
Abstract
Background
Dyschromatosis symmetrica hereditaria (DSH) is an autosomal dominantly inherited skin disease associated with mutations of ADAR1, the gene that encodes a double-stranded RNA-specific adenosine deaminase. The purpose of this study was to investigate the potential mutations in ADAR1 in seven Chinese families with DSH.
Methods
All the coding exons including adjacent intronic as well as 5′ and 3′ untranslated region (UTR) of ADAR1 were screened by direct sequencing. Moreover, quantitative reverse-transcription polymerase chain (qRT-PCR) and Western blot were applied to determine the pathogenic effects associated with the mutations.
Results
Molecular genetic investigations detected five novel mutations (c.556C > T, c.3001C > T, c.1936_1937insTG, c.1065_1068delGACA and c.1601G > A resulting in p.Gln186X, p.Arg1001Cys, p.Phe646LeufsX16, p.Asp357ArgfsX47 and p.Gly471AspfsX30 protein changes, respectively) as well as two previously reported (c.2744C > T and c.3463C > T causing p.Ser915Phe and p.Arg1155Trp protein changes, respectively). Among them, we found that the substitution c.1601G > A at the last nucleotide of exon 2 compromised the recognition of the splice donor site of intron 2, inducing an aberrant transcript with 190-bp deletion in exon 2 and causing an approximately 50% reduction of ADAR1 mRNA level in affected individual. In addition, consistent with the predicted results, the expression patterns of other novel mutations were detected by Western blot.
Conclusion
We identified five novel and two recurrent mutations of the ADAR1 gene in seven Chinese families with DSH and investigated potential effects of the novel mutations in this study. Our study expands the database on mutations of ADAR1 and for the first time, demonstrates the importance of exonic nucleotides at exon-intron junctions for ADAR1 splicing.
Similar content being viewed by others


Background
Dyschromatosis symmetrica hereditaria (DSH, MIM #127400), also called symmetric dyschromatosis of the extremities, is characterized by hyperpigmented and hypopigmented macules on the face and dorsal aspects of the extremities. The skin lesions usually appear from infancy or early childhood, commonly stop spreading before adolescence and last for life [1]. Generally showing an autosomal dominant pattern of inheritance with high penetrance, DSH has been reported in various parts of the world, but predominantly in Japan and China [2, 3]. RNA-specific adenosine deaminase 1 (ADAR1) gene, located on chromosome 1q21.3, was identified to be responsible for DSH [2].
ADAR1, also called DSRAD (double-stranded RNA-specific adenosine deaminase), spans 30 kb and contains 15 exons [4]. It encodes RNA-specific adenosine deaminase 1 composed of 1226 amino acid residues, with a calculated molecular mass of 139 kDa. Mammalian ADAR1 shows ubiquitous expression and is supposed to be one of the housekeeping genes. The enzyme catalyses the deamination of adenosine (A) to isosine (I) in double-stranded RNA substrates, which results in the creation of alternative splicing sites or alternations of codon and thus lead to functional changes in proteins [4].
In this study, we investigated seven families with DSH in Chinese Han population and found five novel and two recurrent mutations. We applied in vivo mRNA assays and in vitro expression assays to investigate the effects of these novel mutations and discussed their potential pathogenic mechanisms.
Methods
Sample collection and ethics statement
Seven unrelated multi-generation DSH families exhibited an autosomal dominant inherence patterns (Figure 1). All affected individuals had typical macules on the dorsal aspects of the hands and feet. These lesions, with irregular shapes and sizes, appeared from early childhood, generally ranging from 3 to 10 years (Figure 2A, 2B). The skin lesions became more pronounced after sun exposure. No patient had extracutaneous symptoms. In family 3, the affected individuals had hypopigmented and hyperpigmented macules around ankle and in family 4 and 5, small freckle-like pigmented macules disturbed on the faces of affected individuals. DSH was diagnosed by experienced dermatologists based on the typical manifestations. The study was approved by the Research Ethics Committee of China Medical University and all the participants gave written informed consent.

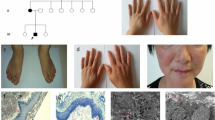
Family Pedigrees in this study. Seven multi-generation families with DSH illustrated autosomal dominant inherence pattern with 29 affected individuals. (A-G) Pedigrees of the studied DSH family 1-7. Affected family members are presented by black symbols. The probands are arrowed.

Typical skin lesions and mutations of the ADAR1 gene. (A, B) A mixture of hyperpigmented and hypopigmented macules distributed on the dorsal aspects of hands and feet of the proband of family 4 (III1); (C-G, I-J) Direct DNA sequencing showing the five novel mutations in the family 1–5 (C-G) and the two recurrent mutations in family 6 and 7 (I, J), respectively; (H) ClustaIW multiple alignment of various species around residue 1001 of human ADAR1.
Extraction of genomic DNA and PCR
Genomic DNA samples were extracted from peripheral blood using the Universal Genomic DNA Extraction Kit (TaKaRa, Dalian, China). The primers that flanked all 15 coding exons and intron-exon boundaries of the ADAR1 were designed (Table 1). Using Taq DNA polymerase, all fragments were amplified as follows: 1 min at 94°C, 30 cycles (30 sec at 94°C, 30 sec at 58°C, 1 min at 72°C) and 5 min at 72°C. After the amplification, the products were purified on agarose gels and subjected to sequencing.
RNA splicing analysis
Fresh peripheral blood samples were collected from the affected individual and 3 healthy controls. Total RNA was extracted using the TRIzol reagent according to the manufacturer’s protocols. After quantification, 1 μg of RNA sample was reversely transcribed into cDNA using an AMV reverse transcriptase kit from TaKaRa Company according to the manufacturer’s instructions. To analyze the expression of the mutant ADAR1 allele, RT-PCR was performed with primers E2F/E3R (Table 2) [5]. The cDNA fragments were then subcloned into pMD 18-T vector and subjected with sequencing. Moreover, RT-PCR was carried out with primer pairs E1F/E3R and E1F/E4R to determine whether there was exon skipping in affected individual.
Real-time quantitative RT-PCR
To quantify the relative mRNA expression of ADAR1 in affected individual, real-time quantitative RT-PCR was carried out using primer pairs E9-10 F/E10-11R (Table 2). The amplicon of ADAR1 was 140 bp in length, normalized by a 120-bp glyceraldehyde-3-phosphate dehydrogenase (GAPDH) fragment. Reactions were performed with SYBR Green PCR Master Mix (TaKaRa) [5]. Three replicates of each reaction were carried out and the 2-ΔΔCT method was used to calculate relative changes in ADAR1 expression. Melting curve analysis confirmed that only the expected product was amplified [5].
In vitroexpression assay
The wild-type coding sequence of ADAR1 was amplified from pCMV6-ADAR1 (Origene) by PCR and subcloned into pFLAG-CMV-2 (Sigma-Aldrich, St. Louis, MO, USA) at its Not I restriction site by standard procedure. To generate mutant plasmids, site-directed mutagenesis using PCR-based method was applied. Concisely, primers were designed for mutagenesis and fragments amplified by PCR were subcloned into pFLAG-CMV-2 vector using a standard subcloning protocol, respectively. After confirmed by sequencing, all plasmids were transfected into human embryonic kidney (HEK-293) cells, respectively [6]. The whole cellular lysates were extracted at 48 h post-transfection and immunoblotting was performed as reported previously [7].
Results
Mutation analysis
Among 7 Chinese families with DSH, we found 5 novel mutations in ADAR1 (Table 3). The spectrum of mutations included one nonsense mutation, two missense mutations, one small insertion and one small deletion, all of which were heterozygous (Figure 2C-G). Searching the Human Gene Mutation Database (HGMD) and PubMed, we did not find the reports about these variants. All the mutations segregated affected individuals from 100 normal Chinese subjects, suggesting that neither of them was a polymorphism (For point mutations c.556C > T, c.3001C > T and c. 1601G > A, PCR-RFLP was applied with restriction endonuclease Bsp1407I, Bgl II and Sca I while for small insertion/deletion c.1936_1937insTG and c.1065_1068delGACA, SDS-PAGE was applied after PCR amplification, respectively).
In the family 1, the nonsense mutation was detected in exon 2 of ADAR1 and defined as c.556C > T (Figure 2C). This mutation was suggested leading to a truncated ADAR1 protein lacking 1040 amino acid residues. In the family 2, the missense mutation in exon 11, c.3001C > T, was identified by sequencing (Figure 2D). This mutation replaced arginine residue with cysteine, which was shown as a highly conserved residue within the functional domain of the ADAR1 (Figure 2H).Moreover, nucleotides TG were found to insert between c.1936 and c.1937 in the family 3 and this 2-bp small insertion induced a frameshift from codon 646 and expected to produce a premature termination codon (PTC) at codon 661 (Figure 2E). In addition, one 4-bp small deletion, c.1065_1068delGACA, was detected in the family 4. This mutation might shift the reading frame and generate a PTC at codon 403 (Figure 2F).
Furthermore, direct sequencing of ADAR1 revealed a missense substitution, c. 1601G > A, at the end of exon 2 in the family 5 (Figure 2G). Since this G nucleotide belongs to the conserved exon-intron junction, we performed splicing predictions with web-based resources. Analysis with SpliceView (http://zeus2.itb.cnr.it/~webgene/wwwspliceview_ex.html) and Human Splicing Finder (http://www.umd.be/HSF/) indicated that the recognition of such splice donor site was potentially attenuated in the presence of c.1601G > A, supporting that this mutation might play a role in splicing process.
Moreover, two recurrent mutations, c.2744C > T and c.3463C > T, were detected in family 6 and 7, respectively (Figure 2I, 2J). Both of them were missense mutations, suggesting the replacements of serine 915 by phenylalanine and arginine 1155 by tryptophan, respectively [8, 9].
Molecular analysis of expression patterns of the novel mutations
Furthermore, we attempted to determine the exact consequence of the c.1601G > A mutation in splicing with in vivo mRNA assays. RT-PCR analysis revealed differential splicing patterns between affected individual and healthy control. While predicted 780-bp PCR fragments of normal splicing were shown and confirmed by sequencing analysis both in the affected and healthy individual, an additional about 600-bp PCR product was shown in the presence of mutant and subsequently subcloned into pMD18-T (Figure 3A, 3B). Direct sequencing of TA clones revealed that this mutation caused a splicing error in ADAR1. With functional loss of the native intron 2 donor site, this mutation activated the cryptic splicing donor site GT lying upstream of the normal splicing donor site in exon 2 and thus caused a 190-bp deletion in mRNA sequence (Figure 3C). Hence, this c.1601G > A mutation was expected to shift the reading frame from codon 471 with 29 aberrant amino acids and generate premature termination at codon 500. Moreover, no additional band was detected in RT-PCR assays using the primer pairs E1F/E3R and E1F/E4R, excluding the possibility of exon skipping in affected individual due to this mutation (data not shown).

Aberrant splicing pattern of ADAR1 caused by c.1601G > A mutation. (A) Locations of primers used in RT-PCR; (B) RT-PCR analysis revealed a transcript abnormality in ADAR1; (C) cDNA sequencing and predicted amino acid sequence of the aberrant (left) and normal (right) transcripts of ADAR1; (D) The relative quantities of ADAR1 mRNA level derived from real-time quantitative RT-PCR.
Upon the worthwhile observation that the aberrant transcript was present at much reduced level in the affected individual, mRNA quantification was evaluated with real-time quantitative RT-PCR. By validating with the 2-ΔΔCT method, the relative quantities were 0.525, 0.639 and 0.467 (mean ± SD, 0.54 ± 0.09) for individual carrying with c.1601G > A mutation compared with healthy subject, indicating an approximately 50% reduction of the ADAR1 mRNA expression in affected individual.
Since fresh blood samples from affected individuals of family 1 to 4 were not available, we continued to investigate the expression patterns of these mutants with in vitro expression assays. Briefly, immunoblottings were performed with lysates of HEK-293 cells overexpressing wild-type or mutants of ADAR1. Consistent with the predicted results, the truncated ADAR1 proteins were detected as 21 kDa in cells overexpressing c.556C > T mutation, 75 kDa in cells overexpressing c.1936_1937insTG mutation and 46 kDa in cells overexpressing c.1065_1068delGACA mutation, respectively (Figure 4). Moreover, as a missense mutation, c.3001C > T (p.Arg1001Cys) mutant demonstrated the same molecular weight as the wild-type ADAR1 protein.

In vitro expression assays. Total cellular lysates of HEK-293 cells transfected with FLAG-tagged wild-type or mutant plasmids were immunoblotted with FLAG antibody. beta-tubulin was used as an internal control.
Discussion
A-to-I RNA editing is one of the post-transcriptional modifications widespread in mammals, which modifies adenosine (A) base in pre-mRNA to yield inosine (I) [10]. Since I is recognized as guanosine during translation, A-to-I RNA editing in coding sequences probably leads to amino acid changes which often entails changes in protein function [10]. Moreover, editing A-to-I modification might alter splice patterns as well as RNA structures and stabilities [1, 10, 11]. A-to-I editing is catalyzed by ADARs in vertebrate and till now, three ADAR family members, ADAR 1–3, have been identified. Among them, ADAR1 was confirmed to be responsible for DSH in 2003 [1]. To date, the mechanisms by which ADAR1 mutations cause DSH still remain unclear [10, 11].
Human ADAR1 contains, from N- to C-terminals, two adenosine deaminase Z-alpha (Zα) domains, three double-stranded RNA binding motifs (DSRMs) and a tRNA-specific and double-stranded RNA adenosine deaminase (ADEAMc) domain, which encodes by exon 2, 2–7 and 9–15 of ADAR1, respectively [5]. Although ADEAMc is the catalyze domain for A-to-I modification, Zα and DSRM domains are also essential for the activity of ADAR1 [5, 11–14].
In this study, we reported five novel mutations and two recurrent mutations. Among them, the nonsense mutation c.556C > T (p.Gln186X) and small deletion c.1065_1068delGACA (p.Asp357ArgfsX47) are expected to cause ADAR1 truncations lacking the entire three DSRMs and ADEAMc domain while c.1936_1937insTG (p.Phe646LeufsX16), the small insertion identified in our study, supposes to change the reading frame from the second DSRM and induces truncated ADAR1 protein without the last DSRM and ADEAMc domain (Figure 5). In addition, Figure 4 suggested that the mutations c.556C > T and c.1065_1068delGACA might reduce the expression of mutant proteins and we speculated that the nonsense-mediated mRNA decay (NMD) might be one of the possibilities. For the missense mutations c.3001C > T (p.Arg1001Cys), c.2744C > T (p.Ser915Phe) and c.3463C > T (p.Arg1155Trp), the transitions of nucleotides change codons in the highly conserved region of ADEAMc domain, which possibly alter the activity of ADAR1 directly, or by disturbing the formation of wild-type ADAR1 homodimers [9]. In family 5, the aberrant splicing decreased approximately half ADAR1 mRNA level, potentially caused by NMD, supporting that haploinsufficiency of ADAR1 produces DSH [5, 9]. In addition, we compared the clinical features with the mutations indentified in all the families but failed to find any clear relationship between phenotypes and genotypes as other groups have reported [2, 13].

Novel mutations of ADAR1 identified in this study. The functional domains (Zα, DSRM, ADEAMc) were indicated in the figure. Zα: Z-DNA binding domains; DSRM: double stranded RNA binding domains; ADEAMc: deaminase domain.
Till now, about 145 ADAR1 mutations have been reported in DSH patients, which consist of 80 missense and nonsense mutations, 11 splicing mutations, 37 small deletions, 14 small insertions, 2 small indels and 1 gross insertion [15–29]. So far, most known missense mutations (49 in 57, 86%) are located within the ADEAMc domain encompassing amino acids 839–1222 [3]. The missense mutations identified in our study are also located in this domain. These data support that the ADEAMc domain is critical for this enzyme and indicates that this region might be hot spot for mutation within ADAR1[10, 16, 17]. To date, 23 nonsense mutations have been reported in ADAR1 and they appear to distribute throughout the gene without apparent unifying connection. Till now, 11 splicing mutations have been identified, all of which situate in ADAR1 introns. Among them, only 4 mutations were further studied with cDNA sequence analysis and detected the aberrant splicing products [20, 21, 27], in which 3 splicing mutations led to skipping one or more exons near to the mutations [27], and the other one was a in-frame mutation which generated a 4-amino acid deletion in ADAR1 protein [21]. To our knowledge, the c.1601G > A mutation identified in our study is the first report about the exonic splicing mutation in ADAR1.
Although ADAR1 expressed ubiquitously in mammalian tissues and playing various roles such as viral host defense, embryonic development, tumor growth and miRNA processing [22–25], only a few its target genes including the neurotransmitter receptors in the central nervous system and the hepatitis virus antigen in the liver are identified [9]. Therefore, mutations in the ADAR1 might associate with neurological dysfunction such as dystonia and mental deterioration [26]. However, like most affected individuals reported previously, patients in our study only show pigmentary abnormality in skin without obvious dysfunction in extracutaneous tissues. Recently, mutations identified in ADAR1 were reported to be relevant with autoimmune disorder, hair color change and dental abnormality [15, 27]. Thus, further investigation on the mechanisms by which ADAR1 regulates its target genes in different tissues will facilitate the understanding of ADAR1 functions.
Driven by different promoter, ADAR1 has been characterized into two isoforms, an interferon-inducible 150 kDa protein (p150) and a constitutively-expressed 110 kDa protein (p110), in mammalian cells [1, 2]. The p150 isoform, translated from the methionine initiation codon, is the full length protein mainly distributed in cytosol while the p110 isoform initially translates from the AUG at codon 296 and locates in the nucleus [28]. Till now, including p.Gln186X identified in our study, four mutations of ADAR1 have been reported at the 5′ side of codon 296 [2, 29]. However, the distinct roles of the isoforms are obscure. Recently, Zhang et al. reported that the small deletion p.R91fsX123 could eliminate the expression of the p150 transcript through NMD, while did not significantly alter the expression of p110 in patients of DSH, indicating that p150 isoform of ADAR1 might be the determinant of DSH [29].
Conclusion
In conclusion, we identified five novel and two recurrent mutations of the ADAR1 gene in seven Chinese families with DSH and investigated potential effects of the novel mutations in this study. Together with previous related studies, these results might give insight into the still unknown mechanisms leading to DSH. Additionally, our results enrich the ADAR1 mutation database and will contribute further to the understanding of DSH genotype/phenotype correlations as well as to the pathogenesis of this disease.
Abbreviations
- DSH:
-
Dyschromatosis symmetrica herediaria
- ADAR1:
-
RNA-specific adenosine deaminase 1
- PTC:
-
Premature termination codon
- NMD:
-
Nonsense-mediated mRNA decay.
References
Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, Tomita Y: Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 2003, 73: 693-699.
Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Horikawa T, Fujiwara S, Shiko A, Matsunaga K, Aoyama Y, Tosaki-Ichikawa H, Tomita Y: Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria. J Invest Dermatol. 2007, 127: 309-311.
Kondo T, Suzuki T, Mitsuhashi Y, Ito S, Kono M, Komine M, Akita H, Tomita Y: Six novel mutations of the ADAR1 gene in patients with dyschromatosis symmetrica hereditaria: histological observation and comparison of genotypes and clinical phenotypes. J Dermatol. 2008, 35: 395-406.
Hou YX, Chen JJ, Gao M, Zhou FS, Du WH, Shen KJ, Yang S, Zhang XJ: Five novel mutations of RNA-specific adenosine deaminase gene with dyschromatosis symmetrica hereditaria. Acta Derm Venereol. 2007, 87: 18-21.
Liu Q, Jiang L, Liu WL, Kang XJ, Ao Y, Sun M, Luo Y, Song Y, Lo WH, Zhang X: Two novel mutations and evidence for haploinsufficiency of the ADAR gene in dyschromatosis symmetrica hereditaria. Br J Dermatol. 2006, 154: 636-642.
Gao JL, Liu Q, Liu X, Ji CY, Qu SQ, Wang SS, Luo Y: Cyclin G2 suppresses estrogen-mediated osteogenesis through inhibition of Wnt/β-catenin signaling. PLoS One. 2014, 9 (3): e89884-
Liu Q, Liu X, Gao J, Shi X, Hu X, Wang SS, Luo Y: Overexpression of DOC-1R inhibits cell cycle G1/S transition by repressing CDK2 expression and activation. Int J Biol Sci. 2013, 9 (6): 541-549.
Li CR, Li M, Ma HJ, Luo D, Yang LJ, Wang DG, Zhu XH, Yue XZ, Chen WQ, Zhu XY: A new arginine substitution mutation of DSRAD gene in a Chinese family with dyschromatosis symmetrica hereditaria. J Dermatol Sci. 2005, 37 (2): 95-99.
Murata I, Hozumi Y, Kawaguchi M, Katagiri Y, Yasumoto S, Kubo Y, Fujimoto W, Horikawa T, Kondo T, Kono M, Tomita Y, Suzuki T: Four novel mutations of the ADAR1 gene in dyschromatosis symmetrica hereditaria. J Dermatol Sci. 2009, 53: 76-77.
Wang Q: RNA editing catalyzed by ADAR1 and its function in mammalian cells. Biochemistry (Mosc). 2011, 76: 900-911.
Bass BL: RNA editing by adenosine deaminases that act on RNA Annu. Rev Biochem. 2002, 71: 817-846.
Brooks PJ: The case for 8, 5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007, 145: 1407-1417.
Liu Q, Liu W, Jiang L, Sun M, Ao Y, Zhao X, Song Y, Luo Y, Lo WH, Zhang X: Novel mutations of the RNA-specific adenosine deaminase gene (DSRAD) in Chinese families with dyschromatosis symmetrica hereditaria. J Invest Dermatol. 2004, 122: 896-899.
Cho DS, Yang W, Lee JT, Shiekhatter R, Murray JM, Nishikura K: Requirement of dimerization for RNA editing activity of adenosine deaminase acting on RNA. J Biol Chem. 2003, 278: 17093-17102.
Kantaputra PN, Chinadet W, Ohazama A, Kono M: Dyschromatosis symmetrica hereditaria with long hair on the forearms, hypo/hyperpigmented hair, and dental anomalies: report of a novel ADAR1 mutation. Am J Med Genet A. 2012, 158A (9): 2258-2265.
Murata I, Hayashi M, Hozumi Y, Fujii K, Mitsuhashi Y, Oiso N, Fukai K, Kuroki N, Mori Y, Utani A, Tomita Y, Fujita Y, Suzuki T: Mutation analyses of patients with dyschromatosis symmetrica hereditaria: five novel mutations of the ADAR1 gene. J Dermatol Sci. 2010, 58: 218-220.
Wang XP, Liu Y, Wang JM, Xiao SX: Two novel splice site mutations of the ADAR1 gene in Chinese families with dyschromatosis symmetrica hereditaria. J Dermatol. 2010, 37: 1051-1052.
Li M, Jin C, Yang L, Lai M, Yao Z: A novel complex insertion-deletion mutation in ADAR1 gene in a Chinese family with dyschromatosis symmetrica hereditaria. J Eur Acad Dermatol Venereol. 2011, 25: 743-746.
Wang XP, Wang WJ, Wang JM, Liu Y, Xiao SX: Four novel and two recurrent mutations of the ADAR1 gene in Chinese patients with dyschromatosis symmetrica hereditaria. J Dermatol Sci. 2010, 58: 217-218.
Liu H, Fu XA, Yu YX, Yu GQ, Yan XX, Liu HX, Tian HQ, Zhang FR: Identification of two novel splice mutations of the ADAR1 gene in two Chinese families with dyschromatosis symmetrica hereditaria. Clin Exp Dermatol. 2011, 36: 797-799.
Kono M, Akiyama M, Kondo T, Suzuki T, Suganuma M, Wataya-Kaneda M, Lam J, Shibaki A, Tomita Y: Four novel ADAR1 gene mutations in patients with dyschromatosis symmetrica hereditaria. J Dermatol. 2012, 39: 819-821.
Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Lizasa H, Davuluri RV, Nishikura K: ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013, 153 (3): 575-589.
Qiu W, Wang X, Buchanan M, He K, Sharma R, Zhang L, Wang Q, Yu J: ADAR1 is essential for intestinal homeostasis and stem cell maintenance. Cell Death Dis. 2013, 4: e599-
Nemlich Y, Greenberg E, Ortenberg R, Besser MJ, Barshack I, Jacob-Hirch J, Jacoby E, Rivkin L, Eyal E, Prieto VG, Chakravarti N, Duncan LM, Kallenberg DM, Galun E, Bennett DC, Amariglio N, Bar-Eli M, Schachter J, Rechavi G, Markel G: MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J Clin Invest. 2013, 123 (6): 2703-2718.
Nishikura K, Sakurai M, Ariyoshi K, Ota H: Antagonistic and stimulative roles of ADAR1 in RNA silencing. RNA Biol. 2013, 10 (8): 1240-1247.
Tojo K, Sekijima Y, Suzuki T, Suzuki N, Tomita Y, Yoshida K, Hashimoto T, Ikeda S: Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family with ADAR1 mutation. Mov Disord. 2006, 21: 1510-1513.
Rice GI, Kasher PR, Fort GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desquerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaquchi M, Kunar R, Lin JP, Lourenco CM, Male AM, Marques W, et al: Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012, 44: 1243-1248.
Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Fukai K, Takama H, Sato K, Ishikawa O, Abe M, Shimizu H, Kawai M, Horikawa T, Yoshida K, Matsumoto K, Terui T, Tsujioka K, Tomita Y: Mutation analysis of the ADAR1 gene in dyschromatosis symmetrica hereditaria and genetic differentiation from both dyschromatosis universalis hereditaria and acropigmentatio reticularis. J Invest Dermatol. 2005, 124: 1186-1192.
Zhang JY, Chen XD, Zhang Z, Wang HL, Guo L, Liu Y, Zhao XZ, Cao W, Xing QH, Shao FM: The adenosine deaminase acting on RNA 1 p150 isoform is involved in the pathogenesis of dyschromatosis symmetrica hereditaria. Br J Dermatol. 2013, 169 (3): 637-644.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/15/69/prepub
Acknowledgments
We would like to thank all of the family members and control individuals in this study for their interest and cooperation. This work was supported by National Natural Science Foundation of China (No.:81100419 and 81170543) and National Basic Research Program of China (No.:2012CB944600).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
QL participated in the design of this study, performed the statistical analysis and drafted the manuscript. QL, YHW and LHC carried out the molecular genetic study. XSX and QZT performed sequence alignment. ZW, HWM and SFZ diagnosed the patients and collected clinical data. YL conceived of the study, participated in the design and coordination and helped the draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Liu, Q., Wang, Z., Wu, Y. et al. Five novel mutations in the ADAR1 gene associated with dyschromatosis symmetrica hereditaria. BMC Med Genet 15, 69 (2014). https://doi.org/10.1186/1471-2350-15-69
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-15-69