
Abstract
T-cell prolymphocytic leukemia (T-PLL) is a rare and aggressive subtype of chronic lymphocytic leukemia. Usually it presents in older people with a median age of 61 years. T-PLL is characterized by elevated white blood cell (WBC) count with anemia and thrombocytopenia, hepatosplenomegaly, and lymphadenopathy; less common findings are skin infiltration and pleural effusions. The most frequent chromosomal abnormalities in T-PLL include 14q11.2, chromosome 8, and 11q rearrangements. Also deletions in the short arm of a chromosome 9 are reported in ~30% of T-PLL together with other aberrations. Here we report a childhood T-PLL case with a de novo del(9)(p13) as sole acquired anomaly leading to monosomy of the tumor suppressor gene CDKN2A (cyclin-dependent kinase inhibitor 2A). Also, to the best of our knowledge this is the first case of a childhood T-PLL with this aberration.
Similar content being viewed by others


Introduction
T-cell prolymphocytic leukemia (T-PLL) is an aggressive lymphoproliferative disorder that represents in 2% of all mature lymphocytic leukemias in adults; also it has been classified as an aggressive subtype of chronic lymphocytic leukemia (CLL) [1]. T-PLL causes major problems due to negative effects on the immune system, thus predisposing the affected patient to a variety of infections, and possibly death [2]. T-PLL affects mainly adults (median age 61 years) and is more common in male (male/female =2:1) [3].
T-PLL characterized by several clinical features like splenomegaly (75% of patients), generalized lymphadenopathy (50%), and skin infiltration (25%) with skin nodules, maculopapular rash, or more rarely erythroderma. Also serous effusions, particularly pleural effusions (15%) are regularly seen [4, 5]. The majority of T-PLL patients present with severely increased lymphocyte count (>100,000/μl). Anemia and thrombocytopenia are present in half of the patients, and lactate dehydrogenase (LDH) levels are usually elevated [6]. The clinical course of the disease is usually aggressive, with poor or no response to therapy [7].
The immunophenotype of T-PLL cells resembles that of mature post-thymic T-cells with expression of CD2, CD3, and CD7. The T-cell receptor (TCR) beta/gamma genes can be clonally rearranged [1]. The most frequent chromosomal abnormalities in T-PLL include 14q11.2-aberrations, chromosome 8 rearrangements and/or 11q abnormalities, the latter leading to the deletion of the ataxia-telangiectasia mutated (ATM) and mixed lineage leukemia (MLL) genes, as well as abnormalities e.g. in 5q, 6q, 9p, 12p, and 13q [8–11].
Here we report an untreated childhood T-PLL case with do novo del(9)(p13) as sole abnormality, which leads to monoallely of tumor suppressor gene CDKN2A (cyclin-dependent kinase inhibitor 2A).
Material and methods
Case report
A 16 year old male patient without significant personal or familial medical chronically presented with a 1 month history of dyspnea, fatigue, loss of weight, fever and pleural effusion in right lung. Pleural fluid examination revealed an elevated white blood cell (WBC) count of 218.4 × 109/l (70% were lymphocytes and 15% were blasts), and LDH level of 3,486 U/l (normal up to 480 U/l). On physical examination enlarged liver was suggested whereas, CT scan and echography of the abdomen revealed normal liver size; several skin nodules (1–2 cm) in different locations such as neck, mandible and armpit were detected (data not shown). Routine peripheral blood test showed WBC count of 70.6 × 109/l (74.6% were lymphocytes). The red blood cell (RBC) count was 4.52 × 106/mm3 with a hemoglobin level of 11.8 g/dl and platelets count of 0.277 × 109/l. Biochemistry analyses revealed LDH level of 1,377 U/l; alanine aminotransferase (ALT) level was 142 U/l (normal up to 40U/l); aspartate aminotrasferase (AST) level 51 U/l (normal up to 40U/l); total serum protein 6 g/dl (normal 6.4-8.3 g/dl); serum albumin 4 g/dl (normal 3.5-5.2 g/dl); and serum calcium value 9.7 mg/dl (normal 8.5-10.5 mg/dl). Bone marrow smear showed approximately 95% of cells were blasts. Unfortunately the patient died two months after diagnosis from the disease due to respiratory arrest.
Cytogenetic analysis
Cytogenetic analysis using GTG-banding was performed according to standard procedures [12]. A minimum of 20 metaphase cells derived from unstimulated bone marrow culture were analyzed. Karyotypes were described according to the International System for Human Cytogenetic Nomenclature (ISCN 2009).
Molecular cytogenetics
FISH using a whole chromosome painting (WCP) probe for chromosome 9 (MetaSystems/Germany) and a locus specific probe for CDKN2A gene (LSI p16 in 9p21) with a probe for centromere 9 (Abbott Molecular/Vysis, Abbott Park, IL, USA) were applied according to manufacturer’s instructions [12]. Also a multicolor banding probe (MCB) sets based on microdissection derived region-specific libraries for chromosome 9 was applied as previously described [13]. A minimum of 20 metaphase spreads were analyzed, using a fluorescence microscope (AxioImager.Z1 mot, Carl Zeiss Ltd., Hertfordshir, UK) equipped with appropriate filter sets to discriminate between a maximum of five fluorochromes plus the counterstain DAPI (4',6- diamino-2-phenylindole). Image capture and processing were performed using an ISIS imaging system (MetaSystems).
Flow cytometry
Flow cytometry of leukemic blasts was performed using a general panel of fluorescent antibodies against the following antigens typical for different cell lineages and cell types: CD1a, CD2, CD3, CD4, CD5, CD8, CD10, CD11b, CD11c, CD13, CD14, CD15, CD16, CD19, CD20, CD22, CD23, CD32, CD33, CD34, CD38, CD41a, CD45, CD56, CD57, CD64, CD103, CD117, CD123, CD209, CD235a and CD243; in addition antibodies against Kappa and Lambda light Chains, sIgD, sIgM, and HLADr were applied (BD Biosciences). Four-color immunophenotyping on peripheral blood specimen was performed. Samples were stained and analyzed on a BD FACSCalibur™ flow cytometer according to BD Biosciences manuals and products insert sheets. Autofluorescence, viability, and isotype controls were included. Flow cytometric data acquisition and analysis were conducted by BD Cellquest™ Pro software.
Results
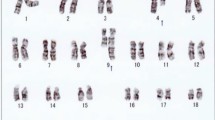
Karyotyping was performed before initiation of any treatment and GTG banding revealed a karyotype of 46,XY,del(9)(p?) (20) (Figure 1). Dual color FISH was performed to confirm presence of the aberration. A probe specific for CDKN2A confirmed that the deletion encompassed subband 9p21 (Figure 2A). Finally, MCB9 probe set (Figure 2B) revealed the following karyotype: 46,XY,del(9)(p13) (20).Flow cytometric analysis of bone marrow specimen characterized this case as a T-PLL (Figures 3A-C). The abnormal cell population (97% of tested cells) was positive for CD45, CD2, CD4, CD7, and expressed CD5 heterogeneously and at low levels. Also the population was negative for TdT, CD1a, CD3, CD8, CD34, and HLA-DR.

GTG-banding revealed a deletion of the short arm of a derivative chromosome 9 del(9)(p?). A derivative chromosome is marker by arrowhead.

Karyotype and chromosomal aberrations were confirmed using molecular cytogenetic approaches. (A) The deletion of CDKN2A was identified on the der(9). (B) The application of MCB 9 characterized the del(9)(p13) comprehensively. Abbreviations: # = chromosome; der = derivative chromosome.

Flow cytometric dot plots showing the abnormal population of T-cells. (A) dim CD45 expression. (B) CD4 expression. (C) CD2 and CD7 coexpression.
Discussion
T-PLL primarily affects older adults (median age at presentation, 61 years) with male predominance [3] and is considered as an aggressive leukemia with poor or no response to therapy [7].
T-cell CLL is now reclassified as T-PLL according to the WHO/revised form of European-American Classification of lymphoid neoplasm classification for lymphoid malignancies because of aggressive clinical behavior [14]. T-PLL represents approximately 2% of all mature lymphocytic leukemia in adults and 3% of T-cell malignancies overall [1, 15].
Concerning the immunophenotype of the present case, it fits into the known picture of T-PLL. Immunphenotype of T-prolymphocytes is consistent with that of a mature post-thymic T-cell. As such it was negative for CD1a and TdT, but positive for pan T-cell markers, such as CD2, CD3, CD5, and CD7. CD7 is expressed with stronger intensity than seen in normal T-cells and other mature T-cell malignancies, but at levels comparable to those seen in T-acute lymphocytic leukemia (T-ALL) [16]. Most of the cases (65%) have CD4+/CD8- cells, 21% of cases are CD4+/CD8+, and 13% of cases are CD4-/CD8+ [17]. Surface expression of CD3 and TCR-α/ß may not be detected in up to 20% of cases, but their expression is always seen in the cytoplasm [16].
As already mentioned T-PLL is a rare entity; here we describe a childhood T-PLL case with de novo del(9)(p13) leading to monoallelic deletion of the tumor suppressor gene CDKN2A. To the best of our knowledge, the present case is unique in two ways: it is the only one ever seen with this kind of aberration in a childhood case, and it is the only one with a del(9)(p13) as the sole aberration (for comparison see [18]).
The most frequent chromosomal abnormalities in T-PLL include 14q11.2-aberrations involving TCR-region, and 11q-rearrangements leading to deletions of ATM or MLL genes. Other yet reported abnormalities involve various chromosomal regions like 5, 6, 8, 9p, 7q, 12p, 13q, 14q, 16q, 17, 21 and 22 [8–11, 19, 20].
T-PLL occurs in enhanced frequencies in ataxia telangiectasia (AT) families [21]. This observation supports possible association between ATM mutations and T-PLL. Accordingly Friedenson [22] described ATM mutations in 44-66% of T-PLL patients.
CDKN2A and CDKN2B are tumor suppressor genes located in 9p21. They belong to the family of inhibitors of cyclin-dependent kinases. CDKN2A gene encodes for the two proteins p14ARF and p16INK4a, and the CDKN2B gene for p15 INK4b protein; all three are key regulators of G1 phase cell-cycle arrest and senescence [23]. It has recently been observed that these genes are inactivated in a wide range of human cancers by epigenetic mechanisms [23, 24],
Loss of heterozygosity of chromosome arm 9p, including the CDKN2A locus, is one of the most frequent genetic events in childhood ALL, suggesting inactivation of the second allele or, possibly, haploinsufficiency [25]. Haploinsufficiency of a tumor suppressor gene (e.g. CDKN2A), has been shown to be adequate to promote tumor progression [26]. Homozygous deletion of CDKN2A has been suggested as the dominant mechanism of its inactivation in leukemogenesis [27].
FOXA1 is a key transcription (TF) factor for CDKN2A expression [24, 28] and a member of fork head gene family. TFs have remarkable pioneering activities to open chromatin for its subsequent cooperation with DNA-interacting proteins, especially important during embryogenesis and organ development [29, 30].
Our case fulfills the clinical, cytogenetic, molecular cytogenetic and flow cytometry criteria for a T-PLL case. It is important to recognize T-PLL because it has a more aggressive clinical course than mature T-cell leukemia. As del(9)(p13) was the only cytogenetic aberration in this aggressive childhood T-PLL case alterations of CDKN2A gene may be an early or even the primary event in T-PLL formation.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
References
Maljaei SH, Brito-Babapulle V, Hiorns LR, Catovsky D: Abnormalities of chromosomes 8, 11, 14, and X in T-prolymphocytic leukemia studied by fluorescence in situ hybridization. Cancer Genet Cytogenet 1998, 103: 110–116. 10.1016/S0165-4608(97)00410-X
Oliveira F, Tone LG, Simones BP, Rego EM, Marinato AF, Jacomo RH, Falcao RP: Translocations t(X;14)(q28;q11) and t(Y;14)(q12;q11) in T-cell prolymphocytic leukemia. Int J Lab Hematol 2008, 31: 453–456.
Dearden C: How I treat prolymphocytic leukemia. Blood 2012, 120: 538–551. 10.1182/blood-2012-01-380139
Matutes E, Brito-Babapulle V, Yullie MR, Catovsky D: Prolymphocytic leukemia of B- and T-cell types. In Chronic Lymphoid Leukemias. Edited by: Cheson BD. New York, Basel: Marcel Dekker, Inc; 2001:525–541.
Catovsky D, Ralfkiaer E, Muller-Hermelink HK: T-cell prolymphocytic leukaemia. In World Health Organization Classification of Tumours: Tumours of Haematopoietic and Lymphoid Tissues. Edited by: Jaffe ES, Harris NL, Stein H, Vardiman JW. Lyon: IARC Press; 2001:195–196.
Pawson R, Schulz TF, Matutes E, Catovsky D: The human T-cell lymphotropic viruses type 1/11 are not involved in T Prolymphocytic leukaemia and large granular lymphocytic leukaemia. Leukemia 1997, 11: 1305–1311. 10.1038/sj.leu.2400725
Brito-Bapapelle V, Pomfret M, Matutes E, Catovsky D: Cytogenetic studies on prolymphocytic leukemia. II. T-cell pro-lymphocytic leukemia. Blood 1987, 70: 926–931.
Durig J, Bug S, Klein-Hitpass L, Boes T, Jons T, Subero-Martin JI, Harder L, Baudis M, Duhrsen U, Siebert R: Combined single nucleotide polymorphism-based genomics mapping and global gene expression profiling identifies novel chromosomal imbalances, mechanisms and candidate genes important in the pathogenesis of T-Cell prolymphocytic leukemia with inv(14)(q11q32). Leukemia 2007, 21: 2153–2163. 10.1038/sj.leu.2404877
Nowak D, Stern MH, Kawamata N, Akagi T, Dyer M, Hofmann WK, Ogawa S: Molecular allelokaryotyping of T-cell prolymphocytic leukemia cells with high density single nucleotide polymorphism arrays identifies novel common genomic lesions and acquired uniparental disomy. Haematologica 2009, 94: 518–527. 10.3324/haematol.2008.001347
Das D, Pathan S, Joneja M, Al-Musawi F, John B, Mirza K: T-Cell Prolymphocytic Leukemia (T-PLL) with overlapping cytomorphological features with T-CLL and T-ALL: a case initially diagnosed by fineneedle aspiration cytology and lmmunocytochemistry. Diagn Cytopathol 2013,41(4):360–365. 10.1002/dc.21781
Soulier J, Pierron G, Vecchione D, Garand R, Brizard F, Sigaux F, Stern MH, Aurias A: A complex pattern of recurrent chromosomal losses and gains in T-cell prolymphocytic leukemia. Genes Chromosomes Cancer 2001,31(3):248–254. 10.1002/gcc.1141
AL-achkar W, Wafa A, Nweder MS: A complex translocation t(5;9;22) in Philadelphia cells involving the short arm of chromosome 5 in a case of chronic myelogenous leukemia. J Exp Clin Cancer Res 2007, 26: 411–415.
Liehr T, Heller A, Starke H, Rubtsov N, Trifonov V, Mrasek K, Weise A, Kuechler A, Claussen U: Microdissection based high resolution multicolor banding for all 24 human chromosomes. Int J Mol Med 2002, 9: 335–339.
Costa D, Queralt R, Aymerich M, Carrio A, Rozman M, Vallespi T, Colomer D, Nomdedeu B, Montserrat E, Campo E: High levels of chromosomal imbalances in typical and small-cell variants of T-Cell prolymphocytic leukemia. Cancer Genet Cytogenet 2003,147(1):36–43. 10.1016/S0165-4608(03)00161-4
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter KC, Grogan TM, Isaacson PG, Knowles DM, Mason DY, Muller-Hermelink HK, Pileri SA, Piris MA, Ralfkiaer E, Warnke RA: A revised European-American classification of lymphoid neoplasms: a proposal from the international lymphoma study group. Blood 1994, 84: 1361–1392.
Bartlett NL, Longo DL: T-small lymphocyte disorders. Semin Hematol 1999, 36: 164–170.
Ginaldi L, Matutes E, Farahat N, De Martinis M, Morilla R, Catovsky D: Differential expression of CD3 and CD7 in T-cell malignancies: a quantitative study by flow cytometry. Br J Haematol 1996, 93: 921–927. 10.1046/j.1365-2141.1996.d01-1720.x
Matutes E, Brito-Babapulle V, Swansbury J, Ellis J, Morilla R, Dearden C, Sempere A, Catovsky D: Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood 1991, 78: 3269–3274.
Mitelman F, Johansson B, Mertens F: Mitelman Database of Chromosome Aberrations in Cancer. 2014. " http://cgap.nci.nih.gov/Chromosomes/Mitelman
Salomon-Nguyen F, Brizard F, Le Coniat M, Radford I, Berger R, Brizard A: Abnormalities of the short arm of chromosome 12 in T cell prolymphocytic leukaemia. Leukemia 1998, 12: 972–975. 10.1038/sj.leu.2401034
Stankovic T, Kidd A, Sutcliffe A, McGuire G, Robinson P, Weber P, Bedenham T, Bradwell A, Easton D, Lennox G, Haites N, Byrd P, Taylor A: ATM mutations and pheotypes in ataxia-telangiectasia families in the British isles: expression of mutant ATM and the risk of leukemia, lymphoma and breast cancer. Am J Hum Genet 1998, 62: 334–335. 10.1086/301706
Friedenson B: The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer 2007, 7: 152. 10.1186/1471-2407-7-152
Collado M, Blasco MA, Serrano M: Cellular senescence in cancer and aging. Cell 2007, 130: 223–233. 10.1016/j.cell.2007.07.003
Zhang Y, Tong T: FOXA1 antagonizes EZH2-mediated CDKN2A repression in carcinogenesis. Biochem Biophys Res Commun 2014, 453: 172–178. 10.1016/j.bbrc.2014.09.092
Irving JA, Bloodworth L, Bown NP, Case MC, Hogarth LA, Hall AG: Loss of heterozygosity in childhood acute lymphoblastic leukemia detected by genome-wide microarray single nucleotide polymorphism analysis. Cancer Res 2005, 65: 3053–3058.
Chapman EJ, Harnden P, Chambers P, Johnston C, Knowles MA: Comprehensive analysis of CDKN2A status in microdissected urothelial cell carcinoma reveals potential haploinsufficiency, a high frequency of homozygous co-deletion and associations with clinical phenotype. Clin Cancer Res 2005, 11: 5740–5747. 10.1158/1078-0432.CCR-05-0411
Ohnishi H, Kawamura M, Ida K, Sheng XM, Hanada R, Nobori T, Yamamori S, Hayashi Y: Homozygous deletions of p16/MTS1 gene are frequent but mutations are infrequent in childhood T-cell acute lymphoblastic leukemia. Blood 1995, 86: 1269–1275.
Li Q, Zhang Y, Fu J, Han L, Xue L, Lv C, Wang P, Li G, Tong T: FOXA1 mediates p16(INK4a) activation during cellular senescence. EMBO J 2013, 32: 858–873. 10.1038/emboj.2013.35
Jozwik KM, Carroll JS: Pioneer factors in hormone-dependent cancers. Nat Rev Cancer 2012, 12: 381–385. 10.1038/nrc3263
Katoh M, Igarashi M, Fukuda H, Nakagama H, Katoh M: Cancer genetics andgenomics of human FOX family genes. Cancer Lett 2013, 328: 198–206. 10.1016/j.canlet.2012.09.017
Acknowledgments
We would like to thank Prof. Ibrahim Othman, the Director General of Atomic Energy Commission of Syria (AECS), and Dr. Nizar MirAli, Head of Molecular Biology and Biotechnology Department, for their support. This work was supported by the AECS, in parts by the DAAD and KAAD.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AW provided the case and/or did primary cytogenetic and main part of the FISH-tests; AA did the flow cytometry analysis; TL, MAKO and AE did detailed FISH studies. WA supervised the cytogenetic analysis as Director of the MBBD HGD. AW and TL drafted the paper and all authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wafa, A., Aljapawe, A., Othman, M.A. et al. Do novo del(9)(p13) in a childhood T-cell prolymphocytic leukemia as sole abnormality. Exp Hematol Oncol 3, 28 (2014). https://doi.org/10.1186/2162-3619-3-28
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2162-3619-3-28