
Abstract
Combined quantum mechanics/molecular mechanics approaches are used to determine the mechanisms of organophosphate hydrolysis in an active site of Pseudomonas diminuta phosphotriesterase. For a substrate with a good leaving group, the reaction proceeds through two elementary stages with low energy barriers, and a gain in energy is observed. With a poor leaving group, only the formation of an unstable reaction intermediate is possible, and hydrolysis is incomplete. A comparison of the resulting reaction mechanisms explains the experimental kinetic data, according to which the enzyme hydrolyzes only substrates with good leaving groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Organophosphorus compounds (OPC), which are triesters of phosphates, are widely used as pesticides, insecticides, and fire retardants. OPCs can accumulate in soil and water and also have toxic effects on plants, animals and humans [1–4]. Phosphotriesterases are enzymes that catalyze hydrolysis of organophosphorus compounds; the one most studied experimentally is phosphotriesterase from Pseudomonas diminuta (Pd-PTE). According to kinetic data [5], Pd-PTE hydrolyzes paraoxon (kcat = 2230 s−1) and dibutyl-4-nitrophenyl phosphate (kcat = 570 s−1) and does not exhibit catalytic activity with such compounds as dibutylphenyl phosphate and triphenyl phosphate. The main difference between these organophosphates is the different structure of their leaving groups: the first two compounds are organophosphates with a good leaving group, the other two have poor leaving groups. We therefore compared the hydrolysis reactions of two organophosphates with the same side substituents but different leaving groups: one with a good (dibutyl-4-nitrophenylphosphate (1)) leaving group and one with a poor (dibutylphenylphosphate (2)) leaving group (Fig. 1).

Structures of substrates: (a) dibutyl 4-nitrophenyl phosphate (1) with a good leaving group and (b) dibutyl phenyl phosphate (2) with a poor leaving group.
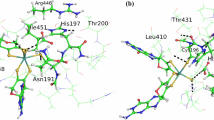
According to X-ray diffraction analysis data [6], the active center of Pd-PTE contains two metal cations at the distance of 3.9 Å (Fig. 2). The native enzyme contains zinc cations. The bridging ligands for the binuclear metal center are the hydroxide anion and the carboxylated residue Lys169, whose oxygen atoms are at a distance of around 2 Å from the metal cations. In addition, zinc cation \({\text{Zn}}_{\alpha }^{{2 + }}\) is coordinated in the active site by two amino acid residues of histidine (His55, His57) and aspartate (Asp301). The second zinc cation \({\text{Zn}}_{\beta }^{{2 + }}\) is also coordinated by two histidine residues (His201, His230). The remaining free space in its coordination sphere is occupied by the substrate.

Left: Structure of phosphotriesterase from the bacterium Pseudomonas diminuta (Pd-PTE). Active site atoms are shown as spheres. Right: substrate in the active site of Pd-PTE. The dotted lines show the coordination bonds of zinc cations, and the dashed line shows the hydrogen bond of the catalytic hydroxide anion and amino acid residue Asp301.
The exact mechanism of hydrolysis of organophosphorus compounds in the active site of Pd-PTE has not been established. It is known that the hydrolysis of organophosphates in the active site of Pd-PTE phosphotriesterase proceeds via a two-stage mechanism. During the first stage, a nucleophilic attack of the phosphorus atom by the hydroxide anion occurs with the formation of an intermediate with pentacoordinated phosphorus [7–18] (Fig. 3). The main differences are associated with the description of the second stage of the reaction, during which the bond between the phosphorus atoms and the oxygen of the P-OLG leaving group is broken to form an enzyme-product complex. At the moment, there are several computational works [8–10, 12, 16, 17] that discuss the mechanism of organophosphate hydrolysis using the example of paraoxon in the active site of Pd-PTE phosphotriesterase. The authors of [9, 17] stated that the formation of an enzyme-product complex is accompanied by the transfer of a proton from the hydroxide anion to aspartic acid Asp301 (Fig. 3b), while other works [8, 10, 12] showed that the proton remains on the oxygen atom of the nucleophilic particle and the protonated form of the organophosphorus product subsequently forms (Fig. 3a). The relative stability of the deprotonated and protonated forms of the product was assessed in [12]. Analysis of the mean force potential profile shows that the protonated state of the hydrolysis product is 5.5 kcal/mol more stable than the deprotonated state.

Mechanisms proposed in the literature for the hydrolysis of OPC by phosphotriesterase Pd-PTE. Both mechanisms assume a nucleophilic attack by a hydroxide anion on the phosphorus atom to form a pentacoordinated intermediate. Further detachment of the leaving group is accompanied by the formation of a P‒OH bond (in mechanism (a)) or a P‒O– bond and transfer of a proton to aspartic acid (in mechanism (b)).
It should be noted that among the above works, only in [9] was the enzyme–product complex stabilized relative to the enzyme–substrate complex. A two-level approach was used in [8, 10, 12] that consisted of obtaining enzyme configurations using such semi-empirical means as AM1 and PM3, and adjusting the obtained energies further using the Kohn–Sham approach with the B3LYP functional. However, it is known that the AM1 and PM3 models do not work well for phosphorus-containing compounds [19, 20] or metalloenzymes [21]. The development of today’s computing resources allows us to use a wider arsenal of DFT techniques to describe a growing number of systems [22].
In this work, we obtain the mechanisms of the reaction for the hydrolysis of organophosphates (1) and (2) in the active site of Pd-PTE phosphotriesterase and compare them using modern approaches.
EXPERIMENTAL
The initial coordinates were taken from crystallographic data for the Pd-PTE dimer (PDB ID: 3CAK) [23], obtained with a resolution of 1.8 Å. Cobalt cations were replaced with zinc cations because zinc is native to the Pd-PTE enzyme. Hydrogen atoms were added to the protein structure using the Reduce program [24] so that the protonated states of amino acid residues corresponded to neutral pH. Special attention was given to the amino acid residues of histidine, since protonation can occur on different nitrogen atoms (Nδ or Nε) or on two at once, depending on the environment. In both monomers of the Pd-PTE enzyme, all histidine residues except His254 were assumed to be neutral. In accordance with X-ray diffraction data, the bridged amino acid residue Lys169 was modified to a carboxylated form. This modification allowed us to stabilize both \({\text{Zn}}_{\alpha }^{{2 + }}\) and \({\text{Zn}}_{\beta }^{{2 + }}\) cations in the active site of phosphotriesterase. Substrates dibutyl 4-nitrophenyl phosphate (1) and dibutyl phenyl phosphate (2) were in the active site of the enzyme, as in the structure of the Pd-PTE enzyme complex with paraoxon [6]. The resulting two enzyme-substrate complexes were solvated, and three chlorine ions were added to neutralize the system. All-atomic models were prepared and their structures were analyzed using the VMD software package [25].
Classical molecular dynamics (MD) modeling with fixed enzyme and substrate atoms was done for relaxation of the solvation shell using the NAMD software package [26]. The trajectory was 1 ns long. An unconstrained MD model 1 ns long was then created to relax the protein environment. The CHARMM36 force field was used to describe the enzyme [27]; CGenFF [28] was used for substrates (1) and (2), and the carboxylated form of Lys169; and TIP3P was used for the water molecule [29]. The size of the system was 68 × 101 × 90 Å3.
Intermediates of the hydrolysis reaction of organophosphates (1) and (2) in the active site of Pd-PTE were obtained next. To relax the structure into such a state, we calculated a molecular dynamics trajectory with potentials of combined quantum and molecular mechanics (QM/MM-MD) with a duration of 5 ps. Quantum mechanics was used to describe the active site of phosphotriestrease (Fig. 2), which includes zinc cations Zn2+, hydroxide anion OH─, side chains of amino acid residues His55, His57, carboxylated Lys169, His201, His230, His254, Asp301, and organophosphate. The calculations of the quantum part used the electron density functional with hybrid functional PBE0 [30], dispersion correction D3 [31], and the 6‑31G** basis. Zinc cations were described using LANL2DZ pseudopotentials [32]. The CHARMM36 force field was used to describe the molecular mechanical part. The Pd-PTE enzyme complex with substrates dibutyl-4-nitrophenylphosphate (1) had a quantum part that contained 127 atoms. With dibutylphenylphosphate (2), it had 125 atoms. The total charge of the quantum subsystem was +2. The interaction between quantum chemical package TeraChem [33] and molecular dynamics program NAMD [26] was allowed by a special interface of the NAMD program [34].
All molecular dynamics calculations were made in the canonical NPT ensemble at p = 1 atm and T = 300 K, which were maintained using a Nosé–Hoover barostat [35] and a Langevin thermostat [36] with an integration step of 1 fs.
A series of sequential optimizations of geometries with a fixed value of the reaction coordinate was performed at regular intervals within the stationary QM/MM technique in order to obtain a cross section of the potential energy surface (PES) along the reaction coordinate. Since the hydrolysis reaction of organophosphates in the active center of phosphotriesterase proceeds in two stages, the distances between phosphorus and oxygen of the nucleophile were chosen as reaction coordinates (P-ONu) and between the phosphorus and oxygen of the leaving group (P-OLG) for the first and second stages, respectively.
We used QM/MM-MD to take intermediates of the hydrolysis reaction of compounds (1) and (2) in the active center of Pd-PTE inside a solvation shell limited by water molecules at a distance of no more than 4 Å from the protein and substrate macromolecule. QM/MM optimization was performed using Tcl ChemShell software [37] with the efficient DL-FIND optimizer [38] and the TURBOMOLE quantum chemistry software package [39]. The quantum subsystem and ways of describing it were chosen as in the previous stage.
RESULTS AND DISCUSSION
Figure 4 shows potential energy profiles along the reaction coordinate obtained by via QM/MM for the hydrolysis reaction of two selected substrates in the Pd-PTE active site. The hydrolysis of dibutyl-4-nitrophenylphosphate (1), an organophosphate with a good leaving group, proceeds with low barriers. The barrier to intermediate formation is 5.2 kcal/mol, and the one to transition from intermediate to product is 1.6 kcal/mol. In addition to low barriers, the efficiency of the reaction is ensured by a drop in the relative energy of each subsequent intermediate. There is no hydrolysis of dibutylphenyl phosphate (2), an organophosphate with a poor leaving group, and it is impossible to localize the energy minimum corresponding to the enzyme-product complex: starting from a value of the reaction coordinate P-OLG equal to 3.2 Å, the relative energy values reach a plateau and total 18–19 kcal/mol. Pd-PTE phosphotriesterase thus exhibits catalytic efficiency only toward an organophosphate with a good leaving group. These results are consistent with kinetic data in [5], which show that the catalytic activity of Pd-PTE for the hydrolysis of dibutyl phenyl phosphate is reduced by 106 times, relative to hydrolysis of dibutyl 4-nitrophenyl phosphate.

Potential energy profile for the hydrolysis reactions of: (a) dibutyl-4-nitrophenyl phosphate (1) and (b) dibutyl phenyl phosphate (2) in the active center of Pd-PTE phosphotriesterase. ES is the enzyme substrate complex, TS1 and TS2 are transitional states, INT is the intermediate product, and EP is the enzyme-product complex.
The established mechanism of the hydrolysis reaction of dibutyl-4-nitrophenylphosphate is shown in Fig. 5. Hydrolysis begins with nucleophilic attack by the hydroxide anion on the phosphorus atom of the organophosphate, resulting in the formation of a pentacoordinated phosphorus intermediate (INT). The P-OLG bond then breaks with the transfer of a proton from the oxygen of the hydroxide anion to the aspartic acid residue (Asp301). This mechanism is consistent with the one in [9, 17]. The kinetic role of Asp301 is consistent with [18], where a comparison of kinetic parameters for the wild-type enzyme and mutant forms showed that replacing charged aspartic acid with uncharged alanine or asparagine lowers the catalytic activity. Based on the experimental data and the established mechanism, we may assume that proton transfer to aspartic acid is an important part of the reaction mechanism for the hydrolysis of organophosphates in the active site of Pd-PTE phosphotriesterase.

Mechanism of the hydrolysis reaction of dibutyl-4-nitrophenyl phosphate in the active site of Pd-PTE phosphotriesterase. ES is the enzyme substrate complex, TS1 and TS2 are transitional states, INT is the intermediate, and EP is the enzyme-product complex.
Figure 6 shows stationary points for the first stage of hydrolysis of dibutylphenyl phosphate. The structures of enzyme-substrate complexes and intermediates for hydrolysis of dibutyl-4-nitrophenylphosphate and dibutylphenylphosphate are similar. In enzyme-substrate complexes, the distance of a nucleophilic attack is 2.6 and 2.7 Å, respectively. The formation of intermediates is accompanied by a reduced distance between the phosphorus and oxygen of the hydroxide anion to 1.7 and 1.8 Å, and the P-OLG distance is 2.0 Å. Considerable differences are observed in the structures of the first transition state: with dibutyl phenyl phosphate, the nucleophile approaches at a shorter distance (1.9 Å) than with dibutyl 4-nitrophenyl phosphate (2.4 Å). Despite this, there is no further breakage of the P‑OLG bond in dibutylphenyl phosphate.

First stage of the hydrolysis reaction of dibutylphenyl phosphate in the active site of Pd-PTE phosphotriesterase. ES is the enzyme substrate complex, TS1 is the transitional state, and INT is the intermediate.
CONCLUSIONS
We constructed the potential energy profiles of the hydrolysis reaction of organophosphates and Pd-PTE phosphotriesterase. For a substrate with a good leaving group, the reaction proceeds in two stages with low energy barriers. Each subsequent minimum on the potential energy surface is stabilized relative to the previous one, ensuring the experimentally observed efficient proceeding of the chemical reaction. With a non-hydrolyzable substrate and a poor leaving group, the potential energy profile is characterized by a destabilized state corresponding to a reaction intermediate. The second stage of the reaction does not occur because breaking of the bond with the leaving group raises the energy by 18–19 kcal/mol relative to the enzyme-substrate complex (reagents).
REFERENCES
P. C. Tsai, N. Fox, A. N. Bigley, et al., Biochemistry 51, 6463 (2012). https://doi.org/10.1021/bi300811t
T. Reemtsma, M. García-López, I. Rodríguez, et al., Trends Anal. Chem. 27, 727 (2008). https://doi.org/10.1016/j.trac.2008.07.002
J. Du, H. Li, S. Xu, et al., Environ. Sci. Pollut. Res. 26, 22126 (2019). https://doi.org/10.1007/s11356-019-05669-y
W. A. Stubbings, E. D. Schreder, M. B. Thomas, et al., Environ. Pollut. 238, 1056 (2018). https://doi.org/10.1016/j.envpol.2018.03.083
D. F. Xiang, A. N. Bigley, Z. Ren, et al., Biochemistry 54, 7539 (2015). https://doi.org/10.1021/acs.biochem.5b01144
J. L. Vanhooke, M. M. Benning, F. M. Raushel, and H. M. Holden, Biochemistry 35, 6020 (1996). https://doi.org/10.1021/bi960325l
J. K. Grimsley, B. Calamini, J. R. Wild, and A. D. Mesecar, Arch. Biochem. Biophys. 442, 169 (2005). https://doi.org/10.1016/j.abb.2005.08.012
X. Zhang, R. Wu, L. Song, et al., J. Comput. Chem. 30, 2388 (2009). https://doi.org/10.1002/jcc.21238
Sh.-L. Chen, W.-H. Fang, and F. Himo, J. Phys. Chem. B 111, 1253 (2007). https://doi.org/10.1021/jp068500n
K.-Y. Wong and J. Gao, Biochemistry 46, 13352 (2007). https://doi.org/10.1021/bi700460c
C. J. Jackson, J.-L. Foo, H.-K. Kim, et al., J. Mol. Biol. 375, 1189 (2008). https://doi.org/10.1016/j.jmb.2007.10.061
V. López-Canut, J. J. Ruiz-Pernía, R. Castillo, et al., Chem.-Eur. J. 18, 9612 (2012). https://doi.org/10.1002/chem.201103615
A. N. Bigley and F. M. Raushel, Biochim. Biophys. Acta 1834, 443 (2013). https://doi.org/10.1016/j.bbapap.2012.04.004
J. Kim, P.-Ch. Tsai, Sh.-L. Chen, et al., Biochemistry 47, 9497 (2008). https://doi.org/10.1021/bi800971v
C. Jackson, H.-K. Kim, P. D. Carr, et al., Biochim. Biophys. Acta 1752, 55 (2005). https://doi.org/10.1016/j.bbapap.2005.06.008
R. P. Bora, M. J. L. Mills, M. P. Frushicheva, and A. Warshel, J. Phys. Chem. B 119, 3434 (2015). https://doi.org/10.1021/jp5124025
Yuzhuang Fu, F. Fan, B. Wang, and Z. Cao, Chem.: Asian J. 17, e202200439 (2022). https://doi.org/10.1002/asia.202200439
S. D. Aubert, Y. Li, and F. M. Raushel, Biochemistry 43, 5707 (2004). https://doi.org/10.1021/bi0497805
K. Nam, Q. Cui, J. Gao, and D. M. York, J. Chem. Theory Comput. 3, 486 (2007). https://doi.org/10.1021/ct6002466
X. Lopez and D. M. York, Theor. Chem. Acc. 109, 149 (2003). https://doi.org/10.1007/s00214-002-0422-2
M. Bräuer, M. Kunert, E. Dinjus, et al., J. Mol. Struct.: THEOCHEM. 505, 289 (2000). https://doi.org/10.1016/S0166-1280(99)00401-7
N. Mardirossian and M. Head-Gordon, Mol. Phys. 115, 2315 (2017). https://doi.org/10.1080/00268976.2017.1333644
J. Kim, P. C. Tsai, S. L. Chen, et al., Biochemistry 47, 9497 (2008). https://doi.org/10.1021/bi800971v
J. M. Word, S. C. Lovell, J. S. Richardson, and D. C. Richardson, J. Mol. Biol. 285, 1735 (1999). https://doi.org/10.1006/jmbi.1998.2401
W. Humphrey, A. Dalke, and K. Schulten, J. Mol. Graph. 14, 33 (1996). https://doi.org/10.1016/0263-7855(96)00018-5
J. C. Phillips, R. Braun, W. Wang, et al., J. Comput. Chem. 26, 1781 (2005). https://doi.org/10.1002/jcc.20289
R. B. Best, X. Zhu, J. Shim, et al., J. Chem. Theory Comput. 8, 3257 (2012). https://doi.org/10.1021/ct300400x
K. Vanommeslaeghe, E. Hatcher, C. Acharya, et al., J. Comput. Chem. 31, 671 (2009). https://doi.org/10.1002/jcc.21367
W. L. Jorgensen, J. Chrasekhar, J. D. Madura, et al., J. Chem. Phys. 79, 926 (1983). https://doi.org/10.1063/1.445869
C. Adamo and V. Barone, J. Chem. Phys. 110, 6158 (1999). https://doi.org/10.1063/1.478522
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, J. Chem. Phys. 132, 154104 (2010). https://doi.org/10.1063/1.3382344
P. J. Hay and W. R. Wadt, J. Chem. Phys. 82, 299 (1985). https://doi.org/10.1063/1.448975
S. Seritan, C. Bannwarth, B. S. Fales, et al., WIREs Comput. Mol. Sci. 11, e1494 (2021). https://doi.org/10.1002/wcms.1494
M. C. R. Melo, R. C. Bernardi, T. Rudack, et al., Nat. Methods 15, 351 (2018). https://doi.org/10.1038/nmeth.4638
G. J. Martyna and M. L. Klein, J. Chem. Phys. 97, 2635 (1992). https://doi.org/10.1063/1.463940
K. Singer and W. Smith, Mol. Phys. 64, 1215 (1988). https://doi.org/10.1080/00268978800100823
Y. Lu, M. R. Farrow, P. Fayon, et al., J. Chem. Theory Comput. 15, 1317 (2019). https://doi.org/10.1021/acs.jctc.8b01036
J. Kästner, J. M. Carr, T. W. Keal, and W. Thiel, J. Phys. Chem. A 113, 11856 (2009). https://doi.org/10.1021/jp9028968
R. Ahlrichs, M. Bar, M. H. Iser, et al., Chem. Phys. Lett. 162, 165 (1989). https://doi.org/10.1016/0009-2614(89)85118-8
ACKNOWLEDGMENTS
Our calculations were made on equipment at Moscow State University’s shared resource center of ultra–high-performance computing resources and the Russian Academy of Sciences’ interdepartmental supercomputer center.
Funding
This work was supported by the Russian Foundation for Basic Research, project no. 21-33-70001.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Translated by Sh. Galyaltdinov
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mulashkina, T.I., Kulakova, A.M., Nemukhin, A.V. et al. Comparison of the Mechanisms of Hydrolysis of Organophosphates with Good and Poor Leaving Group by Phosphotriesterase from Pseudomonas diminuta. Russ. J. Phys. Chem. (2024). https://doi.org/10.1134/S0036024424020146
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1134/S0036024424020146