-methyl-glutaryl-coenzyme A reductase
- HSPA8:
-
Heat shock protein family A (Hsp70) member 8
- Hsp90α:
-
Heat shock protein 90-α
- IL15:
-
Interleukin 15
- IPA:
-
Ingenuity pathway analysis
- IQR:
-
Interquartile range
- IR:
-
Ionizing radiation
- IRF1:
-
Interferon regulatory factor 1
- JUN:
-
Jun proto-oncogene, AP-1 transcription factor subunit
- LDIR:
-
Low dose of ionizing radiation
- LFC:
-
Log2 fold-change
- LIN9:
-
Lin-9 homolog
- LINC00601:
-
Long intergenic non-protein coding ribonucleic acid 601
- LINC02257:
-
Long intergenic non-protein coding ribonucleic acid 2257
- MDM2:
-
Mouse double minute 2
- mRNA:
-
Messenger ribonucleic acid
- MSH6:
-
MutS homolog 6
- MYBL2:
-
MYB proto-oncogene like 2
- MYO7B:
-
Myosin VIIB
- N0:
-
Fibroblasts of cancer-free controls
- N1:
-
Fibroblasts of donors with a first primary neoplasm in childhood
- N2+:
-
Fibroblasts of donors with a first primary neoplasm in childhood and at least one second primary neoplasm
- NaN:
-
Not a number
- ncRNA:
-
Non-coding ribonucleic acid
- NF-kB:
-
Nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells
- NUP153:
-
Nucleoporin 153
- p53:
-
Tumor protein 53
- PATZ1:
-
POZ/BTB and AT hook containing zinc finger 1
- PCNA:
-
Proliferating cell nuclear antigen
- PGR:
-
Progesterone receptor
- PIDD1:
-
P53-induced death domain protein 1
- POLH:
-
Deoxyribonucleic acid polymerase eta
- PPM1D:
-
Protein phosphatase, Mg2 + /MN2+ dependent 1D
- PPRC1:
-
Peroxisome proliferator-activated receptor gamma coactivator-related protein 1
- qPCR:
-
Quantitative real-time polymerase chain reaction
- RAB41:
-
Member RAS oncogene family
- RAD9A:
-
Cell cycle checkpoint control protein RAD9A
- RIPK1:
-
Receptor interacting serine/threonine kinase 1
- RNA:
-
Ribonucleic acid
- RNA-Seq:
-
Messenger ribonucleic acid-sequencing
- SESN1:
-
Sestrin 1
- SESN2:
-
Sestrin 2
- SLK:
-
STE20-like serine/threonine-protein kinase
- SOD1:
-
Superoxide dismutase 1
- SPDYE3:
-
Speedy/RINGO cell cycle regulator family member E3
- SRF:
-
Serum response factor
- TBC1D10A:
-
TBC1 domain family member 10A
- TFCP2:
-
Transcription factor CP2
- TIAF1:
-
Transforming growth factor beta-induced anti-apoptotic factor 1
- TNFRSF10A:
-
Tumor necrosis factor receptor superfamily member 10a
- TP53INP1:
-
Tumor protein P53 inducible nuclear protein 1
- ZNF2:
-
Zinc finger protein 2
- ZNF226:
-
Zinc finger protein 226
References
Albrecht H, et al. Transcriptional response of ex vivo human skin to ionizing radiation: comparison between low- and high-dose effects. Radiat Res. 2012;177(1):69–83.
Alsbeih G, et al. Gender bias in individual radiosensitivity and the association with genetic polymorphic variations. Radiother Oncol. 2016;119(2):236–43.
Benjamini Y, et al. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B (methodol). 1995;57(1):289–300.
Berglund SR, et al. Transient genome-wide transcriptional response to low-dose ionizing radiation in vivo in humans. Int J Radiat Oncol Biol Phys. 2008;70(1):229–34.
Berrington de Gonzalez A, et al. Relationship between paediatric CT scans and subsequent risk of leukaemia and brain tumours: assessment of the impact of underlying conditions. Br J Cancer. 2016;114(4):388–94.
Bolger AM, et al. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Brackmann LK, et al. Comparison of time and dose dependent gene expression and affected pathways in primary human fibroblasts after exposure to ionizing radiation. Mol Med. 2020;26(1):85.
Cardarelli JJ, et al. It is time to move beyond the linear no-threshold theory for low-dose radiation protection. Dose-Response. 2018;16(3):1559325818779651.
Chaudhry MA, et al. Micro RNA responses to chronic or acute exposures to low dose ionizing radiation. Mol Biol Rep. 2012;39(7):7549–58.
Claudia Hachenberger AT-S, Löbke-Reinl A, Peter J. Umweltradioaktivität und Strahlenbelastung—Jahresbericht 2016. Bonn: Bundesministerium für Umwelt, Naturschutz, Bau und Reaktorsicherheit (BMUB); 2018 November 2018.
Diallo I, et al. Frequency distribution of second solid cancer locations in relation to the irradiated volume among 115 patients treated for childhood cancer. Int J Radiat Oncol Biol Phys. 2009;74(3):876–83.
Ding LH, et al. Gene expression profiles of normal human fibroblasts after exposure to ionizing radiation: a comparative study of low and high doses. Radiat Res. 2005;164(1):17–26.
Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Dracham CB, et al. Radiation induced secondary malignancies: a review article. Radiat Oncol J. 2018;36(2):85–94.
El-Saghire H, et al. Gene set enrichment analysis highlights different gene expression profiles in whole blood samples X-irradiated with low and high doses. Int J Radiat Biol. 2013;89(8):628–38.
Ghandhi SA, et al. Global gene expression analyses of bystander and alpha particle irradiated normal human lung fibroblasts: synchronous and differential responses. BMC Med Genomics. 2008;1:63.
Ghandhi SA, et al. Radiation dose-rate effects on gene expression for human biodosimetry. BMC Med Genomics. 2015b;8(1):22.
Graupner A, et al. Genotoxic effects of high dose rate X-ray and low dose rate gamma radiation in Apc(Min/+) mice. Environ Mol Mutagen. 2017;58(8):560–9.
Helm JS, et al. Adverse outcome pathways for ionizing radiation and breast cancer involve direct and indirect DNA damage, oxidative stress, inflammation, genomic instability, and interaction with hormonal regulation of the breast. Arch Toxicol. 2020;94(5):1511–49.
Hodgson D, et al. Breast cancer after childhood, adolescent, and young adult cancer: it’s not just about chest radiation. Am Soc Clin Oncol Educ Book. 2017;37:736–45.
Hollander MC, et al. Akt1 deletion prevents lung tumorigenesis by mutant K-ras. Oncogene. 2011;30(15):1812–21.
Hou J, et al. Gene profiling characteristics of radioadaptive response in AG01522 normal human fibroblasts. PLoS ONE. 2015;10(4): e0123316.
Inskip PD, et al. Radiation-related new primary solid cancers in the childhood cancer survivor study: comparative radiation dose response and modification of treatment effects. Int J Radiat Oncol Biol Phys. 2016;94(4):800–7.
International Agency for Research on C. International Childhood Cancer Day: Much remains to be done to fight childhood cancer. 2016.
Jen KY, et al. Identification of novel p53 target genes in ionizing radiation response. Cancer Res. 2005;65(17):7666–73.
Ji K, et al. Research progress on the biological effects of low-dose radiation in China. Dose Response. 2019;17(1):1559325819833488.
Kaatsch P, Grabow D, Spix C. German Childhood Cancer Registry - Annual Report 2017 (1980-2016). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz, 2018.
Kabacik S, et al. Gene expression following ionising radiation: identification of biomarkers for dose estimation and prediction of individual response. Int J Radiat Biol. 2011;87(2):115–29.
Kamburov A, et al. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2012;41(D1):D793–800.
Krämer A, et al. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30(4):523–30.
Kukurba KR, et al. RNA Sequencing and Analysis. Cold Spring Harb Protoc. 2015;2015(11):951–69.
Kutanzi KR, et al. Pediatric exposures to ionizing radiation: carcinogenic considerations. Int J Environ Res Public Health. 2016;13(11):1057.
Law CW, et al. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29.
Liao Y, et al. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
Linet MS, et al. Cancer risks associated with external radiation from diagnostic imaging procedures. CA Cancer J Clin. 2012;62(2):75–100.
Little MP, et al. Leukaemia and myeloid malignancy among people exposed to low doses (<100 mSv) of ionising radiation during childhood: a pooled analysis of nine historical cohort studies. Lancet Haematol. 2018;5(8):e346–58.
Liu S, et al. METTL13 methylation of eEF1A increases translational output to promote tumorigenesis. Cell. 2019;176(3):491-504.e21.
Luan XF, et al. The miR-28–5p-CAMTA2 axis regulates colon cancer progression via Wnt/β-catenin signaling. J Cell Biochem. 2019;122:945.
Luckey TD. Physiological benefits from low levels of ionizing radiation. Health Phys. 1982;43(6):771–89.
Ma S, et al. Biological effects of low-dose radiation from computed tomography scanning. Int J Radiat Biol. 2013;89(5):326–33.
Maier P, et al. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci. 2016;17(1):102.
Marron M, et al. Identification of genetic predispositions related to ionizing radiation in primary human skin fibroblasts from survivors of childhood and second primary cancer as well as cancer-free controls: protocol for the nested case-control study KiKme. JMIR Res Protoc. 2021;10(11): e32395.
Martin LM, et al. DNA mismatch repair and the DNA damage response to ionizing radiation: making sense of apparently conflicting data. Cancer Treat Rev. 2010;36(7):518–27.
Mavragani IV, et al. Key mechanisms involved in ionizing radiation-induced systemic effects. A current review. Toxicol Res (camb). 2016;5(1):12–33.
Meadows AT, et al. Bone sarcoma as a second malignant neoplasm in children: influence of radiation and genetic predisposition for the Late Effects Study Group. Cancer. 1980;46(12):2603–6.
Mezentsev A, et al. Global gene expression responses to low- or high-dose radiation in a human three-dimensional tissue model. Radiat Res. 2011;175(6):677–88.
Mollet IG, et al. Integrator of stress responses calmodulin binding transcription activator 1 (Camta1) regulates miR-212/miR-132 expression and insulin secretion. J Biol Chem. 2016;291(35):18440–52.
Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet. 1986;73(4):320–6.
Nikitaki Z, et al. Systemic mechanisms and effects of ionizing radiation: a new ‘old’ paradigm of how the bystanders and distant can become the players. Semin Cancer Biol. 2016;37–38:77–95.
Pan G, et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276(5309):111–3.
Pearce MS, et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. The Lancet. 2012;380(9840):499–505.
Plesingerova H, et al. COBLL1, LPL and ZAP70 expression defines prognostic subgroups of chronic lymphocytic leukemia patients with high accuracy and correlates with IGHV mutational status. Leuk Lymphoma. 2017;58(1):70–9.
Plešingerová H, et al. Expression of COBLL1 encoding novel ROR1 binding partner is robust predictor of survival in chronic lymphocytic leukemia. Haematologica. 2018;103(2):313–24.
Pordanjani SM, et al. The role of NF-kB inhibitors in cell response to radiation. Curr Med Chem. 2016;23(34):3951–63.
Ray M, et al. Comparison of low and high dose ionising radiation using topological analysis of gene coexpression networks. BMC Genomics. 2012;13:190.
Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7): e47.
Ron E, et al. Tumors of the brain and nervous system after radiotherapy in childhood. New Engl J Med. 1988;319(16):1033–9.
Ruijter JM, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37(6): e45.
Sahlberg SH, et al. The influence of AKT isoforms on radiation sensitivity and DNA repair in colon cancer cell lines. Tumor Biol. 2014;35(4):3525–34.
Saletta F, et al. Genetic causes of cancer predisposition in children and adolescents. Transl Pediatrics. 2015;4(2):67–75.
Scholz-Kreisel P, et al. Second malignancies following childhood cancer treatment in Germany from 1980 to 2014. Dtsch Arztebl International. 2018;115(23):385–92.
Seidlitz A, et al. Practice points for radiation oncology. In: Kerr DJ, Haller DG, van de Velde CJH, Baumann M, editors., et al., Oxford Textbook of Oncology. 3rd ed. Oxford University Press; 2017.
Smith-Bindman R, et al. Rising use of diagnostic medical imaging in a large integrated health system. Health Affairs (project Hope). 2008;27(6):1491–502.
Sokolov M, et al. Global gene expression alterations as a crucial constituent of human cell response to low doses of ionizing radiation exposure. Int J Mol Sci. 2015;17(1):55.
Song K, et al. The transcriptional coactivator CAMTA2 stimulates cardiac growth by opposing class II histone deacetylases. Cell. 2006;125(3):453–66.
Spector LG, et al. Genetic and Nongenetic Risk Factors for Childhood Cancer. Pediatr Clin North Am. 2015;62(1):11–25.
Spycher BD, et al. Background ionizing radiation and the risk of childhood cancer: a census-based nationwide cohort study. Environ Health Perspect. 2015;123(6):622–8.
Stecca C, et al. Adaptive response to DNA-damaging agents: a review of potential mechanisms. Biochem Pharmacol. 1998;55(7):941–51.
Stoyanova T, et al. DDB2 decides cell fate following DNA damage. Proc Natl Acad Sci. 2009;106(26):10690–5.
Sugasawa K. Chapter four—mechanism and regulation of DNA damage recognition in mammalian nucleotide excision repair. In: Zhao L, Kaguni LS, editors. The Enzymes, vol. 45. Academic Press; 2019. p. 99–138.
Supek F, et al. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6(7): e21800.
Sutou S. Low-dose radiation from A-bombs elongated lifespan and reduced cancer mortality relative to un-irradiated individuals. Genes Environ. 2018;40:26.
Thun MJ, et al. Cancer Epidemiology and Prevention. 2017.
Tilton SC, et al. Identification of differential gene expression patterns after acute exposure to high and low doses of low-LET ionizing radiation in a reconstituted human skin tissue. Radiat Res. 2016;186(5):531–8.
Travis LB, et al. Second malignant neoplasms and cardiovascular disease following radiotherapy. JNCI. 2012;104(5):357–70.
Tucker MA, et al. Therapeutic radiation at a young age is linked to secondary thyroid cancer. The Late Effects Study Group. Cancer Res. 1991;51(11):2885–8.
Tukenova M, et al. Radiation therapy and late mortality from second sarcoma, carcinoma, and hematological malignancies after a solid cancer in childhood. Int J Radiat Oncol Biol Phys. 2011;80(2):339–46.
Velegzhaninov IO, et al. Differential molecular stress responses to low compared to high doses of ionizing radiation in normal human fibroblasts. Dose Response. 2015;13(1).
Vera M, et al. The translation elongation factor eEF1A1 couples transcription to translation during heat shock response. Elife. 2014;3:e03164.
Victor A, et al. Searching for gene expression differences in primary fibroblasts between patients with one and two neoplasms in childhood. Pediatr Hematol Oncol. 2013a;30(1):33–45.
Victor A, et al. Searching for gene expression differences in primary fibroblasts between patients with one and two neoplasms in childhood. Pediatric Hematol Oncol. 2013b;30:1–13.
Vogelstein B, et al. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–99.
Wahba A, et al. Radiation-induced translational control of gene expression. Translation. 2016;5(1):e1265703.
Wang L-X, et al. Sestrin2: its potential role and regulatory mechanism in host immune response in diseases. Front Immunol. 2019;10(2019).
Warters RL, et al. Differential gene expression in primary human skin keratinocytes and fibroblasts in response to ionizing radiation. Radiat Res. 2009;172(1):82–95.
Weis E, et al. Reduced mRNA and protein expression of the genomic caretaker RAD9A in primary fibroblasts of individuals with childhood and independent second cancer. PLoS ONE. 2011;6(10): e25750.
Ye J, et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13(1):134.
Yunis R, et al. Genomic characterization of a three-dimensional skin model following exposure to ionizing radiation. J Radiat Res. 2012;53(6):860–75.
Zitouni S, et al. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014;15(7):433–52.
Zuehlke AD, et al. Regulation and function of the human HSP90AA1 gene. Gene. 2015;570(1):8–16.
Acknowledgements
The authors gratefully acknowledge the assistance from Peter Kaatsch, Franziska Himmelsbach, Cornelia Becker, Ilona Kerenyi, and Marianne Brömmel from the German Childhood Cancer Registry. For his work as the coordinator of the ISIBELa consortium, we thank Peter Scholz-Kreisel. Moreover, we thank Maria Blettner for her contributions to the conceptualization, design, and conduction of this study. We are very thankful for the local support of all participating dermatologists in Germany, Austria, and Switzerland, and Ursula Disque-Kaiser for the excellent technical laboratory assistance. The authors acknowledge resources and support from the Bioinformatics Core Facility at the University Medical Center Mainz. TH and CK acknowledge the expert technical work of the staff of the Nucleic Acids Core Facility, Faculty of Biology, JGU Mainz. The study would not have been possible without the voluntary collaboration of all participants who participated in the extensive examinations. We further thank Heiko Karle for his help in establishing the method for fibroblast irradiation.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was funded by the Federal Ministry of Education and Research in Germany under contract No. 02NUK016A, 2NUK042A, 2NUK042B, 2NUK042C, and 2NUK042D and conducted among other research projects as part of the research consortia ISIMEP (Intrinsic radiation sensitivity: Identification, mechanisms and epidemiology, principal investigator: Maria Blettner) and ISIBELa (Intrinsic radiation sensitivity: Identification of biological and epidemiological long-term effects, principal investigators: Maria Blettner and Heinz Schmidberger).
Author information
Authors and Affiliations
Contributions
MM conceptualized the research idea on differential gene expression at different time points after exposure to high and low doses of ionizing radiation and designed the experiments. MM developed the KiKme study and its design as principal investigator. LKB and MM implemented the KiKme study. LKB, IS, DG, and MM conducted the participant recruitment, which was organized and planned by LKB, IS, and MM. Former childhood cancer patients were identified, matched and contacted by the German Childhood Cancer Registry. Doctors responsible for sample drawing (lead by PD) were trained and supervised by MM and HS. The method of fibroblast sampling was established by DG and HS. HSZ takes care of the project´s biobank and controls the quality of all biosamples. Cell culture and radiation exposure of primary fibroblasts were established by SZ and DG. IS conducted the work in the laboratory, including the processing of skin biopsies and radiation experiments. LKB and SZ were responsible for the pseudonymization of all biosamples. CK and TH led the RNA-Seq data production team. The analysis pipeline for the project was developed by MM, MH, AP, and HB. Analysis data of biosamples was processed by AP and TH. LKB and WHB were responsible for the data management. CLG, LKB, and AP conducted the statistical analysis. The final analysis of data concerning content and the hypothesis was done by CLG. CLG, LKB, and MM prepared the initial manuscript. AP, DGW, DG, FM, JM, MH, TH, and SZ revised the manuscript critically for important intellectual content, approved the final version, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We certify that all applicable institutional and governmental regulations concerning the ethical use of human volunteers were followed during this research. Approval by the Ethics Committee of the Medical Association of Rhineland-Palatinate was obtained (no. 837.262.12 (8363-F), no. 837.103.04 (4261), and no. 837.440.03 (4102). Study participants will not undergo any procedures unless they give consent for examinations, collection of samples, subsequent analysis and storage of personal data and collected samples. Study subjects can consent to single components of the study while abstaining from others.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
All results of the differential gene expression analyses. AF1a. Complete list of differentially expressed genes. Model 1 considers age at sampling and sex; model 2 considers age at sampling, sex, age at and year of first diagnosis, and tumor type (not used with data from cancer-free controls). Abbreviations: Gy = Gray, N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm, FDR = p-value adjusted for false discovery rate. AF1b. Differential expression computed with the interaction of radiation and donor group. The comparison-column refers to the donor groups that were compared in the analysis for interactions of the effect of radiation dose and donor group to identify genes showing a differential reaction to radiation exposure between donor groups. Abbreviations: Gy = Gray, N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm, FDR = p-value adjusted for false discovery rate.
Additional file 2.
Comparisons of FDR and LFC across donor groups. Comparison of differential expression (DE) and its direction between donor groups.
Additional file 3.
Primer for and results of qRT-PCR. AF3a. Constructed and purchased primer sequences for the qPCR. AF3b. Log10 relative expression measured in quantitative polymerase reverse transcriptase chain reaction (qRT-PCR) for selected genes compared to TATA-Box Binding Protein (TBP), stratified by group (each n = 6, three replicates per individuals) and radiation dose. Significance stars imply the p-value thresholds of two-sided t-tests after adjustment for false discovery rate (* p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001). N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm. AF3c. Log10-relative expression measured in quantitative polymerase reverse transcriptase chain reaction (qRT-PCR) for mouse double minute 2 (MDM2) and Cyclin Dependent Kinase Inhibitor 1A (CDKN1A) compared to TATA-Box Binding Protein (TBP). Data adapted from Brackmann et al. 2020, coloured by group (each n = 2, three replicates per individual) and stratified by radiation dose. Significance stars imply the p-value thresholds of two-sided t-tests (* p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001). N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm.
Additional file 4.
IPA settings.
Additional file 5.
All results of IPA and GO analyses. AF5a. Complete list of predicted upstream regulators. Model 1 considers age at sampling and sex; model 2 considers age at sampling, sex, age at and year of first diagnosis, and tumor type (not used with data from cancer-free controls). Abbreviations: Gy = Gray, N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm, FDR = p-value adjusted for false discovery rate. AF5b. Complete list of all affected pathways. Model 1 considers age at sampling and sex; model 2 considers age at sampling, sex, age at and year of first diagnosis, and tumor type (not used with data from cancer-free controls). Abbreviations: Gy = Gray, N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm, FDR = p-value adjusted for false discovery rate. AF5c. Complete list of all predicted downstream diseases and biofunctions. Model 1 considers age at sampling and sex; model 2 considers age at sampling, sex, age at and year of first diagnosis, and tumor type (not used with data from cancer-free controls). Abbreviations: Gy = Gray, N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm, FDR = p-value adjusted for false discovery rate. rate. AF5d. Complete list of all molecular networks. Model 1 considers age at sampling and sex; model 2 considers age at sampling, sex, age at and year of first diagnosis, and tumor type (not used with data from cancer-free controls). Abbreviations: Gy = Gray, N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm. AF5e. Complete list of Gene Ontology (GO) terms for the top 50 genes (with regard to the p-value) from the gene expression models comparing the reaction to treatment based on phenotype, considering age at sampling and sex (model 1). Abbreviations: Gy = Gray, N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm, FDR = p-value adjusted for false discovery rate. AF5f. List of genes used as background for the Gene Ontology over-representation analysis.
Additional file 6.
All enriched pathways after 2 Gray. Heat map showing all pathways that were significantly enriched in one of the three donor groups (false discovery rate adjusted p-value < 0.05) in the differential gene expression data after exposure to 2 Gray. Model 1 considers age at sampling and sex, model 2 additionally considers age at and year of diagnosis of the first neoplasm, and tumor type. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm, * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001.
Additional file 7.
Heat maps for differentially expressed genes in affected/modulated pathways.
Additional file 8.
Molecule activity prediction using Ingenuity Pathway Analysis.
Additional file 9.
Heat map of genes in relevant molecular networks. Heat map showing all genes and their respective log2 fold-change that were associated with molecular networks with a network score > 10 in any of the three donor groups in the differential gene expression data after exposure to 0.05 or 2 Gray. Model 1 considers age at sampling and sex; model 2 considers age at sampling, sex, age at and year of first diagnosis, and tumor type (not used with data from cancer-free controls). Tiles with black frames show genes that were part of the top networks for that dose/group/model combination. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm.
Additional file 10.
Results of GO over-representation analyses in gene expression models with interaction terms for donor group. AF10a. Heat maps displaying p-value and log2 fold-change of the top 5 genes per radiation dose, the direction of log2 fold-change per group-wise comparison for expression models including group-dose interaction (with regard to p-value). Log2 fold-change values can be found inside the tiles. Only the results for model 1 (considering age at sampling and sex) are displayed. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of donors with a first primary neoplasm, N2+ = fibroblasts of donors with at least one second primary neoplasm. * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001 (adjusted for false discovery rate). AF10b. Tree maps displaying clustered top gene ontology terms for the top 50 genes (with regard to p-value) for expression models including group-dose interaction (with regard to p-value) for each group-wise comparison. Only the results for model 1 (considering age and sex) are displayed. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of donors with a first primary neoplasm, N2+ = fibroblasts of donors with at least one second primary neoplasm.
Additional file 11.
Results of differential gene expression and pathway analyses stratified by sex. AF11a. Bar charts showing the number and proportion of up- and downregulated genes stratified by dose, donor group, and sex. Differentially expressed genes were computed comparing irradiated to sham-irradiated fibroblasts after exposure to 0.05 and 2 Gray (false discovery rate adjusted p-value < 0.05), considering age at sampling. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm; * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001. AF11b. Upper Half: Volcano plots showing differential expression after 0.05 Gray stratified by donor group and sex, considering age at sampling. Lower Half: Heat map showing top five ranking genes with regard to p-value per group considering age at sampling. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm; * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001. N.S. = not significant (p-value > 0.05). AF11c. Upper Half: Volcano plots showing differential expression after 2 Gray stratified by donor group and sex, considering age at sampling. Lower Half: Heat map showing top five ranking genes with regard to p-value per group considering age at sampling. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm; * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001. N.S. = not significant (p-value > 0.05). AF11d. Overview of affected (false discovery rate adjusted p-value < 0.05) and (in-) activated pathways (|z|≥ 2), predicted upstream effectors, downstream biofunctions and diseases, and observed molecular networks after irradiation with a low (0.05 Gray) or a high dose (2 Gray) ordered by p-value, stratified by sex and considering age at sampling. For molecular networks, the network score instead of a p-value and no z-score was calculated by Ingenuity Pathway Analysis. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm; *p-value < 0.05, ** p-value < 0.01, ***p-value < 0.001. AF11e. Heat map showing all pathways from Ingenuity Pathway Analysis that were significantly enriched in one of the three donor groups (false discovery rate adjusted p-value < 0.05) using results from the differential gene expression data stratified by sex and considering age at sampling after exposure to 2 Gray. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm; * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001.
Additional file 12.
Results of differential gene expression and pathway analyses after exclusion of self-reported non-Caucasian donors. AF12a. Bar charts showing the proportion of up- and downregulated genes stratified by dose, and donor group after exclusion of self-reported non-Caucasian participants. Differentially expressed genes in irradiated compared to sham-irradiated fibroblasts from N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm. Model 1 considers age at sampling and sex; model 2 considers age at sampling, sex, as well as age at and year of diagnosis of the first neoplasm, and tumor type. AF12b. Overview of affected (false discovery rate adjusted p-value < 0.05) and (in-) activated pathways (|z|≥ 2), predicted upstream effectors, downstream biofunctions and diseases, and observed molecular networks after irradiation with a low (0.05 Gray) or a high dose (2 Gray) ordered by p-value, after exclusion of self-reported non-Caucasian participants. For molecular networks, the network score instead of a p-value and no z-score was calculated by Ingenuity Pathway Analysis. Model 1 considers age at sampling and sex. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm; * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001. AF12c. Heat map showing all pathways from Ingenuity Pathway Analysis that were significantly enriched in one of the three donor groups (false discovery rate adjusted p-value < 0.05) in the differential gene expression data after exclusion of self-reported non-Caucasian participants after exposure to 2 Gray. Model 1 considers age at sampling and sex. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm; * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001.
Additional file 13.
Results of pathway analysis after exclusion of genes with less than 20% up- or downregulation.Overview of affected (false discovery rate adjusted at p-value < 0.05) and (in-) activated pathways (|z| ≥ 2), predicted upstream effectors, downstream biofunctions and diseases after irradiation with a low (0.05 Gray) or a high dose (2 Gray) ordered by p-value.* p--value < 0.05, ** p--value < 0.01, *** p--value < 0.001. Model 1: considering age at sampling and sex, model 2: considering age at sampling, sex, age at and year of diagnosis of the first neoplasm, and tumor type (not used with data from N0), N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood-cancer survivors, N2+ = fibroblasts of childhood-cancer survivors with at least one second primary neoplasm.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Grandt, C.L., Brackmann, L.K., Poplawski, A. et al. Radiation-response in primary fibroblasts of long-term survivors of childhood cancer with and without second primary neoplasms: the KiKme study. Mol Med 28, 105 (2022). https://doi.org/10.1186/s10020-022-00520-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10020-022-00520-6