
Abstract
Background
Differential expression analysis is usually adjusted for variation. However, most studies that examined the expression variability (EV) have used computations affected by low expression levels and did not examine healthy tissue. This study aims to calculate and characterize an unbiased EV in primary fibroblasts of childhood cancer survivors and cancer-free controls (N0) in response to ionizing radiation.
Methods
Human skin fibroblasts of 52 donors with a first primary neoplasm in childhood (N1), 52 donors with at least one second primary neoplasm (N2 +), as well as 52 N0 were obtained from the KiKme case–control study and exposed to a high (2 Gray) and a low dose (0.05 Gray) of X-rays and sham- irradiation (0 Gray). Genes were then classified as hypo-, non-, or hyper-variable per donor group and radiation treatment, and then examined for over-represented functional signatures.
Results
We found 22 genes with considerable EV differences between donor groups, of which 11 genes were associated with response to ionizing radiation, stress, and DNA repair. The largest number of genes exclusive to one donor group and variability classification combination were all detected in N0: hypo-variable genes after 0 Gray (n = 49), 0.05 Gray (n = 41), and 2 Gray (n = 38), as well as hyper-variable genes after any dose (n = 43). While after 2 Gray positive regulation of cell cycle was hypo-variable in N0, (regulation of) fibroblast proliferation was over-represented in hyper-variable genes of N1 and N2+. In N2+, 30 genes were uniquely classified as hyper-variable after the low dose and were associated with the ERK1/ERK2 cascade. For N1, no exclusive gene sets with functions related to the radiation response were detected in our data.
Conclusion
N2+ showed high degrees of variability in pathways for the cell fate decision after genotoxic insults that may lead to the transfer and multiplication of DNA-damage via proliferation, where apoptosis and removal of the damaged genome would have been appropriate. Such a deficiency could potentially lead to a higher vulnerability towards side effects of exposure to high doses of ionizing radiation, but following low-dose applications employed in diagnostics, as well.
Similar content being viewed by others


Introduction
Application of high-dose ionizing radiation (HDIR) in radiotherapy can cause acute (e.g., inflammation) or late adverse reactions, such as the development of a second primary neoplasm (Tukenova et al. 2011; Spector et al. 2015; Inskip et al. 2016; Scholz-Kreisel et al. 2018), the risk for which is additionally increased if exposure to HDIR occurs at a young age (Hodgson et al. 2017). However, it is still unclear why only a fraction of childhood cancer survivors, regardless of therapy, develops second primary neoplasms later in life (Kutanzi et al. 2016). An explanation may be provided by the high degree of individual biological variability in pathways like radiation-response, which are needed in reaction to stressors such as radiotherapy (Smirnov et al. 2012; Hornhardt et al. 2014; Borràs-Fresneda et al. 2016; Seibold et al. 2019). Individual genetic variations such as single nucleotide polymorphisms and copy number alterations affect the expression variability (EV) (Li et al. 2010), which subsequently modulates the translation into cellular function. Moreover, non-sequence-based genetic factors like epigenetic modifications (e.g., methylation) also impact gene expression magnitude and EV while posing as a link between environment, lifestyle, and genome (A I et al. 2013; Bashkeel et al. 2019). After several decades of differential expression analyses and testing for differences in mean expression, the variability of an expressed gene may additionally impact phenotypes, modulate fitness, be indicative for disease (Ho et al. 2008; Li et al. 2010; Mar et al. 2011; Corrada Bravo et al. 2012; Alemu et al. 2014), and underlie evolutionary selection (Feinberg and Irizarry 2010; Zeller et al. 2010; Bashkeel 2019). To date, only a few studies focused on more elaborate methodologies to estimate the EV (Oleksiak et al. 2002; Storey et al. 2007; Ho et al. 2008; Li et al. 2010; Stranger et al. 2012; Breschi et al. 2016). However, some of these approaches perform poorly when estimating the EV of lowly expressed genes (Alemu et al. 2014; Simonovsky et al. 2019). To address this issue, the study group of Alemu and colleagues was the first to define the term ‘EV’, establishing it as a measurement that separates expression variability from overall expression levels (Alemu et al. 2014) by defining EV as the ratio of varianceobserved to varianceexpected. As their approach was still sensitive to outliers and moderately biased towards lowly expressed genes (Simonovsky et al. 2019), others thus modified it by using the median absolute deviation (MAD) instead of the standard deviation (SD) to add robustness against outliers (Wu et al. 2014; Bashkeel et al. 2019). Bashkeel et al. (2019) additionally filtered for bimodally expressed genes and used bootstrapping to compute the ‘observed MAD’. Similar to Corrada et al. (2012), genes were then classified based on a defined range of the EV-metric and classifications were then cross-validated to reduce sampling error and to provide further robustness. In cancer, the EV bears information concerning oncogenesis (Afsari et al. 2014) and methylation-mediated changes in EV can explain the heterogeneity between tumour subtypes (Hansen et al. 2011; Landau et al. 2014; Ecker et al. 2015), as well as adverse clinical outcomes (Landau et al. 2014; Yard et al. 2016). Corrada et al. (2012) showed that an increased EV of specific genes enabled machine-learning-based distinction between healthy and tumour tissue samples. To this day, no study has examined the EV in healthy tissue from subjects with a history of childhood cancer. However, the information on EV might explain why some long-term survivors of childhood cancer develop further primary neoplasms and some do not. This work, therefore, aims to identify the expression variability in primary skin fibroblasts from long-term survivors of childhood cancer without (N1) and with at least one second primary malignancy (N2+), as well as from cancer-free controls (N0), after exposure to a high (2 Gray, HDIR) or low (0.05 Gray, LDIR) dose of ionizing radiation. To do so, gene expression data, previously examined for differential expression (Grandt et al. 2022) were examined to identify patterns in EV potentially related to the participants’ onset of first and second primary neoplasms, adapting the pipeline proposed by Bashkeel et al. (2019) and furthermore to explore whether there are variability patterns that might be used to differentiate N1 and N2+ through application of machine learning algorithms.
Methods
Study design, participants, and differential gene expression
The KiKme nested case–control study was conducted to identify genetic predispositions associated with paediatric cancer and second primary neoplasms, potentially initialised by exposure to ionizing radiation during radiation therapy for the first cancer in childhood or radiation diagnostics prior to the first childhood cancer. For this purpose, biosamples, as well as data on lifestyle, medical history, and history of radiation exposure were collected. The KiKme study design (Marron et al. 2021), a detailed description of the establishment of the radiation experiments (Brackmann et al. 2020), the differential expression analysis, as well as a description of the study sample used in this work (Grandt et al. 2022), can be found in detail elsewhere. In short, the median age of donors with at least one second primary neoplasm was 32.0 years, and 32.5 years of donors with only a first neoplasm in childhood, respectively (interquartile range 28.0–38.2 years). The median age of cancer-free controls was 33.0 years (interquartile range 27.8–38.0) at the time of sampling. Half (51.9%) of the participants were female. All subjects included in this study were matched by age at recruitment and sex. The long-term survivors of childhood cancer were additionally matched by first cancer site, as well as age at and year of the first cancer diagnosis. Primary skin fibroblasts were sampled from skin biopsies of 156 donors with cancer in childhood without a second primary neoplasm (N1, n = 52), donors with cancer in childhood and at least one second primary neoplasm (N2 + , n = 52), and cancer-free controls (N0, n = 52).
RNA-sequencing and processing
In short, fibroblasts were cultured for ~ 14 days, then irradiated as triplets with fibroblasts of the matched donors with 0, 0.05 (LDIR), or 2 (HDIR) Gray, respectively. RNA was then isolated 4 h after exposure. Samples with an RNA integrity number < 7 were not used for the subsequent library preparation. The libraries were processed on a HiSeq2500 instrument (Illumina, San Diego, California, USA) which was set to high-output mode (Nucleic Acids Core Facility, Faculty of Biology, Univ. Mainz). The reads were then generated using the TruSeq Single Read Cluster Kit v3 and the TruSeq SBS Kit v3 (Illumina, San Diego, California, USA). Here, single-end reads had a length of 51 base pairs using single indices (8). The base calling was performed by Real-Time Analysis (Version 1.8.4) and the resulting data were then converted into FASTQ format using bcl2fastq (Version 1.8.4, Illumina, San Diego, California, USA). The raw reads were separated from the adapter sequences using Trimmomatic (Bolger et al. 2014) and the processed reads were aligned to the human reference genome (GRCh38) using STAR (Dobin et al. 2013). The expression per gene was then computed as the number of aligned reads per gene, quantified using FeatureCounts (Liao et al. 2014). The data were then normalized using the voom method (Law et al. 2014) for the detection of differentially expressed genes (adjusted for age at recruitment and sex) via limma, (Ritchie et al. 2015) (Additional file 1a) and DESeq2 (Love et al. 2014) for the EV pipeline.
Computation of expression variability and classification of genes
We used a modified version of the pipeline by Bashkeel et al. (2019) for computing the EV and the subsequent classification into hypo-, non-, and hyper-variable genes. As with that pipeline, this work also solely examined the EV of genes showing a unimodal distribution across all three donor groups per experiment. Hereby the assumption was that a high EV represented the widened ranges of count values across the median. Genes where the expression showed multimodal patterns, thus having more than one discrete state (e.g., differential expression between the phenotypes) were examined in another work using differential expression analysis (Grandt et al. 2022). Contrary to Bashkeel et al., (2019) who used microarray data and thus computed bimodal genes using Gaussian distribution, we used SIBERG (version 2.0.3) to calculate a bimodal index for RNA-Sequencing data (Wang et al. 2009) with default settings (zeroPercentThr = 0.2, base = exp(1) and eps = 10). To account for the normalization of mRNA data, a vector, containing the respective normalization factors for each sample calculated with DESeq2 (Love et al. 2014), was provided to the SIBERG algorithm. Raw RNA-sequencing counts of bimodal expression distributions were analysed for all donor groups (N2 + , N1, and N0) using a log-normal mixture model (Tong et al. 2013). Moreover, as the same genes may show different expression patterns depending on the radiation dose, data of each experiment (sham irradiation, LDIR, and HDIR) were analysed separately. Thus, genes with a bimodal distribution, identified by a bimodal index > = 1.1, were removed from the data for further analyses (Additional file 1b) (Tong et al. 2013). Assuming that the EV and the participants’ cancer history could be associated, the following computational steps were applied to the data stratified by donor group and radiation dose. First, the MADoberserved was calculated for each gene per donor group and radiation treatment as the median of 1,000 bootstrap iterations. Second, the MADpredicted was estimated by fitting a non-parametric local polynomial regression (loess) function. The EV for each gene was then calculated as the difference MADoberserved—MADpredicted. Genes were classified as hypo-, non, or hyper-variable if their EV was below, within, or above the interval: MedianEV ± 3 \(\cdot\) MADEV. Here, x̃EV is the median EV in the N0 group. The MADEV was again computed as the median of 1000 bootstrap iterations. Assuming that the EV of the N0 group is associated with the non-cancer phenotype, EV in the N2 + and N1 groups were not classified by their internally defined ranges, but by the range defined through expressional data of the N0 group. The resulting classifications were then cross-validated for each treatment by randomly splitting each donor group (N0, N1, N2 +) in half and repeating the complete classification pipeline 10 times. Next, classification of genes was confirmed using a binomial test, where the success was defined as a concordant classification in the first (split 1) and in the second (split 2) half of each donor group, as well as in the whole dataset. Here, the alternative hypothesis was defined as the true probability of success being greater than 0.5. The resulting p-values from the binomial tests were then adjusted for false discovery at a rate of 0.05. The resulting data sets (Additional file 3) were then analysed for overlaps and displayed using upsetR (Conway et al. 2017). Genes whose classification as hypo- or hyper-variable was verified by cross-validation and that were part of unique gene sets after the set-based analysis for overlap with upsetR (e.g., genes that were only classified as hypo-variable in N0 after LDIR) were then subjected to the Gene Ontology (GO) over-representation analysis (see below).
Sensitivity analyses
This workflow was also applied to subsets of the data after stratifying by sex. Moreover, to examine the EV without potential confounding introduced by tobacco smoking or alcohol consumption, we repeated the analysis pipeline after excluding donor triplets where at least one donor (i) had smoked tobacco for over ten pack years (23 of 52 triplets remaining) and/or (ii) consumed more than 2 alcoholic beverages per day (38 of 52 triplets remaining). Due to an otherwise too strongly reduced sample size, we only filtered for the values representing the questionnaire categories for the highest consumption of tobacco smoking and alcohol, respectively. For all sensitivity analyses, we applied an appropriate higher threshold of 1.3 for the bimodal index due to the reduced sample sizes (nFemale: 81, nMale: 75, as well as nhealthy: 60) as recommended by Tong et al. (2013).
Comparison of methods to compute variability estimates
To evaluate the performance of EV as a measurement of gene expression variability, we also calculated the coefficient of variation (CV = SD/mean) and the standardized MAD (d = MAD/median) for each gene per dose and donor group. We then compared the influence of expression magnitude on all three metrics using Kendall’s Tau. This is a measure robust to departure from linear associations, to assess the correlation between the variability metric and the median expression level.
Construction of the candidate gene list
To reduce the high complexity of our data, we also constructed a candidate list, containing genes assumed to be associated with the radiation response. To do so, we collected genes curated in the RadAtlas (Xu et al. 2020), genes known to be involved in DNA repair (Knijnenburg et al. 2018), as well as genes annotated to the GO terms response to stress and response to radiation (The Gene Ontology Consortium 2018), and the top 100 genes with regard to p-value from the differential gene expression analysis in reaction to LDIR and HDIR we have previously conducted (Grandt et al. 2022). We then filtered the genes in our data that were classified as hypo- and hyper-variable in any combination of donor group and radiation dose for genes on the candidate list. Furthermore, we acquired information in the literature on genes whose methylation may be potentially affected by radiation and examined these regarding their respective EV in reaction to the different radiation doses (Antwih et al. 2013; Miousse et al. 2017).
Gene Ontology over-representation analysis
Genes whose classification as hypo- or hyper-variable confirmed by cross-validation were additionally analysed for over-representation using the ConsensusPathDB (Kamburov et al. 2012). According to best practice for pathway/functional analyses (e.g., the over-representation analysis used in this work), a gene set is compared to a given list of total genes measured in an experiment, called background (Wijesooriya et al. 2022). As such, gene lists per radiation dose after exclusion of bimodally expressed genes were used (Additional file 3). Resulting GO terms of the category biological process were filtered for adjusted p-values < 0.05 (Benjamini–Hochberg procedure (Benjamini and Hochberg 1995)). This was done for (i) each combination of radiation dose, EV-classification, and donor group, as well as (ii) unique gene sets identified in the upsetR-analysis. The complete list of GO terms for (i) can be found in Additional file 4. These GO term results for each radiation dose were also examined for overlap between the donor groups per radiation dose and EV classification and filtered for the top 5 GO terms regarding the adjusted p-value. For (ii), respective GO terms were summarized into clusters using REVIGO (Supek et al. 2011) with an allowed semantic similarity of GO terms set to 0.7 and the database set to Homo sapiens. The results were then extracted and plotted as tree maps using the R script provided by the platform. In these, tile sizes of the tree maps were defined to represent the adjusted p-value of each respective GO term (Additional file 6).
Application of classification algorithms
We further used selected gene sets uniquely classified as hyper-variable in reaction to LDIR in N2 + to distinguish between N1 and N2 + , using N1 as the reference class. For this purpose, MLSEQ (Goksuluk et al. 2019) was employed; an R package designed to apply a plethora of classifiers to RNASeq data (Additional file 2d). The models were trained on a randomly assigned 20% training split of the data and their respective accuracy was then examined using the remaining 80% of the data. The models were trained with the “validation method” set to 10 times repeated fivefold cross-validation and the tune length (number of values used for the tuning parameter, if the respective classifier had a tuning parameter) set to 10, according to the readme of the package. In sparse models (e.g., voomNSC) that set parameters of the model to 0 if not relevant for the outcome prediction accuracy, the selectedGenes-function was used to identify potential biomarker genes that contributed most to the discrimination function of the classifier.
Results
Bimodal genes, computation of expression variability, and gene classification
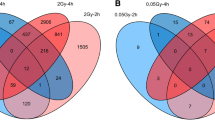
The data contained expression information for 14,756 genes. Of these, 97 genes (0.66%) in the sham irradiation data set, 511 genes (3.46%) in the LDIR data set, and 164 genes (1.11%) in the HDIR data set had a bimodal index higher or equal to 1.1 and were excluded before computation of the EV (Additional file 1a). The comparison of EV, the CV, and the standardized MAD (d) with the median expression showed negative correlation values which were more pronounced for CV and d (tauCV = − 0.622/− 0.619/− 0.623, taud = − 0.596/− 0.591/− 0.598 for 0, 0.05, and 2 Gy, respectively) than for EV (tauEV = − 0.019/− 0.018/− 0.021 for 0, 0.05, and 2 Gy, respectively), whereas the EV was more dispersed in very high expression levels. The distribution of EV-values was similar across donor groups and radiation doses (Fig. 1b). The number of hypo- and hyper-variable genes decreased in all data sets after cross-validation (Fig. 1c). After all radiation doses, most genes were classified as hypo- (sham irradiation: 1059, LDIR: 1049, HDIR: 1037) and hyper- (sham irradiation: 808, LDIR: 795, HDIR:790) variable in N0, while there were about 20% fewer genes classified as hypo- or hyper-variable in N1 and N2 +. Comparing gene classifications, we identified 461 hypo- and 333 hyper-variable genes that had identical classifications in fibroblasts of all donors at any radiation dose (Additional files 2 and 5a). The following 4 intersect groups consisted exclusively of N0-data sets. These were hypo-variable genes after sham irradiation (n = 49 genes), LDIR (n = 41 genes), and HDIR (n = 38 genes). Additionally, 43 genes were only hyper-variable in N0 after any radiation dose. Of these 171 genes in total, 49 (sham irradiation: BAG5, CCND1, DNAJB2, EIF2AK2, FH, GNAQ, LAMB2, OSMR, POLR2C, PPIG, PSMA3, PSMB3, TOLLIP, VDAC3, and WDR48; LDIR: ABCF3, ARID1A, CCM2, CIRBP, GTF2H1, PPP1R10, PRDX5, PSMD4, PSMD5, RPS19, SIGMAR1, SRPK2, and STK25; HDIR: LASP1, CST3, GLB1, MSH6, RAD21, RAD23A, SIN3A, TXNRD1, ULK1, and YBX3; Hyper-variable after all doses: ATF4, CAMK1D, DKK2, FBLN5, IL1R1, KLF2, LOXL3, MSRB3, RCN3, SREBF2, and TRAIL) were also present in our candidate list, which consisted originally out of 4,810 genes.

Comparison of methods for quantification of expression variability and overview of distribution and subsequent variability classification: A Comparison of different metrics that are typically used for the characterization of expressional variability. Scatter plots showing the association of the coefficient of variation (CV, standard deviation/mean), standardized median absolute deviation (d, median absolute deviation (MAD)/median), and expression variability (EV, computed as MADobserved-MADexpected) on the respective y-axes with the median expression on the x-axis. Kendall’s correlation coefficient (τ) for the correlation of each variability metric with the median gene expression is provided per facet. The yellow stripes on the left show the value distribution of each variability metric. The colours of the dots represent the donor groups (N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood cancer survivors without a second primary neoplasm, N2 + = fibroblasts of childhood cancer survivors with at least one second primary neoplasm) Below, the distribution of the median gene expression is presented as box- and density plots. B Density plots showing the distribution of the EV (truncated using the smallest Q01 (N0 after LDIR) EV = − 427.9 to the largest Q99 (N0 after 2 Gray) EV = 1204.8 for reasons of legibility), also stratified by donor group, and radiation dose. Dashed lines imply the ranges employed for the classification into hypo-, non-, and hyper-variable, whereas genes with an EV lower than the left dashed lines were classified as hypo-variable and genes with an EV higher than the right dashed line were conversely classified as hyper-variable. Genes with an EV between the dashed lines were classified as non-variable. C Bar charts showing the number of genes that were classified as hypo-, non-, and hyper-variable, using the whole data set and the number of genes with stable classification confirmed by cross-validation. All data were stratified by exposed radiation dose (0, 0.05, and 2 Gray)
Sensitivity analyses
Bimodal genes and differential gene expression
To examine whether genes were bimodally expressed e.g., because they were differentially expressed between phenotypes, we compared the excluded bimodally expressed genes (Additional file 1a) with their respective data on differential expression status between donor groups (Additional file 1b). After exposure to the sham-irradiation (n = 97) and HDIR (n = 164), half of the identified bimodally expressed genes were also differentially expressed comparing N1 and N2 + with N0 (Additional file 7a). After LDIR, about one-third of the 511 bimodal genes were also differentially expressed genes comparing donor groups (Additional file 7b). Genes showing the highest values for the bimodal index (≥ 2.5) were mostly genes that were not differentially expressed between donor groups (Additional file 7a).
Sex
To examine differences in EV we stratified the data by sex and repeated the analysis pipeline. After sham-irradiation, 281 genes from the female and 35 from the male data set were excluded, compared to 97 genes in the combined data (Additional file 8a). In the LDIR data set, 138 genes from the female and 1553 from the male data were excluded (511 genes in the complete data set). In the HDIR data, 68 genes from the female and 151 genes from the male data had a bimodal index over 1.3 and were excluded (complete data: 164). The values for the EV were comparable between males and females (adjusted r2: 0.779; Kendall’s tau: 0.786; Additional file 8b). The number of genes per classification was comparable between sexes (Additional File 8c), and the overlap of hypo- and hyper-variable genes, stratified by radiation dose, sex, and donor group was persistent along the classification as hyper- (n = 531) and hypo- (n = 400) variable (Additional file 8d).
Exclusion of smokers and participants with heavy alcohol consumption
After filtering out donor triplets containing participants with strong smoking and drinking behaviour, as well as individuals with missing values on these variables, 20 triplets remained for this sensitivity analysis. Thus, after the exclusion of donor triplets with missing values, participants without any heavy smokers or drinkers in their respective triplets summed to a total of 60, and donors from triplets with at least one heavy drinker or smoker added up to 54 persons. The number of bimodally expressed genes was much higher here than in the analysis with all participants (nSI: 1280, nLDIR: 1726, and nHDIR: 527; Additional file 8e). Subsequently, the number of genes classified as hypo-/hyper-variable was lower than in the whole data set (Additional file 8f). Computed EV values from donor triplets termed healthy, meaning not heavy drinker, nor heavy smoker and unhealthy based on smoking and alcohol consumption were different (adjusted r2: 0.377; Kendall’s tau: 0.683, Additional file 8g). A comparison of data from triplets without heavy smoking and alcohol consumption showed the largest overlaps across all hyper- and all hypo-variable sets of genes, respectively (Additional file 8h). We further examined whether the number of bimodally expressed genes was explained by the exclusion of participants termed unhealthy based on smoking and alcohol consumption. Thus, we randomly generated samples with comparable large (n = 96) and small (n = 60) subsamples. These showed the same fluctuation in the number of bimodally expressed genes irrespective of metadata, solely explained by sample size (Additional file 8i).
Candidate genes and methylation status
Based on our literature research, we identified 59 genes that would potentially show radiation-dependent methylation patterns. Of these, 50 genes were present in our data and 35 of these 50 genes were classified as non-variable in all combinations of donor group and radiation doses. The remaining 15 genes are depicted in Additional file 9. Here, YWHAQ, YWHAG, YWHAE, YWHAB, as well as RAD23B were classified as hypo-; CDH13 and IGFBP3 as hyper-variable in all donor group- and radiation dose-combinations. CCND1 had a heterogeneous EV at sham-irradiation that aligned at LDIR and HDIR across all donor groups. CDKN1A was non-variable in all donor groups after sham irradiation and hypo-variable in all donor groups after HDIR. ASPH was hypo-variable in N0 after LDIR and in N0 and N1 after HDIR. The EV of IGF1R was dispersed across donor groups, whereas its expression was hyper-variable after sham-irradiation in N1, after LDIR in N0 and N2 + , and after HDIR in N1 and N2 + . Comparing the classification of genes between different donor groups and radiation doses, we identified 22 genes (ALDOA, ANPEP, CCNG1, CD63, CDKN1A, ENO1, GLUL, IL6ST, IMPDH2, LIMS1, LRP1, MTCH1, MXRA8, MYH10, PLS3, RPLP0, RPS18, RPS27L, SPTBN1, THY1, TMEM119, and TRAM2) that showed a large enough difference in EV to be classified as hypo-variable in any donor group and hyper-variable in at least one of the other donor groups (Fig. 2a). Of these genes, 11 (CCNG1, CD63, CDKN1A, ENO1, GLUL, IL6ST, LRP1, MYH10, PLS3, RPS27L, and THY1) were also present in our candidate list.

Heat maps of genes with functional interest: which were A classified both as hyper- and hypo-variable in any data set and B associated with the GO term response to radiation and -showed a classification range from to hyper-variable across all donor groups. Genes in A were additionally filtered for genes found in the RadAtlas database for radiation-associated genes, genes associated with DNA repair functions, or that were annotated to the Gene Ontology (GO) terms response to stress or response to radiation (see facet Candidate List). N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood cancer survivors without a second primary neoplasm, and N2 + = fibroblasts of childhood cancer survivors with at least one second primary neoplasm
Gene Ontology analysis
A comparison of GO term results showed a term consistency across variability-classifications (Additional file 5). 385 terms were over-represented in hyper-, and 276 GO terms were over-represented for hypo-variable genes of all donor groups across all radiation doses. Additional 76 GO terms were over-represented after all radiation doses for both, hypo- and hyper-variable genes. The GO term cellular response to radiation was associated with hypo-variable genes of all donor groups after all radiation treatments (Additional file 4). Its child-term cellular response to ionizing radiation was associated with the hypo-variable genes of N1 after HDIR and in N2 + data after all radiation doses, including the sham irradiation. Corresponding hypo- and hyper-variable genes are shown in Fig. 2b. Among the top 5 GO terms over-represented in hypo-variable gene sets of all donor groups, protein/macromolecule localization and intracellular transport were present after all radiation doses (Fig. 3).Prominent GO terms that were over-represented for hyper-variable gene sets of all donor groups after all radiation doses were extracellular structure/matrix organization, system development, multicellular organism development, and anatomical structure development (Fig. 4). Next, we analysed the unique hypo-variable genes for N0 (Additional file 3b). After sham irradiation, these genes were clustered as protein modification process (Additional file 6a). In reaction to LDIR, the GO terms for the uniquely hypo-variable genes of N0 were transcription initiation-coupled chromatin remodelling, regulation of hematopoietic progenitor cell differentiation, RNA catabolic process, and negative regulation of hydrolase activity (Additional file 6b). After HDIR, clusters were negative regulation of catalytic activity and regulation of cellular process (Additional file 6c) Besides the unique hypo-variable genes, 43 genes were hyper-variable only in N0 across all radiation doses. These were functionally clustered into lung development, cellular response to laminar fluid shear stress, and neutrophil migration, Following the unique genes for N0, the next largest set of genes was among the hypo-variable genes in N2 + after HDIR (n = 30). The associated GO term was N-glycan processing (Additional file 6e). Additionally, there were 30 genes uniquely classified as the hyper-variable genes for N2 + after LDIR (n = 30). Among this identified set of genes, the associated GO terms were clustered into establishment of protein localization to organelle, translational initiation, ERK1 and ERK2 cascade, among others containing ERK1 and ERK2 cascade (RRAS, ANGPT1, TNFAIP8L3, NRP1), protein kinase B signaling (RRAS, FGF5, ANGPT1, TNFAIP8L3), MAPK cascade (TNFAIP8L3, ROBO1, RRAS, FGF5, ANGPT1, NRP1), Ras protein signal transduction [(ROBO1, RRAS, RALGPS2, NRP1), Fig. 5].We additionally filtered the data for the expression values for these 30 genes of N1 and N2 + post-LDIR and applied several classification algorithms (Additional file 2d). The voomNSC algorithm performed best with an accuracy of 0.68, sensitivity of 65.71% and a specificity of 70.27% using on average 14 of the 30 genes for the classification (ANGPT1, CCDC71L, EIF3F, FGF5, IER3, KCNK15, NRP1, RGMB, ROBO1, RPL11, RPL13A, RPL3, RPS24, SETBP1). GO terms associated with these genes were amongst others subsumed under the terms translational initiation and cellular response to growth factor stimulus [also containing MAPK cascade, (Additional file 6f)].

Summary of the top 5 Gene Ontology terms regarding the adjusted p-value: stratified by group-wise overlap (Top 5 in A) all groups, B overlap of two donor groups, and C unique in one group), per radiation dose for genes classified as hypo-variable. Inside the tiles, each respective adjusted -log10 (p-value) is provided. Since a p-value was computed per group for A and B, the top 5 GO terms e.g., found only in N0 and N1, but not in N2 + post-HDIR might differ. In this case, the top 5 for N0 and N1 are shown each, resulting in more than 5 terms. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm

Summary of the top 5 Gene Ontology terms regarding the adjusted p-value: stratified by group-wise overlap (Top 5 in A all groups, B overlap of two donor groups, and C unique in one group), per radiation dose for genes classified as hyper-variable. Inside the tiles, each respective adjusted − log10 (p-value) is provided. Since a p-value was computed per group for A) and B), the top 5 GO terms e.g., found only in N0 and N1, but not in N2 + post-HDIR might differ. In this case, the top 5 for N0 and N1 are shown each, resulting in more than 5 terms. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm

Tree map summarizing over-represented Gene Ontology terms for genes that were only classified as hyper-variable (n = 30) in fibroblasts of long-term survivors of childhood-cancer with at least one second primary neoplasm in reaction to exposure to 0.05 Gray
Discussion
In this study, we adapted a pipeline for the computation of expression variability of microarray expression data to next-generation RNA-sequencing data to compare the EV in primary fibroblasts of a collective of cancer-free controls and childhood cancer survivors with and without at least one second primary neoplasm after treatment with different doses of ionizing radiation. This method was less affected by low expression values than metrics usually employed to describe expression variability. Regardless of the radiation dose, we found the highest total and unique number of hypo- and hyper-variable genes in fibroblasts of cancer-free controls. Genes only classified as hypo-variable genes in this donor group were associated with regulatory processes and the stress response. Genes that were only hyper-variable in fibroblasts of childhood cancer survivors with at least one second primary neoplasm after the low dose were associated with the cell fate decision. In long-term survivors of childhood cancer without any second primary neoplasms, no prominent pathways were predicted to be uniquely affected by genes with hypo- or hyper-variable expression.
Computation of bimodally expressed genes and EV
Bimodally expressed genes were either differentially expressed genes between donor groups or genes that showed a bimodal distribution across all donor groups. Interestingly, after LDIR, the total number of bimodal genes was far higher and the proportion of differentially expressed genes was lower than after the sham irradiation and HDIR. The observed substantial number of bimodal genes after LDIR may be explained by an intrinsic threshold for radiation-response, potentially irrespective of the donor group. Regarding genes that were not excluded due to their bimodal expression, the distribution of EV was similar across donor groups, but across all radiation doses, more genes were classified as hypo- and hyper-variable in N0 than in N1 and N2 + . N0 showed distinct and unique hypo-variable genes, which were associated with regulatory and stabilizing functions after all doses.
Reaction to LDIR
Most notably, we identified genes to be only hyper-variable in N2 + post-LDIR that were associated with the cell fate decision. Among these were the ERK1 and ERK2 cascade, that are essential regulators of cell proliferation, differentiation, as well as the response to stress (Guo et al. 2020). Additionally, RAS protein signal transduction, also important for cell growth, division, and differentiation (Molina and Adjei 2006) and Protein Kinase B signaling, whereas its three isoforms are associated with the promotion of proliferation and increased cell survival (Nicholson and Anderson 2002), were identified exclusively for N2+ post-LDIR. We have previously reported that disturbances in the proper damage recognition and subsequent cell fate decision post-LDIR may be a cancer-driving factor in N2 + (Grandt et al. 2022). The best performing machine learning algorithm for the classification of N1 and N2 + based on the 30 genes that were only hyper-variable in N2 + post-LDIR showed an accuracy of only 0.68. We explain this due to the computational nature of the classification, since these usually rely on metrics such as mean, variance, and their relationship. As such metrics are, as shown by this work, sensitive to low expression levels and/or not the perfect fit for RNASeq-data distribution, novel algorithms might be needed to incorporate information such as the EV. However, since the EV is a metric that is computed on the group level, this is not straightforward to implement and requires further efforts. Interestingly, isoform one (AKT1) of protein kinase B was observed to halt apoptosis and induce cell-cycle progression only in N2 + through its upregulation post-HDIR (Grandt et al. 2022), further highlighting the role of the protein kinase B for radiation induced transfer of DNA-damage to the next cellular generations through incorrect cell fate decisions post-LDIR and -HDIR.
Reaction to HDIR
The importance of the adequate regulation of the cell fate decision was further underlined by two findings. First, only in N0 positive regulation of cell cycle was a function of all hypo-variable and can be presumed to be only tightly regulated in N0. Second, in N1 and N2 + but not N0, regulation of fibroblast proliferation and fibroblast proliferation were GO terms over-represented in hyper-variable genes, and thus only deregulated in the long-term survivors of childhood cancer post-HDIR.
Radiation-induced methylation
Genes that were reported to be affected by radiation-induced changes in methylation (Miousse et al. 2017) were either classified as non-variable or at least did not change classification across radiation treatments. Assuming that methylation has a radiation-dependent influence on EV and may be measured by our analysis pipeline presented, it is possible that 4 h after irradiation changes in global methylation may not be present in the early phase of DNA damage response (Maierhofer et al. 2017).
Inter-individual variation and the response to ionizing radiation
Tumour treatment, and radiotherapy in particular, can lead to acute normal tissue toxicities or late effects such as secondary primary malignancies in long-term survivors of childhood cancer. The occurrence of acute radiogenic normal tissue toxicities is related to alterations of pathways such as DNA damage response, cell cycle, chromatin organization, and RNA metabolism, which are also thought to be responsible for radiogenic late effects (Gomolka et al. 2019). Thus, it is hypothesized that cellular processes span beyond these well-established pathways that regulate cellular survival after radiation (Yard et al. 2016). Although this study could not use data on immediate adverse reactions to ionizing radiation in donors such as inflammation of tissues, we observed donor group-specific variability in functions associated with adverse reactions to ionizing radiation to be hypo-variable only in fibroblasts of cancer-free controls.
Strengths and limitations
While other studies examined small samples sizes (Storey et al. 2007) or used cell lines (Stranger et al. 2012) which are not able to fully depict the true variability in a population, we analysed a large donor group (N = 156) with a unique profile of cancer-free controls and childhood cancer survivors with and without second primary neoplasms. To reduce technical variation (commonly referred to as noise), all experiments were conducted in the same lab using matched triplets, consisting of one N0, one N1, and one N2 + donor. These triplets were then simultaneously subjected to the same treatment under identical lab conditions. Moreover, we used a pipeline that accounted for sampling error by incorporating bootstrapping and cross-validation in the classification process. The method used is more robust to outliers compared to other proposed methods (Storey et al. 2007; Li et al. 2010; Stranger et al. 2012). Despite all our efforts to reduce the technical variation to a minimum, we cannot fully exclude the possibility that other factors e.g., differences in sampling location (N2 + and N1 were predominantly sampled from inside the elbow by local dermatologists for this study, while N0 donors were predominantly sampled from the knee and feet in the course of elective surgery) between cancer survivors and tumour-free donors may have had an impact on expressional variability. Nevertheless, we expect that filtering of bimodal genes excluded such genes (e.g., the various HOX-cluster and their expression dependent on the developmental axes (Rinn et al. 2007)). We compared identical cell types and matched the donors by central parameters that affect EV, such as sex and age, which have been previously shown to be the most influential factor for the EV (Bashkeel et al. 2019), to successfully identify subtle group differences in EV between donor groups that may be associated with their respective cancer status, especially via sex-specific modulation of immune-pathways as one recent work reported (Khodursky et al. 2022). Thus, we applied analyses stratified by sex to examine these potential differences in more detail. Our analyses did identify sex-specific differences in the number of bimodally expressed genes, but not in EV and its subsequent variability classification. A difference in bimodal expression is unsurprising as the sex-specific expression can be found across all tissues and are of immense pathophysiological relevance (Gershoni and Pietrokovski 2017) and impact the radiation response (Narendran et al. 2019). To further elaborate on this, we further accounted for EV-modulating factors such as smoking and drinking habits in the additional sensitivity analyses. Smoking and excessive consumption of alcohol have been shown to affect gene expression in active vs never smokers (Cao et al. 2015), as well as smokers compared to non-tobacco consumers (Arimilli et al. 2017). However, the genes proposed as biomarkers for smoking individuals (CYP1A1, CYP1B1, YWHAZ and PTPRD, MAX, and USF1) did not differ in classification between the samples termed healthy and those termed unhealthy, irrespective of the radiation dose. Nevertheless, further work with more adequate sample sizes is needed to elaborate on these findings.
Conclusion
For the first time, we adapted a comprehensive method to compute and characterize the expression variability unaffected by the expression magnitude in fibroblasts of childhood cancer survivors and cancer-free controls in response to a low and a high dose of ionizing radiation. Our results suggest that cancer-free controls exhibit different variability structure in gene expression with more hypo- and hyper-variable genes than long-term survivors of childhood cancer. The fibroblasts of these former childhood cancer patients with at least one second primary neoplasm showed deregulated mechanisms essential for cell fate decision. This could partially explain a subsequently occurring second primary neoplasm. Based on these results we encourage future works to focus on pathways for cell fate decision post-LDIR to shed further light on the carcinogenesis of potentially radiation induced second primary neoplasms.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author upon reasonable request. An exemplary code to reproduce the classification pipeline can be found on the GitHub-repository: https://github.com/clg1990/KiKme_mrna_variability.
Abbreviations
- ABCF3:
-
Atp Binding Cassette Subfamily F Member 3
- ALDOA:
-
Aldolase, Fructose-Bisphosphate A
- ANGPT1:
-
Angiopoietin 1
- ANPEP:
-
Alanyl Aminopeptidase, Membrane
- ARID1A:
-
At-Rich Interaction Domain 1a
- ASPH:
-
Aspartate Beta-Hydroxylase
- ATF4:
-
Activating Transcription Factor 4
- BAG5:
-
Bag Cochaperone 5
- CAMK1D:
-
Calcium/Calmodulin Dependent Protein Kinase Id
- CCM2:
-
Ccm2 Scaffold Protein
- CCND1:
-
Cyclin D1
- CCNG1:
-
Cyclin G1
- CD44:
-
Cd44 Molecule (Indian Blood Group)
- CD63:
-
Cd63 Molecule
- CDH13:
-
Cadherin 13
- CDKN1A:
-
Cyclin Dependent Kinase Inhibitor 1a
- CIRBP:
-
Cold Inducible Rna Binding Protein
- CST3:
-
Cystatin C
- CV:
-
Coefficient of variation
- CYP1A1:
-
Cytochrome P450 Family 1 Subfamily A Member 1
- CYP1B1:
-
Cytochrome P450 Family 1 Subfamily B Member 1
- DKK2:
-
Dickkopf Wnt Signaling Pathway Inhibitor 2
- DNA:
-
Deoxy-Ribonucleic Acid
- DNAJB2:
-
Dnaj Heat Shock Protein Family (Hsp40) Member B2
- EIF2AK2:
-
Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2
- ENO1:
-
Enolase 1
- EV:
-
Expression variability
- ERK1:
-
Ras-dependent extracellular signal-regulated kinase 1
- ERK2:
-
Ras-dependent extracellular signal-regulated kinase 2
- FBLN5:
-
Fibulin 5
- FGF5:
-
Fibroblast growth factor 5
- FH:
-
Fumarate Hydratase
- GLB1:
-
Galactosidase Beta 1
- GLUL:
-
Glutamate-Ammonia Ligase
- GNAQ:
-
G Protein Subunit Alpha Q
- GO:
-
Gene Ontology
- GTF2H1:
-
General Transcription Factor IIH Subunit 1
- HDIR:
-
High Dose Ionizing Radiation
- IGFBP3:
-
Insulin Like Growth Factor Binding Protein 3
- IL1R1:
-
Interleukin 1 Receptor Type 1
- IL6ST:
-
Interleukin 6 Cytokine Family Signal Transducer
- IMPDH2:
-
Inosine Monophosphate Dehydrogenase 2
- KLF2:
-
Krüppel Like Factor 2
- LAMB2:
-
Laminin Subunit Beta 2
- LASP1:
-
Lim And Sh3 Protein 1
- LDIR:
-
Low Dose Ionizing Radiation
- LIMS1:
-
Lim Zinc Finger Domain Containing 1
- LOXL3:
-
Lysyl Oxidase Like 3
- LRP1:
-
LDL Receptor Related Protein 1
- MAD:
-
Median Absolute Deviation
- MAPK:
-
Mitogen-activated protein kinase
- MAX:
-
Myc Associated Factor X
- mRNA:
-
Messenger Ribonucleic Acid
- MSH6:
-
Muts Homolog 6
- MSRB3:
-
Methionine Sulfoxide Reductase B3
- MTCH1:
-
Mitochondrial Carrier 1
- MXRA8:
-
Matrix Remodeling Associated 8
- MYH10:
-
Myosin Heavy Chain 10
- N0:
-
Fibroblasts of cancer-free Controls
- NRP1:
-
Neuropilin-1
- N1:
-
Fibroblasts of Donors with a First Primary Neoplasm in Childhood
- N2 + :
-
Fibroblasts of Donors with a First Primary Neoplasm in Childhood and at least one Second Primary Neoplasm
- OSMR:
-
Oncostatin M Receptor
- PLS3:
-
Plastin 3
- POLR2C:
-
Rna Polymerase II Subunit C
- PPIG:
-
Peptidylprolyl Isomerase G
- PPP1R10:
-
Protein Phosphatase 1 Regulatory Subunit 10
- PRDX5:
-
Peroxiredoxin 5
- PSMA3:
-
Proteasome 20 s Subunit Alpha 3
- PSMB3:
-
Proteasome 20 s Subunit Beta 3
- PSMD4:
-
Proteasome 26 s Subunit Ubiquitin Receptor, Non-Atpase 4
- PSMD5:
-
Proteasome 26 s Subunit, Non-ATPase 5
- PTPRD:
-
Protein Tyrosine Phosphatase Receptor Type D
- RAD21:
-
Rad21 Cohesin Complex Component
- RAD23A:
-
Rad23 Homolog A, Nucleotide Excision Repair Protein
- RAD23B:
-
Rad23 Homolog B, Nucleotide Excision Repair Protein
- RALGPS2:
-
Ral GEF with PH Domain and SH3 Binding Motif 2
- Ras:
-
Rat sarcoma virus
- RCN3:
-
Reticulocalbin 3
- ROBO1:
-
Roundabout Guidance Receptor 1
- RPLP0:
-
Ribosomal Protein Lateral Stalk Subunit P0
- RPS18:
-
Ribosomal Protein S18
- RPS19:
-
Ribosomal Protein S19
- RPS19:
-
Ribosomal Protein S19
- RPS27L:
-
Ribosomal Protein S27 Like
- RRAS:
-
Ras-related Protein R-Ras
- SD:
-
Standard deviation
- SIGMAR1:
-
Sigma Non-Opioid Intracellular Receptor 1
- SIGMAR1:
-
Sigma Non-Opioid Intracellular Receptor 1
- SIN3A:
-
Sin3 Transcription Regulator Family Member A
- SIN3A:
-
Sin3 Transcription Regulator Family Member A
- SPEN:
-
Spen Family Transcriptional Repressor
- SPTBN1:
-
Spectrin Beta, Non-Erythrocytic 1
- SREBF2:
-
Sterol Regulatory Element Binding Transcription Factor 2
- SRPK2:
-
SRSF Protein Kinase 2
- STK25:
-
Serine/Threonine Kinase 25
- STK25:
-
Serine/Threonine Kinase 25
- THY1:
-
Thy-1 Cell Surface Antigen
- TMEM119:
-
Transmembrane Protein 119
- TNFAIP8L3:
-
TNF Alpha Induced Protein 8 Like 3
- TOLLIP:
-
Toll Interacting Protein
- TRAF7:
-
TNF Receptor Associated Factor 7
- TRAIL:
-
TNF-related apoptosis-inducing Ligand
- TRAM2:
-
Translocation Associated Membrane Protein 2
- TXNRD1:
-
Thioredoxin Reductase 1
- TXNRD1:
-
Thioredoxin Reductase 1
- UBB:
-
Ubiquitin B
- ULK1:
-
Unc-51 Like Autophagy Activating Kinase 1
- USF1:
-
Upstream Transcription Factor 1
- VDAC3:
-
Voltage Dependent Anion Channel 3
- WDR48:
-
WD Repeat Domain 48
- YBX3:
-
Y-Box Binding Protein 3
- YWHAB:
-
Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein Beta
- YWHAE:
-
Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein Epsilon
- YWHAG:
-
Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein Gamma
- YWHAQ:
-
Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein Theta
- YWHAZ:
-
Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein Zeta
- ZMIZ1:
-
Zinc Finger Miz-Type Containing 1
References
Afsari B, Geman D, Fertig EJ. Learning dysregulated pathways in cancers from differential variability analysis. Cancer Inform. 2014;13(Suppl 5):61–7.
Alemu EY, Carl JW Jr, Corrada Bravo H, Hannenhalli S. Determinants of expression variability. Nucleic Acids Res. 2014;42(6):3503–14.
Antwih DA, Gabbara KM, Lancaster WD, Ruden DM, Zielske SP. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics. 2013;8(8):839–48.
Arimilli S, Madahian B, Chen P, Marano K, Prasad GL. Gene expression profiles associated with cigarette smoking and moist snuff consumption. BMC Genomics. 2017;18(1):156.
Bashkeel N. Human gene expression variability and its dependence on methylation and aging. Ottawa: University of Ottawa; 2019.
Bashkeel N, Perkins TJ, Kærn M, Lee JM. Human gene expression variability and its dependence on methylation and aging. BMC Genomics. 2019;20(1):941.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (methodological). 1995;57(1):289–300.
Bhan A, Hussain I, Ansari KI, Kasiri S, Bashyal A, Manda SS. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J Mol Biol. 2013;425(19):3707–22.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Borràs-Fresneda M, Barquinero J-F, Gomolka M, Hornhardt S, Rössler U, Armengol G, Barrios L. Differences in DNA repair capacity, cell death and transcriptional response after irradiation between a radiosensitive and a radioresistant cell line. Sci Rep. 2016;6(1):27043.
Brackmann LK, Poplawski A, Grandt CL, Schwarz H, Hankeln T, Rapp S, Zahnreich S, Galetzka D, Schmitt I, Grad C, Eckhard L, Mirsch J, Blettner M, Scholz-Kreisel P, Hess M, Binder H, Schmidberger H, Marron M. Comparison of time and dose dependent gene expression and affected pathways in primary human fibroblasts after exposure to ionizing radiation. Mol Med. 2020;26(1):85.
Breschi A, Djebali S, Gillis J, Pervouchine DD, Dobin A, Davis CA, Gingeras TR, Guigó R. Gene-specific patterns of expression variation across organs and species. Genome Biol. 2016;17(1):151.
Cao C, Chen J, Lyu C, Yu J, Zhao W, Wang Y, Zou D. Bioinformatics analysis of the effects of tobacco smoke on gene expression. PLoS ONE. 2015;10(12): e0143377.
Conway JR, Lex A, Gehlenborg N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 2017;33(18):2938–40.
Corrada Bravo H, Pihur V, McCall M, Irizarry RA, Leek JT. Gene expression anti-profiles as a basis for accurate universal cancer signatures. BMC Bioinform. 2012;13(1):272.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Ecker S, Pancaldi V, Rico D, Valencia A. Higher gene expression variability in the more aggressive subtype of chronic lymphocytic leukemia. Genome Med. 2015;7(1):8.
Feinberg AP, Irizarry RA. Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci. 2010;107(suppl 1):1757–64.
Gershoni M, Pietrokovski S. The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol. 2017;15(1):7.
Goksuluk D, Zararsiz G, Korkmaz S, Eldem V, Zararsiz GE, Ozcetin E, Ozturk A, Karaagaoglu AE. MLSeq: machine learning interface for RNA-sequencing data. Comput Methods Programs Biomed. 2019;175:223–31.
Gomolka M, Blyth B, Bourguignon M, Badie C, Schmitz A, Talbot C, Hoeschen C, Salomaa S. Potential screening assays for individual radiation sensitivity and susceptibility and their current validation state. Int J Radiat Biol. 2019; 1–17.
Grandt CL, Brackmann LK, Poplawski A, Schwarz H, Hummel-Bartenschlager W, Hankeln T, Kraemer C, Marini F, Zahnreich S, Schmitt I, Drees P, Mirsch J, Grabow D, Schmidberger H, Binder H, Hess M, Galetzka D, Marron M. Radiation-response in primary fibroblasts of long-term survivors of childhood cancer with and without second primary neoplasms: the KiKme study. Mol Med. 2022;28(1):105.
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007.
Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA, Feinberg AP. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43(8):768–75.
Ho JWK, Stefani M, dos Remedios CG, Charleston MA. Differential variability analysis of gene expression and its application to human diseases. Bioinformatics (oxford, England). 2008;24(13):i390–8.
Hodgson D, van Leeuwen F, Ng A, Morton L, Henderson TO. Breast cancer after childhood, adolescent, and young adult cancer: it’s not just about chest radiation. Am Soc Clin Oncol Educ Book. 2017;37:736–45.
Hornhardt S, Rossler U, Sauter W, Rosenberger A, Illig T, Bickeboller H, Wichmann HE, Gomolka M. Genetic factors in individual radiation sensitivity. DNA Repair (Amst). 2014;16:54–65.
Inskip PD, Sigurdson AJ, Veiga L, Bhatti P, Ronckers C, Rajaraman P, Boukheris H, Stovall M, Smith S, Hammond S, Henderson TO, Watt TC, Mertens AC, Leisenring W, Stratton K, Whitton J, Donaldson SS, Armstrong GT, Robison LL, Neglia JP. Radiation-related new primary solid cancers in the childhood cancer survivor study: comparative radiation dose response and modification of treatment effects. Int J Radiat Oncol Biol Phys. 2016;94(4):800–7.
Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2012;41(D1):D793–800.
Khodursky S, Jiang CS, Zheng EB, Vaughan R, Schrider DR, Zhao L. Sex differences in inter-individual gene expression variability across human tissues. bioRxiv. 2022; 2022.2008.2009.503366.
Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, Liu Y, Akbani R, Feng B, Donehower LA, Miller C, Shen Y, Karimi M, Chen H, Kim P, Jia P, Shinbrot E, Zhang S, Liu J, Hu H, Bailey MH, Yau C, Wolf D, Zhao Z, Weinstein JN, Li L, Ding L, Mills GB, Laird PW, Wheeler DA, Shmulevich I, Monnat RJ Jr, Xiao Y, Wang C. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23(1):239-254.e236.
Kutanzi KR, Lumen A, Koturbash I, Miousse IR. Pediatric exposures to ionizing radiation: carcinogenic considerations. Int J Environ Res Public Health. 2016; 13(11).
Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, Stevenson K, Sougnez C, Wang L, Li S, Kotliar D, Zhang W, Ghandi M, Garraway L, Fernandes SM, Livak KJ, Gabriel S, Gnirke A, Lander ES, Brown JR, Neuberg D, Kharchenko PV, Hacohen N, Getz G, Meissner A, Wu CJ. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26(6):813–25.
Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29.
Li J, Liu Y, Kim T, Min R, Zhang Z. Gene expression variability within and between human populations and implications toward disease susceptibility. PLoS Comput Biol. 2010; 6(8).
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Maierhofer A, Flunkert J, Dittrich M, Müller T, Schindler D, Nanda I, Haaf T. Analysis of global DNA methylation changes in primary human fibroblasts in the early phase following X-ray irradiation. PLoS ONE. 2017;12(5): e0177442.
Mar JC, Matigian NA, Mackay-Sim A, Mellick GD, Sue CM, Silburn PA, McGrath JJ, Quackenbush J, Wells CA. Variance of gene expression identifies altered network constraints in neurological disease. PLoS Genet. 2011;7(8): e1002207.
Marron M, Brackmann LK, Schwarz H, Hummel-Bartenschlager W, Zahnreich S, Galetzka D, Schmitt I, Grad C, Drees P, Hopf J, Mirsch J, Scholz-Kreisel P, Kaatsch P, Poplawski A, Hess M, Binder H, Hankeln T, Blettner M, Schmidberger H. Identification of genetic predispositions related to ionizing radiation in primary human skin fibroblasts from survivors of childhood and second primary cancer as well as cancer-free controls: protocol for the nested case-control study KiKme. JMIR Res Protoc. 2021;10(11): e32395.
Miousse IR, Kutanzi KR, Koturbash I. Effects of ionizing radiation on DNA methylation: from experimental biology to clinical applications. Int J Radiat Biol. 2017;93(5):457–69.
Molina JR, Adjei AA. The Ras/Raf/MAPK pathway. J Thorac Oncol. 2006;1(1):7–9.
Narendran N, Luzhna L, Kovalchuk O. Sex difference of radiation response in occupational and accidental exposure. Front Genet. 2019;10:260.
Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14(5):381–95.
Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression within and among natural populations. Nat Genet. 2002;32(2):261–6.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–23.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7): e47.
Scholz-Kreisel P, Kaatsch P, Spix C, Schmidberger H, Marron M, Grabow D, Becker C, Blettner M. Second malignancies following childhood cancer treatment in germany from 1980 to 2014. Dtsch Arztebl Int. 2018;115(23):385–92.
Seibold P, Auvinen A, Averbeck D, Bourguignon M, Hartikainen JM, Hoeschen C, Laurent O, Noel G, Sabatier L, Salomaa S, Blettner M. Clinical and epidemiological observations on individual radiation sensitivity and susceptibility. Int J Radiat Biol. 2019; 1–16.
Simonovsky E, Schuster R, Yeger-Lotem E. Large-scale analysis of human gene expression variability associates highly variable drug targets with lower drug effectiveness and safety. Bioinformatics (oxford, England). 2019;35(17):3028–37.
Sisakht M, Darabian M, Mahmoodzadeh A, Bazi A, Shafiee SM, Mokarram P, Khoshdel Z. The role of radiation induced oxidative stress as a regulator of radio-adaptive responses. Int J Radiat Biol. 2020;96(5):561–76.
Smirnov DA, Brady L, Halasa K, Morley M, Solomon S, Cheung VG. Genetic variation in radiation-induced cell death. Genome Res. 2012;22(2):332–9.
Spector LG, Pankratz N, Marcotte EL. Genetic and nongenetic risk factors for childhood cancer. Pediatr Clin N Am. 2015;62(1):11–25.
Storey JD, Madeoy J, Strout JL, Wurfel M, Ronald J, Akey JM. Gene-expression variation within and among human populations. Am J Hum Genet. 2007;80(3):502–9.
Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, Price A, Raj T, Nisbett J, Nica AC, Beazley C, Durbin R, Deloukas P, Dermitzakis ET. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8(4): e1002639.
Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6(7): e21800.
The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2018;47(D1):D330–8.
Tong P, Chen Y, Su X, Coombes KR. SIBER: systematic identification of bimodally expressed genes using RNAseq data. Bioinformatics. 2013;29(5):605–13.
Tukenova M, Guibout C, Hawkins M, Quiniou E, Mousannif A, Pacquement H, Winter D, Bridier A, Lefkopoulos D, Oberlin O, Diallo I, De Vathaire F. Radiation therapy and late mortality from second sarcoma, carcinoma, and hematological malignancies after a solid cancer in childhood. Int J Radiat Oncol Biol Phys. 2011;80(2):339–46.
Wang J, Wen S, Symmans WF, Pusztai L, Coombes KR. The bimodality index: a criterion for discovering and ranking bimodal signatures from cancer gene expression profiling data. Cancer Inform. 2009;7:199–216.
Wijesooriya K, Jadaan SA, Perera KL, Kaur T, Ziemann M. Urgent need for consistent standards in functional enrichment analysis. PLoS Comput Biol. 2022;18(3): e1009935.
Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, Quake SR. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11(1):41–6.
Xu H, Liu Y, Li Y, Diao L, Xun Z, Zhang Y, Wang Z, Li D. RadAtlas 1.0: a knowledgebase focusing on radiation-associated genes. Int J Radiat Biol. 2020; 1–8.
Yahyanejad S, Theys J, Vooijs M. Targeting Notch to overcome radiation resistance. Oncotarget. 2016;7(7):7610–28.
Yard BD, Adams DJ, Chie EK, Tamayo P, Battaglia JS, Gopal P, Rogacki K, Pearson BE, Phillips J, Raymond DP, Pennell NA, Almeida F, Cheah JH, Clemons PA, Shamji A, Peacock CD, Schreiber SL, Hammerman PS, Abazeed ME. A genetic basis for the variation in the vulnerability of cancer to DNA damage. Nat Commun. 2016;7(1):11428.
Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, Eleftheriadis M, Sinning CR, Schnabel RB, Lubos E, Mennerich D, Rust W, Perret C, Proust C, Nicaud V, Loscalzo J, Hubner N, Tregouet D, Munzel T, Ziegler A, Tiret L, Blankenberg S, Cambien F. Genetics and beyond–the transcriptome of human monocytes and disease susceptibility. PLoS ONE. 2010;5(5): e10693.
Acknowledgements
The authors gratefully acknowledge the assistance from Peter Kaatsch, Franziska Himmelsbach, Cornelia Becker, Ilona Kerenyi, and Marianne Brömmel from the German Childhood Cancer Registry. For his work as the coordinator of the ISIBELa consortium, we thank Peter Scholz-Kreisel. We are very thankful for the local support of all participating dermatologists in Germany, Austria, and Switzerland, and Ursula Disque-Kaiser for the excellent technical laboratory assistance. The authors acknowledge resources and support from the Bioinformatics Core Facility at the University Medical Center Mainz. TH and CK acknowledge the expert technical work of the staff of the Nucleic Acids Core Facility, Faculty of Biology, JGU Mainz. The study would not have been possible without the voluntary collaboration of all participants who participated in the extensive examinations. We further thank Heiko Karle for his help in establishing the method for fibroblast irradiation.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was funded by the Federal Ministry of Education and Research in Germany under contracts No. 02NUK016A, 2NUK042A, 2NUK042B, 2NUK042C, and 2NUK042D and conducted among other research projects as part of the research consortia ISIMEP (Intrinsic radiation sensitivity: Identification, mechanisms and epidemiology, principal investigator: Maria Blettner) and ISIBELa (Intrinsic radiation sensitivity: Identification of biological and epidemiological long-term effects, principal investigators: Maria Blettner and Heinz Schmidberger).
Author information
Authors and Affiliations
Contributions
MM conceptualized the research idea on gene expression at different time points after exposure to high and low doses of ionizing radiation and designed the experiments. MM developed the KiKme study and its design as principal investigator. LKB and MM implemented the KiKme study. LKB, CS, and MM conducted the participant recruitment, which was organized and planned by LKB and MM. Former childhood cancer patients were identified, matched, and contacted by the German Childhood Cancer Registry. Doctors responsible for sample drawing were trained and supervised by MM and HS. The method of fibroblast sampling was established by DG and HS. HSZ takes care of the project´s biobank and controls for the quality of all biosamples. Cell culture and radiation exposure of primary fibroblasts were established by SZ and DG. LKB and SZ were responsible for the pseudonymization of all biosamples. The analysis pipeline for the project was developed by MM, MH, AP, and HB. Analysis data of biosamples was processed by AP, CK, and TH. CLG and AP conducted the statistical analysis. The final analysis of data regarding content and the hypothesis was done by CLG. All figures were designed and programmed by CLG. CLG, LKB, and MM prepared the initial manuscript. All authors revised the manuscript critically for important intellectual content, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We certify that all applicable institutional and governmental regulations concerning the ethical use of human volunteers were followed during this research. Approval by the Ethics Committee of the Medical Association of Rhineland-Palatinate was obtained (no. 837.262.12 (8363-F), no. 837.103.04 (4261), and no. 837.440.03 (4102). Study participants will not undergo any procedures unless they give consent for examinations, collection of samples, subsequent analysis, and storage of personal data and collected samples. Study subjects can consent to single components of the study while abstaining from others.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Results of the differential gene expression analysis and the bimodal indices. a: Results of the differential expression analysis comparing fibroblasts of long-term survivors of childhood cancer without (N1) and with at least one second primary neoplasm (N2 +) to cancer-free controls (N0). Data were adjusted for age and sex, p-values for false discovery at a rate of 0.05 (FDR). b: Bimodal index (BI) values of the bimodal test, using the SIBERG package for R.
Additional file 2.
Results of the gene expression variability classification. a Gene classifications in whole data set (WDS) and classifications after cross-validation (cv) in fibroblasts of cancer-free controls (N0), childhood cancer survivors without (N1) and with at least one second primary neoplasm (N2 +) after exposure 0 Gray. 1 = Hypo-Variable, 2 = Non-Variable, 3 = Hyper-Variable. NA = Gene excluded by bimodal test/NA in CV = classification from WDS not verified, set to 2 for further analyses. b Gene classifications in whole data set (WDS) and classifications after cross-validation (cv) in fibroblasts of cancer-free controls (N0), long-term survivors of childhood cancer without (N1) and with at least one second primary neoplasm (N2 +) after exposure to 0.05 Gray. 1 = Hypo-Variable, 2 = Non-Variable, 3 = Hyper-Variable. NA = Gene excluded by bimodal test/NA in CV = classification from WDS not verified, set to 2 for further analyses. c Gene classifications in whole data set (WDS) and classifications after cross-validation (cv) in fibroblasts of cancer-free controls (N0), long-term survivors of childhood cancer without (N1) and with at least one second primary neoplasm (N2 +) after exposure to 2 Gray. 1 = Hypo-Variable, 2 = Non-Variable, 3 = Hyper-Variable. NA = Gene excluded by bimodal test/NA in CV = classification from WDS not verified, set to 2 for further analyses. d Results of the various employed algorithms for the classification of N1 and N2 + using 30 genes that were only classified as hyper-variable in N2 + post-LDIR. Models were trained and tested using only N1 and N2 + data post-LDIR.
Additional file 3.
Gene lists that were used as backgrounds in the gene ontology over-representation analyses.
Additional file 4.
Results of the Gene Ontology (GO) term over-representation analyses. Results of the Gene Ontology (GO) term over-representation analyses for all donor groups and radiation doses, filtered for adjusted p-value < 0.05. N0 = fibroblasts of cancer-free donors, N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, and N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm.
Additional file 5.
Intersect graphs of A) genes and B) Gene Ontology (GO) terms. Intersect graphs of A) genes and B) Gene Ontology (GO) terms: Both graphs are stratified by variability-classification, donor group (N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of childhood cancer survivors without a second primary neoplasm, N2 + = fibroblasts of childhood cancer survivors with at least one second primary neoplasm), and radiation dose. Connected rows implicate that A) genes or B) GO terms were identically classified in these data. The bars denote the summed number of identically classified A) genes or B) GO terms among the vertically connected rows of data, e.g., in A) the first column implicates that 416 genes were found to be hypo-variable across all radiation doses and donor groups.
Additional file 6.
Gene lists that were used as backgrounds in the gene ontology over-representation analyses. a: Over-represented Gene Ontology terms for genes only classified hypo-variable (n = 49) in fibroblasts of cancer-free donors after 0 Gray. b: Over-represented Gene Ontology terms for genes classified only hypo-variable (n = 41) in fibroblasts of cancer-free controls after 0.05 Gray. c: Over-represented Gene Ontology terms for genes classified only hypo-variable (n = 38) in fibroblasts of cancer-free donors after 2 Gray. d: Over-represented Gene Ontology terms for genes classified only as hyper-variable (n = 43) in fibroblasts of cancer-free donors after all radiation doses. e: Over-represented Gene Ontology terms for genes classified only hypo-variable (n = 30) in fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm after 2 Gray. f: Over-represented Gene Ontology terms for genes classified only hypo-variable (n = 30) in fibroblasts of long-term survivors of childhood-cancer with at least one second primary neoplasm after 0.05 Gray, filtered for the 14 genes with informational value for classification discrimination between N1 and N2 + .
Additional file 7.
Comparison of the bimodally expressed genes with the information on differential gene expression. Comparison of differential gene expression analysis and results of the bimodal test. A.) Volcano plots comparing bimodal index values and log2 fold-change values, stratified by radiation dose. Here, the expression of fibroblasts of cancer groups (N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm.) was compared to those of cancer-free controls. The dashed line indicates the threshold for bimodal expression set to bimodal index = 1.1. B.) Bar charts showing the total number of bimodally expressed genes per radiation dose and the number of differentially expressed genes thereof per comparison.
Additional file 8.
Stratified_Analyses. a: Violin and jitter plots comparing results of the analyses for bimodally expressed genes stratified by radiation dose and sex. With respect to the reduced sample size due to the stratification, the cut-off was increased to 1.3 (dashed line). Nevertheless, these sample sizes (female: n = 81, male: n = 75) could still not have been sufficient to provide adequate power for the detection of bimodally expressed genes. b: Comparison of expressional variation by sex. The scatterplot compares the sex-specific EV per gene and radiation dose. The black line indicates the linear regression model (adjusted r2: 0.779; Kendall’s tau: 0.786). Only genes with a bimodal index < 1.3 were included in the analysis. c: Bar charts showing the number of genes per classification using the whole data set and the number of genes with stable classification after cross-validation per radiation dose. Data shown here were stratified by sex. Only genes with a bimodal index < 1.3 were included in the analyses. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, and N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm. d: Intersect graphs of overlapping gene classifications: Data are stratified by variability classification, donor group (N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm), sex, and radiation dose. Connected rows implicate that genes were identically classified in these data sets. Bars denote the summed number of identically classified genes among the vertically connected rows of data. e: Violin and jitter plots comparing results of the analyses for bimodally expressed genes stratified by radiation dose and consumption information on smoking and alcohol. Data used here were donor triplets without heavy smokers (> 10 pack years) and/or alcohol consumption (> 2 alcoholic beverages per day), termed “healthy” for brevity; as well as triplets that contained at least one donor with the above-described lifestyle, shortly termed “unhealthy”. To ensure validity, the cut-off was increased to 1.3 due to the reduced sample size. Nevertheless, these sample sizes (Donors from triplets without any heavy smokers or drinkers: n = 60, donors from triplets with at least one heavy drinker or smoker: n = 54) might not be sufficient to provide adequate power for the detection of bimodally expressed genes with the given cut-off. f: Bar charts showing the number of genes per classification using the whole data set and the number of genes with stable classification after cross-validation per radiation dose. Data used here were donor triplets without heavy smokers (> 10 pack years) and/or alcohol consumption (> 2 alcoholic beverages per day), termed”healthy”; as well as triplets that contained at least one donor with the above-described lifestyle, termed “unhealthy” for brevity. Only genes with a bimodal index < 1.3 were included in the analyses. N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, and N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm. g: Comparison of the EV per gene and radiation dose comparison donor triplets with and without heavy smokers (> 10 pack years) and/or alcohol consumption (> 2 alcoholic beverages per day). The red line indicates the linear regression model (adjusted r2: 0.377; Kendall’s tau: 0.683). Only genes with a bimodal index < 1.3 were included in the analysis. h: Intersect graphs of overlapping gene classifications: Data are stratified by variability classification, donor group (N0 = fibroblasts of cancer-free controls, N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm), radiation dose, and additionally computed using only the 20 donor triplets (n = 60) without heavy smokers (> 10 pack years) and/or alcohol consumption (> 2 alcoholic beverages per day). Connected rows implicate that genes were identically classified in these data sets. Bars denote the summed number of identically classified genes among the vertically connected rows of data.
Additional file 9.
Comparison of the bimodally expressed genes with the information on differential gene expression. Expression variation and variability classification of genes that were presumed to be affected in methylation status by ionizing radiation in the literature, stratified by donor group and radiation dose. N0 = fibroblasts of cancer-free donors, N1 = fibroblasts of long-term survivors of childhood cancer without a second primary neoplasm, and N2 + = fibroblasts of long-term survivors of childhood cancer with at least one second primary neoplasm.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Grandt, C.L., Brackmann, L.K., Foraita, R. et al. Gene expression variability in long-term survivors of childhood cancer and cancer-free controls in response to ionizing irradiation. Mol Med 29, 41 (2023). https://doi.org/10.1186/s10020-023-00629-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10020-023-00629-2