
Abstract
Inflammatory bowel disease (IBD) is widespread in industrial countries with every 20th citizen being affected. Dysregulation of the epithelial barrier function is considered to play a key role in IBD. Permeability of the intestinal epithelium depends mostly on its self-renewal potential and the condition of intercellular junctions. Mitochondria are involved in regulating various intracellular processes in addition to their energy function. Recent data implicate mitochondria in intestinal epithelial barrier regulation and IBD. Mitochondrial dysfunction is possibly one of the factors that underlie the structural abnormalities of tight junctions and the cytoskeleton in intestinal epithelial cells and decrease the self-renewal capacity of the epithelium. The barrier function of the intestinal epithelium is consequently distorted, and IBD develops. The mechanisms of these processes are still unclear and require further research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Inflammatory bowel disease (IBD) is widespread in industrial countries [1]. Disorders of unknown etiology, including ulcerative colitis (UC) and Crohn’s disease (CD), are defined as IBD. Its clinical symptoms include intense abdominal pain, fever, nausea, vomiting, diarrhea, rectal bleeding, anemia, and substantial weight loss. Current IBD treatment is exclusively symptomatic and involves anti-inflammatory drugs and a special diet. Surgery is performed in severe cases to remove the affected part of the intestine [2].
Several main factors are recognized in the IBD pathogenesis: genetic predisposition, dysregulation of the immune system, a distorted barrier function of the intestinal epithelium, imbalance of the intestinal microbiota, and environmental factors (including diet and medications) [3, 4].
A distorted barrier function of the intestinal epithelium is thought to contribute substantially to IBD [5–7]. The intestinal epithelial barrier consists of the mucus layer, the glycocalyx, and the epithelial lining itself [8]. Nutrient absorption and the barrier function are two main functions of the intestinal epithelium, which acts to prevent pathogenic entry [6]. The barrier function of the intestinal epithelium depends primarily on the state of contact complexes between epithelial cells [9, 10], as well as on the mucus production by goblet cells [11], secretion of antibacterial peptides by Paneth cells [11], and the self-renewal capacity of the epithelium [12].
Tight junctions play a crucial role in regulating the permeability of the intestinal epithelium. A tight junction is a protein complex that consists of several types of transmembrane proteins (occludin, claudin family proteins, Marvel D3, JAM family adhesion molecules, and tricellulin), which are connected through the tight junction proteins ZO-1, ZO-2, ZO-3, cingulin, and symplekin to the submembrane actomyosin contractile ring [9]. Claudins are the main membrane proteins that determine the permeability of tight junctions [13]. Structural alterations of intercellular junctions substantially increase the paracellular permeability, and pathogens can consequently enter the intestinal wall through gaps between neighbor epithelial cells [14]. Cytokines and growth factors are known to regulate the tight junction permeability [9]. The proinflammatory cytokines interferon γ (IFN-γ) and tumor necrosis factor (TNF) cause a reorganization of many tight junction proteins, including ZO-1, occludin, and claudins 1 and 4, to increase the permeability. In contrast, transforming growth factor β (TGF-β) maintains the integrity of tight junctions and facilitates restoration of the epithelium after injury [15, 16]. The data indicate that intestinal inflammatory processes may induce cytokine-dependent disassembly of intercellular junctions in the intestinal epithelium, thus impairing its barrier function and increasing inflammation.
The tight junction structure is distorted in IBD, and the paracellular permeability of the intestinal epithelium is consequently higher. The thickness of the tight junction zone is reduced in UC patients [17]. Upregulated expression of pore-forming claudin 2 and claudins 1, 12, and 18 was observed in UC. Lower expression levels were detected for claudins 4, 5, 7, and 8, which are involved in decreasing the paracellular permeability; ZO-1; and its binding partner occluding. The change distort the processes that play a key role in maintaining the integrity of tight junctions [8, 18, 19]. CD is also characterized by structural alterations in tight junctions. Disorganization of intercellular tight junctions was observed in the terminal region of the ileum in CD patients [20]. Sigmoid samples from patients with acute CD showed lower expression of claudins 3, 5, and 8 and higher expression of claudin 2 [21, 22].
A distorted barrier function may be a primary cause of IBD or a sequel of inflammatory processes in the intestinal wall. The distortion allow bacteria to enter the mucosa and induce or aggravate inflammation [4, 23]. A vicious circle is likely to arise in this situation. It is thought that bacterial antigens overstimulate the immune system of the deep mucosa. The resulting immune response is accompanied by a release of proinflammatory cytokines, such as tumor necrosis factor-like cytokine 1A (TL1A), TNF, interleukin 1 (IL-1), and INF-γ, which can increase the permeability of the intestinal epithelium, in particular, to antigens that further stimulate the immune system. The process leads to chronic inflammation, which is characteristic of IBD patients [4, 23, 24].
Mutations of approximately 200 genes are associated with IBD risk [25]. Some of the genes are involved in regulating the immune response, while others play a role in maintaining the barrier function of the intestinal epithelium. The fact provides additional evidence that the interaction of the epithelium and immune cells is of importance for the IBD pathogenesis.
Published data are accumulating to indicate that mitochondrial dysfunction contributes to the distorted barrier function of the intestinal epithelium in IBD [26–30]. Mitochondria are involved in regulating many metabolic and signaling pathways, including those that play a crucial role in sustaining the barrier function of the intestinal epithelium [16]. The mitochondrial functions that are most important in this context include the energy function, which consists in producing ATP in necessary amounts, and generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which act as mediators in many signaling pathways. In addition, defects in mitochondrial self-renewal may lead to an accumulation of dysfunctional organelles.
This review considers the roles that the above functions play in the distorted barrier function of the intestinal epithelium in IBD.
MITOCHONDRIA AND DYSFUNCTION OF INTESTINAL EPITHELIUM
Distorted functional activity of mitochondria often underlies dysfunction of the intestinal epithelium and, in turn, may arise in consequence of exposure to various factors (infection, dysbiosis, inflammation, ischemia, and surgical intervention) [31]. As mentioned above, an increasing body of evidence implicates oxidative stress [32] and mitochondrial dysfunction in the distorted barrier function of the intestinal epithelium and IBD [26, 33]. Ultrastructural alterations suggestive of lower functional activity of mitochondria are detectable in intestinal epithelial cells of IBD patients before the tight junction structure changes, indicating that mitochondrial dysfunction plays a role in IBD [34, 35].
Producing ATP as an energy source for the cell is a main function of mitochondria. Apart from performing the energy function, mitochondria are involved in synthesizing nucleotides, fatty acids, and heme; generating ROS; and maintaining Ca2+ homeostasis. Various intracellular signaling pathways, such as induction of programmed cell death, also involve mitochondria [36]. Dynamic rearrangements continuously occur in mitochondria and are necessary for both meeting the energy demands of the cell and sustaining mitochondrial self-renewal. Normal mitochondrial homeostasis is due to mitochondrial chaperones, secretion of mitochondrial vesicles, mitophagy, and biogenesis. Quality control is of special importance to mitochondria because these organelles are continuously exposed to damaging factors, especially in various forms of cell stress [37].
A distorted energy function and impaired homeostasis of mitochondria are observed along with oxidative stress in IBD, occurring in intestinal epithelial cells in particular. Below we consider in detail the roles that these processes play in the IBD pathogenesis.
ENERGY STRESS IN IBD
Haberman et al. [38] performed a transcriptome analysis and revealed mitochondrial dysfunction in rectal biopsy samples from IBD patients. In particular, downregulation was observed for all of the 13 genes that code for subunits of electron transport chain (ETC) complexes and occur in mitochondrial DNA (mtDNA), along with distorted mitochondrial respiration and an altered transmembrane potential.
The ATP level and ETC activity were considerably lower in the colonic mucosa of mice with dextran sulfate sodium (DSS)-induced colitis [39, 40]. Lower ATP levels in the intestinal mucosa were observed in both CD patients, which had defects in the functions of respiratory chain complexes III and IV, and UC patients, which showed reduced activities of complexes II, III, and IV [41, 42]. Complex I was additionally affected in UC patients in the active phase of the disease [38]. A polymorphism of the MT-ND4 gene, which codes for a subunit of ETC complex I, was associated with UC risk [43]. Studies with Caco-2 intestinal epithelial cells showed that inhibition of ETC complex III or ATP synthase with antimycin or oligomycin considerably distorts the distribution of the tight junction protein ZO-1 and that inhibition of ETC complex I with rotenone moderately changes ZO-1 expression [44]. Mitochondrial ROS (mtROS) possibly mediate the effects because antimycin and oligomycin are known to induce ROS generation [45, 46]. The effect of mtROS on the tight junction structure is detailed in the next section of the review. Changes in expression of tight junction proteins were similarly detected when the ATP level decreased in a Caco-2 cell monolayer. In particular, a claudin-7 redistribution was observed along the total membrane surface from the apical to the basolateral cell side, being unusual in normal intestinal epithelial cells [47].
Mitochondrial β-oxidation of fatty acids, especially short-chain ones (butyrate, acetate, and propionate), produced by the intestinal microbiota is thought to provide a main source of energy to intestinal epithelial cells [48]. Deficient fatty acid β-oxidation in the intestinal epithelium may play a critical role in the pathogenesis of IBD, especially in the case of UC. Treatment with sodium 2-bromoctanoate, which is a pharmacological inhibitor of fatty acid β-oxidation, led to spontaneous colitis in rats [49]. In addition, SLC22A5 mutations were associated with CD risk, and SLC22A5 codes for the Na-dependent L-carnitine transporter OCTN2, which is important for fatty acid β-oxidation [48]. Studies in macaques infected with the simian immunodeficiency virus (SIV) similarly demonstrated that fatty acid β-oxidation in mitochondria plays an important role in sustaining the barrier function of the intestinal epithelium. Distorted permeability of the intestinal epithelium in the macaques was associated with lower activity of PPARα, which is involved in fatty acid β-oxidation. Mitochondria decreased in number in cells from infected macaques, and their morphology changed to a fragmented phenotype with poorly developed cristae. In the intestinal epithelium of healthy animals, enterocyte mitochondria occur at the apical end near the tight junction region, possibly reflecting the important role of mitochondria in maintaining functional activity of tight junctions. Restoration of PPARα activity normalized the barrier function of the intestinal epithelium and partly restored the morphology and functions of mitochondria. The changes were confirmed by expression profiling of mitochondrial genes and genes involved in fatty acid β-oxidation [48].
Thus, the above data make it possible to conclude that the function of the ETC complexes, which perform oxidative phosphorylation (OXPHOS), and fatty acid β-oxidation in mitochondria are necessary for maintaining the structure of tight contacts in intestinal epithelial cells.
Stem cells of the intestinal epithelium are characterized by a high OXPHOS intensity [50–52]. Sufficient ATP production in mitochondria is critical for stem cell proliferation and differentiation in the intestinal epithelium. For example, knockdowns in ETC complex genes in stem cells of the Drosophila intestinal epithelium decreased their proliferation and differentiation. The greatest effect was observed when ETC complexes III and IV were inhibited. Suppressed differentiation of stem cells was associated with activity of the FOXO transcription factor [53]. Studies with an experimental animal model showed that the intensity of colitis induced with DSS or 2,3,4-trinitrobenzenesulfonic acid is lower in mice that have higher ATP production and higher OXPHOS activity as compared with mice that have lower activities of the OXPHOS complexes and lower ATP production. An increase in intracellular ATP content increases the intensity of NF-κB-dependent enterocyte proliferation, thus promoting self-renewal of the intestinal epithelium, which is a key process in tissue restoration after exposure to damaging factors [54]. However, the role of NF-κB in regulating the epithelial barrier permeability is ambiguous because synthesis of proinflammatory cytokines is induced by this transcription factor upon excessive activation of the respective signaling pathway. Stem cells of the intestinal epithelium contain a large amount of mitochondria, but display low-level expression of the mitochondrial pyruvate carrier (MPC). Loss of MPC or treatment with a MPC inhibitor led to more intense stem cell proliferation in organoids grown from stem cells of the Drosophila intestinal epithelium. A genetic deletion of the MPC gene in stem cells of the Drosophila intestinal epithelium similarly increased their proliferative activity, while MPC overexpression inhibited their proliferation. Thus, limited pyruvate transport into mitochondria is necessary for sustaining proliferation of stem cells of the intestinal epithelium. A large content of mitochondria in these cells is most likely associated with a high intensity of fatty acid β-oxidation [55]. The findings demonstrate that a distorted energy function of mitochondria in stem cells of the intestinal epithelium can impair its barrier function by altering the self-renewal processes.
Activation of AMP-activated kinase (AMPK) is one of the mechanisms that regulate cell metabolism at a lower ATP level. AMPK is activated in response to an increase in AMP and a simultaneous decrease in APT and then acts to regulate metabolism, the state of the cytoskeleton, protein synthesis, and autophagy. In the intestine, AMPK inhibits ATP-dependent ion transfer and protein synthesis [39]. AMPK activation is thought to facilitate assembly of tight and adherens junctions and to prevent phosphorylation of the myosin light chain and constriction of the actomyosin contractile ring, thus decreasing the paracellular permeability of the intestinal epithelium. AMPK is phosphorylated when the intracellular calcium concentration increases and kinase CaMKK2 is activated [56]. The protective effect of AMPK was demonstrated in several mouse models of induced colitis and in cultured Caco-2 cells [57, 58]. In addition, AMPK was shown to promote intestinal epithelial cell differentiation by inducing expression of the CDX2 transcription factor [59]. To summarize, AMPK is involved in a feedback mechanism in intestinal epithelial cells; i.e., AMPK inhibits the APT-dependent processes and stabilizes tight junctions, thus preventing the progression of epithelial barrier dysfunction in response to energy stress. The mechanism provides additional evidence for the role that the energy function of mitochondria plays in sustaining the barrier function of the intestinal epithelium.
The above data give grounds to conclude that a distorted energy function of mitochondria in the intestinal epithelium in IBD may impair the structure of tight junctions between enterocytes and suppress proliferation and differentiation of stem cells of the intestinal epithelium, while these processes are critical for the barrier function. The experimental data that demonstrate the role of the energy function of mitochondria in IBD are summarized in Table 1.
OXIDATIVE STRESS IN IBD
A major part of ROS produced in mitochondria are generated by ETC complexes I and III. Many factors affect the mtROS production, including the oxygen concentration, the respiration intensity, the Ca2+ concentration, and the mitochondrial membrane potential ΔΨm. Other mitochondrial sources of ROS are dehydrogenases in the matrix, p66Shc (an isoform of Src homology 2 domain-containing transforming protein 1 (SHC1)) in the intermembrane space, and monoamine oxidases in the outer membrane. Electrons continuously leak out of the mitochondrial ETC to form mostly superoxide (\({\text{O}}_{2}^{ - }\)) of the hydroxyl radical (·OH). However, there are antioxidant defense regulatory mechanisms in the cell. Mitochondrial manganese superoxide dismutase (MnSOD) usually eliminates \({\text{O}}_{2}^{ - }\). The MnSOD-driven reaction yields hydrogen peroxide (H2O2) and molecular oxygen; H2O2 is then neutralized by catalase. Other mitochondrial antioxidant systems include the thiol-containing tripeptide glutathione, thioredoxins, glutaredoxins, glutathione reductases, and thioredoxin peroxidases (peroxiredoxins). . The enzymes play an important role in maintaining the redox status of mitochondria and protect the organic molecules from oxidation by ROS [60–63]. An imbalance between mtROS production and neutralization by the antioxidant defense system results in oxidative stress.
Data are accumulating to implicate oxidative stress and mtROS in the IBD pathogenesis. Many studies documented oxidative stress-related damage to mtDNA, cytoskeletal proteins, and lipids of the intestinal mucosa in samples from IBD patients and mouse models, as well as a higher ROS level in the mucosa [64–67]. As was observed by Overhauser-enhanced magnetic resonance imaging, ROS production is intracellular and occurs in the distal and proximal regions of the colon in mice with early DSS-induced colitis, while both intracellular and extracellular ROS production takes place at more advanced stages of the disease. Moreover, a redox imbalance was found to arise before DSS-induced colitis starts developing and to precede inflammatory changes in the colonic mucosa [68]. A higher level of oxidative stress in the colon and signs of developing colitis were observed in mice deficient in glutathione peroxidase 1 (GSHPx-1) and glutathione peroxidase 2 (GPRP-2), which is specific to the intestinal epithelium [69]. Severe DSS-induced colitis developed in mice deficient in uncoupling protein 2 (UCP2), which is a protein of the inner mitochondrial membrane and acts as a negative regulator of mtROS [70].
It seems that mtROS directly affect the tight junctions to play their role in regulating the barrier function of the intestinal epithelium. Studies with models where the integrity of a Caco-2 intestinal epithelial cell monolayer was impaired by DSS treatment and osmotic stress or by mechanical stretching showed that mtROS are involved in a redistribution of occludin and ZO-1 from tight junctions into intracellular compartments, leading to structural alterations of intercellular junctions. Many ROS forms contribute to the disruption of tight junctions by rearranging the actin cytoskeleton, which consequently interacts less efficiently with occludin, ZO-1, and the myosin heavy chain. ROS can directly oxidize and damage actin and tubulin in IBD, thus altering the normal organization of the cytoskeleton [67]. All of these effects increase the paracellular permeability of the epithelium. Hydrogen peroxide, which increases the intracellular ROS level, alters the localization and phosphorylation of occludin and ZO-1, thus impairing the integrity of tight junctions, and affects β-catenin phosphorylation, thus destabilizing the adherens junctions because of an E-cadherin redistribution [71]. As was observed in models where the barrier function of the intestinal epithelium was distorted by osmotic stress and DSS, disruption of tight contacts is prevented when the mtROS level is decreased via a knockdown in the Ca2+ channels Cav1.3 and TRPV6 or treatment with the antioxidant MitoTEMPO [72].
The above data indicate that ROS can distort the normal structure of tight junctions to impair the barrier function of the intestinal epithelium. However, the exact mechanisms of the process are still unknown.
A protective effect of the mitochondrial-targeted antioxidants MitoTEMPO and MitoQ was demonstrated in various IBD models. For example, MitoTEMPO administration prevented distortion of the barrier function of the epithelium and decreased the intensity of intestinal inflammation in DSS-induced colitis. In addition, MitoTEMPO reduced the signs of oxidative stress in intestinal mucosa biopsy samples from CD patients [27]. The antioxidant MitoQ prevented ileocolitis in mice knocked out in glutathione peroxidases 1 and 2 [73] and alleviated DSS-induced colitis in mice. The protective effect of MitoQ was associated with lower activation of the NLRP3 inflammasome in immune cells, and NLRP3 plays a critical role in secretion of the proinflammatory cytokines IL-1β and IL-18 [74]. Mice with NLRP3 deficiency were resistant to DSS-induced colitis [75]. We showed previously that the mitochondrial-targeted antioxidant SkQ1 prevents DSS-induced colitis in mice and disassembly of tight junctions in Caco-2 intestinal epithelial cells [76].
ROS act as secondary messengers in intracellular signaling pathways, including the NF-κB pathway, which plays an important role in the IBD pathogenesis. Activity of NF-κB signaling is elevated in affected colonic tissues of UC patients, and blockage of NF-κB activity is thought to provide an effective means to treat UC [77]. In this case, oxidative stress acts to activate inhibitor κB kinase 2 (IKK2) and to stimulate nuclear translocation of NF-κB, leading to expression of the proinflammatory cytokines TNF-α, IL-1, and IL-8 in intestinal epithelial cells and thereby promoting inflammation [78–80]. It is important to note that a dual role is played by NF-κB in the intestinal epithelium. On the one hand, NF-κB is necessary for normal self-renewal of the intestinal epithelium. On the other hand, excessive or inappropriate NF-κB activation may decrease the barrier function.
The antioxidants ascorbic acid and 2,3,19,23-tetrahydroxyolean-12-en-28-oilc acid (olean-type triterpenoid) decrease DSS-induced colitis in vivo by decreasing NF-κB activity and increasing expression of proteins involved in cell antioxidant defense [81, 82]. This finding provides additional evidence for the association between NF-κB and oxidative stress in IBD.
In addition, ROS can activate MAPK kinases, which also play a critical role in inflammatory signaling. Activation of MAPK kinases p38 and JNK is involved in UC progression [83].
It should be noted that mtROS are necessary for the normal function and self-renewal of the intestinal epithelium apart from contributing to the IBD pathogenesis. For example, concomitant lower activities of the antioxidant enzymes catalase and superoxide dismutase 2 (SOD2) is possibly one of the main causes of apoptotic death in apical enterocytes. Modulation by mtROS was demonstrated for the Notch and AKT signaling pathways, which play an important role in the formation of crypts in intestinal organoid cultures [12].
It is logical to assume from the above that excess mtROS distort the barrier function of the intestinal epithelium and play a role in the IBD pathogenesis by directly affecting the tight junctions and increasing the production of proinflammatory cytokines. The mitochondrial-targeted antioxidants reduce the mtROS amount and exert a protective effect in the intestinal epithelium to restore its barrier function. The agents are therefore possible to consider as promising targeted drugs to treat IBD. The study results that implicate oxidative stress in IBD are summarized in Table 2.
Along with ROS, reactive nitrogen species (RNS) play a substantial role in IBD development. RNS include nitric oxide (NO) and its metabolic products. NO is generated by NO synthase (NOS), which catalyzes conversion of L-arginine to L-citrulline. Several NOS isoforms, including a mitochondrial one, are known. RNS result from the interaction of NO with O\(_{2}^{-}\). Like ROS, RNS damage the cell organic molecules, such as membrane lipids, DNA, and proteins. Excess RNS cause nitrosative stress and are involved in the mechanism of oxidative stress. It should be noted that oxidative and nitrosative stresses underlie many disorders that are associated with inflammation [63, 84–86].
The nitrite/nitrate level in the blood plasma is elevated in IBD patients, and higher NOS activity is observed in biopsy material from UC and CD patients [87]. The findings support the role of RNS in the IBD pathogenesis. RNS affect the permeability of the intestinal barrier by inducing apoptosis and necrosis of enterocytes and distorting the structure of tight junctions [88]. The role of RNS in the IBD pathogenesis was considered in detail in reviews [89–92].
ROLE OF MITOCHONDRIAL DYNAMICS AND HOMEOSTASIS IN IBD
Evidence is accumulating to demonstrate that alterations of the mitochondrial dynamics and homeostasis contribute to the IBD pathogenesis. Mitochondrial homeostasis is determined primarily by the balance between mitophagy and mitochondrial biogenesis and the chaperone-mediated mitochondrial unfolded protein response. Defective mitochondria are eliminated in the processes, and the mitochondrial pool is renewed. Distortions of the processes may lead to an accumulation of defective mitochondria or lack of normal organelles.
Hsieh et al. [35] observed that expression of the chaperone prohibitin is lower in UC remission, in which the disease is restricted to the distal region of the colon. Prohibitin occurs in the inner mitochondrial membrane and regulates many mitochondrial functions, including proper assembly of the ETC complexes. Theiss et al. [93] showed that the prohibitin level in the intestinal epithelium is decreased in IBD patients and several mouse models of experimental colitis. Prohibitin expression was downregulated in oxidative stress, while prohibitin overexpression prevented permeability distortion in Caco2-BBE human intestinal epithelial cells. Jackson et al. [94] found that spontaneous intestinal inflammation develops in mice with prohibitin deficiency in intestinal epithelial cells. Ultrastructural defects in mitochondria and the unfolded protein response developed one week after the prohibitin gene deletion. Activation of the NLRP3 inflammasome by mtROS was observed simultaneously, and secretion of proinflammatory cytokines consequently increased. MitoTEMPO reduced the signs of mitochondrial dysfunction and intestinal inflammation in prohibitin-deficient mice [94]. Thus, the proper function of the mitochondrial chaperone prohibitin ensures the integrity of the epithelial barrier by preventing mtROS production.
When misfolded protein accumulation is too intense and chaperones fail to prevent stress, mitochondria are fragmented and undergo mitophagy subsequently [95]. Dynamin-1-like protein (DNM1L) plays a main role in mitochondrial cleavage. A substantial decrease in ΔΨm and mtROS overproduction are thought to be signs of mitochondrial dysfunction and to provide signals to mitochondrial quality control systems and, in particular, the PINK1/Parkin system, which ensures selective autophagy (mitophagy) of defective mitochondria [60, 96, 97]. PGC-1α is a main regulator of mitochondrial biogenesis and triggers mitochondrial proliferation to restore the amount of mitochondria upon mitophagy. A balance between mitophagy of defective mitochondria and biogenesis of new mitochondria is essential for homeostasis. Maintenance of the mitochondrial pool in a normal state ensures the adequate cell responses to metabolic changes and stress and the adaptive role of mitochondria in cell signaling pathways [95, 98, 99]. Distortions of the process render cells more sensitive to damaging factors and facilitate barrier dysfunction.
Mitochondrial cleavage often precedes mitophagy. The mitochondrial cleavage intensity was shown to increase in the intestinal epithelium of mice with DSS-induced colitis, and inhibition of mitochondrial fragmentation reduced the colitis signs [100].
Bacterial invasion into intestinal epithelial cells distorts mitochondrial homeostasis. In contrast to a noninvasive Escherichia coli strain, an invasive one caused DNM1L-dependent mitochondrial fragmentation, a release of cytochrome c into the cytoplasm, and a decrease in ΔΨm and ATP content in a T84 cell monolayer. Moreover, invasive bacteria downregulated expression of OPA1 and PGC1A, which are necessary for mitochondrial fusion and biogenesis. DNM1L inhibitors, such as Mdivi1 and P110, decreased mitochondrial fragmentation only for a short period of time in this case. A lower content of the OPA1 a PGC-1α factors, which are necessary for biogenesis and fusion of mitochondria, possibly explained why the fused state of the mitochondrial pool could not be maintained during continuous excessive fragmentation and mitophagy. The essential role of bacterial internalization in the model was confirmed by the finding that only live bacteria with type 1 pili, which are necessary for invasion, caused the above effects [101]. Thus, bacterial internalization in enterocytes can distort mitochondrial homeostasis at both morphological and functional levels, thus playing an important role in the early pathogenesis of IBD. The exact mechanism whereby bacteria affect mitochondria remains unknown. It was only demonstrated that EspF, which is characteristic of pathogenic E. coli strains, decreases ΔΨm [102], and a decrease may stimulate mitophagy. Mechanical forces generated by bacteria moving within eukaryotic cells is another possible mechanism that sustains the effect of internalized bacteria on mitochondrial fragmentation. These mechanical forces can induce local activation of the mitochondrial fragmentation system [103].
The above data demonstrate that higher mitochondrial fragmentation and mitochondrial dysfunction develop in IBD and that inhibition of mitochondrial fragmentation may decelerate the development of colitis.
Mitophagy defects are also associated with a higher susceptibility to IBD. For example, an association with IBD was observed for a polymorphism located in IRGM (immunity-related GTPase family M protein), whose product is a GTPase and is involved in regulating mitochondrial fragmentation and mitophagy. Mice with an Irgm deletion are more susceptible to experimental colitis [104, 105]. A polymorphism of ATG16L1, which is involved in total autophagy, is also associated with IBD risk [106]. Given that mitophagy is a mechanism that eliminates dysfunctional mitochondria, the above data indicate that an accumulation of dysfunctional mitochondria contributes to the IBD pathogenesis.
The PGC-1α protein plays a crucial role in mitochondrial biogenesis and is expressed to a high level in the intestinal epithelium. Its expression is lower in IBD patients [107]. However, it should be noted that low PGC-1α expression is characteristic of crypts, which harbor stem cells of the intestinal epithelium. PGC-1α expression in enterocytes is thought to increase in the course of their differentiation to determine high respiratory activity of mitochondria, while the antioxidant defense level is maintained low. This results in mtROS accumulation and activates the apoptotic program in terminally differentiated cells, as is necessary for self-renewal of the intestinal epithelium [108].
In UC, PGC-1α was similarly shown to exert a protective effect and to support the barrier function of the intestinal epithelium. PGC-1α is acetylated and targeted to proteasomal degradation in UC patients and mice with DSS-induced colitis, and its level is consequently lower. Mice with a Pgc1a knockout in the intestinal epithelium were more susceptible to DSS-induced colitis, and their mitochondria displayed signs of dysfunction, including lower activities of ETC complexes I and IV. Pharmacological activation of PGC-1α decreased the colitis signs and restored the mitochondrial morphology. Moreover, a higher permeability of the intestinal epithelium to bacteria was observed in the mutant mice and was associated with distorted expression of tight junction proteins. Based on the findings, Cunningham et al. [28] concluded that a decrease in PGC-1α in the intestinal epithelium promotes structural and functional alterations in mitochondria, thereby impairing the barrier function and facilitating bacterial translocation.
The above data give grounds to thinking that distortion of mitochondrial homeostasis and an accumulation of dysfunctional mitochondria with insufficient biogenesis of new organelles are associated with the IBD pathogenesis. A higher content of damaged mitochondria induces energy and oxidative stresses in intestinal epithelial cells and thus decreases the barrier function of the epithelium. The mitochondrial quality control system is also involved in IBD, but the exact mechanisms of the respective processes are still unknown.
CONCLUSIONS
Thus, the analysis of published data makes it possible to conclude that mitochondria play a critical role in sustaining the barrier function of the intestinal epithelium by regulating the integrity of tight junctions of enterocytes and self-renewal of epithelial cells. Mitochondrial dysfunction is observed in the intestinal epithelium in IBD. Its signs include a distorted energy function, higher ROS production, and altered mitochondrial dynamics and induce fragmentation of mitochondria. Distortion of the energy function of mitochondria decreases ATP production, leading to inflammation and increasing the permeability of the epithelium to bacteria. In addition, the self-renewal capacity of the intestinal epithelium is reduced at a lower ATP level, while self-renewal is necessary for tissue regeneration after injury. Activation of AMPK kinase, which responds to a drop in ATP level, may provide a strategy to treat IBD.
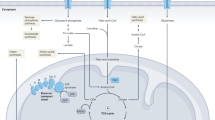
Excessive mtROS generation alters the structure of intercellular junctions in IBD and increases production of proinflammatory cytokines. Treatment with antioxidants, including mitochondrial-targeted ones, can partly restore the distorted barrier function of the intestinal epithelium. A fragmented phenotype is usually characteristic of dysfunctional mitochondria. Mitochondrial dynamics is altered to increase mitochondrial cleavage in IBD. Treatment with agents that suppress mitochondrial fragmentation will help to restore the barrier function of the intestinal epithelium. Mitophagy normally serves to eliminate dysfunctional mitochondria, but is distorted in IBD. Mitophagy-stimulating agents are therefore also possible to consider as potential drugs to treat IBD. Figure 1 illustrates the role that mitochondrial dysfunction plays in distortion of the barrier function of the intestinal epithelium and the IBD pathogenesis. The literature data available now determine the avenues of further research and developments. More detailed investigation of the mechanisms whereby mitochondria affect the IBD development will provide a basis to designing mitochondrial-targeted anti-inflammatory drugs.

Role of mitochondria in the pathogenesis of IBD. When the barrier function of the intestinal epithelium is distorted by various factors (intestinal microbiota, genetic predisposition, diet, drugs, etc.), bacteria invade the epithelial cells. Their invasion affects the state of mitochondria: mtROS generation increases, mitochondrial dynamics shifts towards mitochondrial fragmentation, and biogenesis of mitochondria is inhibited. Damaged fragmented mitochondria undergo mitophagy, which is a protective mechanism that maintains the normal state of the mitochondrial pool. However, an excess accumulation of dysfunctional mitochondria with a lower ΔΨm, lower ATP production, and higher mtROS generation leads to energy and oxidative stresses. Among other factors, this causes structural alterations in intercellular tight junctions and the cytoskeleton in intestinal epithelial cells and decreases the self-renewal capacity of the intestinal epithelium. The permeability of the epithelial barrier to bacteria grows even higher as a result. Bacterial antigens stimulate the immune system of the deep mucosa. Proinflammatory cytokines are released as a result and further increase the permeability of the epithelial barrier. Acting together, these factors form a vicious circle, which allows the disease to become chronic. The figure was prepared using the BioRender.com program (https://www.biorender.com/).
REFERENCES
Kaplan G.G. 2015. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 12 (12), 720–727.
de Lange K.M., Barrett J.C. 2015. Understanding inflammatory bowel disease via immunogenetics. J. Autoimmun. 64, 91–100.
Fiocchi C. 2015. Inflammatory bowel disease pathogenesis: Where are we? J. Gastroenterol. Hepatol. 30 (Suppl. 1), 12–18.
Kucharzik T., Maaser C., Lügering A., Kagnoff M., Mayer L., Targan S., Domschke W. 2006. Recent understanding of IBD pathogenesis: Implications for future therapies. Inflamm. Bowel Dis. 12 (11), 1068–1083.
Coskun M. 2014. Intestinal epithelium in inflammatory bowel disease. Front. Med. 1, 24.
Bischoff S.C., Barbara G., Buurman W., Ockhuizen T., Schulzke J.D., Serino M., Tilg H., Watson A., Wells J.M. 2014. Intestinal permeability—a new target for disease prevention and therapy. BMC Gastroenterol. 14, 189.
Shen L., Turner J.R. 2006. Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: Tight junction dynamics exposed. Am. J. Physiol. Gastrointest. Liver Physiol. 290 (4), G577–G582.
Zolotova N.A., Akhrieva Kh.M., Zayratyants O.V. 2019. Epithelial barrier of the colon in health and patients with ulcerative colitis. Eksp. Klin. Gastroenterol. 162 (2), 4–13.
Suzuki T. 2013. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol. Life Sci. 70 (4), 631–659.
Guttman J.A., Finlay B.B. 2009. Tight junctions as targets of infectious agents. Biochim. Biophys. Acta. 1788 (4), 832–841.
Schoultz I., Keita Å.V. 2020. The intestinal barrier and current techniques for the assessment of gut permeability. Cells. 9 (8), 1909.
Rath E., Moschetta A., Haller D. 2018. Mitochondrial function—gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol. 15 (8), 497–516.
Van Itallie C.M., Holmes J., Bridges A., Gookin J.L., Coccaro M.R., Proctor W., Colegio O.R., Anderson J.M. 2008. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J. Cell. Sci. 121 (Pt. 3), 298–305.
Backert S., Boehm M., Wessler S., Tegtmeyer N. 2013. Transmigration route of Campylobacter jejuni across polarized intestinal epithelial cells: Paracellular, transcellular or both? Cell Commun. Signal. 11, 72.
Peterson L.W., Artis D. 2014. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14 (3), 141–153.
Roda G., Sartini A., Zambon E., Calafiore A., Marocchi M., Caponi A., Belluzzi A., Roda E. 2010. Intestinal epithelial cells in inflammatory bowel diseases. World J. Gastroenterol. 16 (34), 4264–4271.
Schmitz H., Barmeyer C., Fromm M., Runkel N., Foss H.D., Bentzel C.J., Riecken E.O., Schulzke J.D. 1999. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. 116 (2), 301–309.
Oshima T., Miwa H., Joh T. 2008. Changes in the expression of claudins in active ulcerative colitis. J. Gastroenterol. Hepatol. 23 (Suppl. 2), S146–S150.
Landy J., Ronde E., English N., Clark S.K., Hart A.L., Knight S.C., Ciclitira P.J., Al-Hassi H.O. 2016. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 22 (11), 3117–3126.
Marin M.L., Greenstein A.J., Geller S.A., Gordon R.E., Aufses A.H Jr. 1983. A freeze fracture study of Crohn’s disease of the terminal ileum: Changes in epithelial tight junction organization. Am. J. Gastroenterol. 78 (9), 537–547.
Zeissig S., Bürgel N., Günzel D., Richter J., Mankertz J., Wahnschaffe U., Kroesen A.J., Zeitz M., Fromm M., Schulzke J.D. 2007. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 56 (1), 61–72.
Das P., Goswami P., Das T.K., Nag T., Sreenivas V., Ahuja V., Panda S.K., Gupta S.D., Makharia G.K. 2012. Comparative tight junction protein expressions in colonic Crohn’s disease, ulcerative colitis, and tuberculosis: A new perspective. Virchows Arch. 460 (3), 261–270.
Ungaro R., Mehandru S., Allen P.B., Peyrin-Biroulet L., Colombel J.F. 2017. Ulcerative colitis. Lancet. 389 (10080), 1756–1770.
Mayer L. 2010. Evolving paradigms in the pathogenesis of IBD. J. Gastroenterol. 45 (1), 9–16.
Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T., Abedian S., Cheon J.H., Cho J., Dayani N.E., Franke L., Fuyuno Y., Hart A., Juyal R.C., Juyal G., Kim W.H., Morris A.P., Poustchi H., Newman W.G., Midha V., Orchard T.R., Vahedi H., Sood A., Sung J.Y., Malekzadeh R., Westra H.J, Yamazaki K., Yang S.K.; International Multiple Sclerosis Genetics Consortium; International IBD Genetics Consortium; Barrett J.C., Alizadeh B.Z., Parkes M., Bk T., Daly M.J., Kubo M., Anderson C.A., Weersma R.K. 2015. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47 (9), 979–986.
Novak E.A., Mollen K.P. 2015. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 3, 62.
Wang A., Keita Å.V., Phan V., McKay C.M., Schoultz I., Lee J., Murphy M.P., Fernando M., Ronaghan N., Balce D., Yates R., Dicay M., Beck P.L., MacNaughton W.K., Söderholm J.D., McKay D.M. 2014. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am. J. Pathol. 184 (9), 2516–2527.
Cunningham KE, Vincent G, Sodhi CP, Novak EA, Ranganathan S, Egan CE, Stolz D.B., Rogers M.B., Firek B., Morowitz M.J., Gittes G.K., Zuckerbraun B.S., Hackam D.J., Mollen K.P. 2016. Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) protects against experimental murine colitis. J. Biol. Chem. 291 (19), 10184–101200.
Jackson D.N., Theiss A.L. 2020. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut. Microbes. 11 (3), 285–304.
Kłos P., Dabravolski S.A. 2021. The role of mitochondria dysfunction in inflammatory bowel diseases and colorectal cancer. Int. J. Mol. Sci. 22 (21), 11673.
Ho G.T., Theiss A.L. 2022. Mitochondria and inflammatory bowel diseases: Toward a stratified therapeutic intervention. Annu. Rev. Physiol. 10 (84), 435‒459. https://doi.org/10.1146/annurev-physiol-060821-083306
Bourgonje A.R., Feelisch M., Faber K.N., Pasch A., Dijkstra G., van Goor H. 2020. Oxidative stress and redox-modulating therapeutics in inflammatory bowel disease. Trends. Mol. Med. 26 (11), 1034–1046.
Cunningham K., Novak E., Vincent G., Mollen K.P., Chinnder S. 2015. Antibiotic treatment protects against intestinal inflammation in peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) deficient mice in experimental colitis. J. Am. Coll. Surg. 221 (4), S28–S29.
Söderholm J.D., Olaison G., Peterson K.H., Franzén L.E., Lindmark T., Wirén M., Tagesson C, Sjödahl R. 2002. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut. 50 (3), 307–313.
Hsieh S.Y., Shih T.C., Yeh C.Y., Lin C.J., Chou Y.Y., Lee Y.S. 2006. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics. 619), 5322–5331.
Bohovych I., Khalimonchuk O. 2016. Sending out an SOS: Mitochondria as a signaling hub. Front. Cell Dev. Biol. 4, 109.
Pickles S., Vigié P., Youle R.J. 2018. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28 (4), R170–R185.
Haberman Y., Karns R., Dexheimer P.J., Schirmer M., Somekh J., Jurickova I., Braun T., Novak E., Bauman L., Collins M.H., Mo A., Rosen M.J., Bonkowski E., Gotman N., Marquis A., Nistel M., Rufo P.A., Baker S.S., Sauer C.G., Markowitz J., Pfefferkorn M.D., Rosh J.R., Boyle B.M., Mack D.R., Baldassano R.N., Shah S., Leleiko N.S., Heyman M.B., Grifiths A.M., Patel A.S., Noe J.D., Aronow B.J., Kugathasan S., Walters T.D., Gibson G., Thomas S.D., Mollen K., Shen-Orr S., Huttenhower C., Xavier R.J., Hyams J.S., Denson L.A. 2019. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 10 (1), 38.
Heller S., Penrose H.M., Cable C., Biswas D., Nakhoul H., Baddoo M., Flemington E., Crawford S.E., Savkovic S.D. 2017. Reduced mitochondrial activity in colonocytes facilitates AMPKα2-dependent inflammation. FASEB J. 31 (5), 2013–2025.
Xue X, Bredell BX, Anderson ER, Martin A, Mays C, Nagao-Kitamoto H, Huang S., Győrffy B., Greenson J.K., Hardiman K., Spence J.R., Kamada N., Shah Y.M. 2017. Quantitative proteomics identifies STEAP4 as a critical regulator of mitochondrial dysfunction linking inflammation and colon cancer. Proc. Natl. Acad. Sci. U. S. A. 114 (45), E9608–E9617.
Sifroni K.G., Damiani C.R., Stoffel C., Cardoso M.R., Ferreira G.K., Jeremias I.C., Rezin G.T., Scaini G., Schuck P.F., Dal-Pizzol F., Streck E.L. 2010. Mitochondrial respiratory chain in the colonic mucosal of patients with ulcerative colitis. Mol. Cell. Biochem. 342 (1‒2), 111–115.
Restivo N.L., Srivastava M.D., Schafer I.A., Hoppel C.L. 2004. Mitochondrial dysfunction in a patient with crohn disease: Possible role in pathogenesis. J. Pediatr. Gastroenterol. Nutr. 38 (5), 534–538.
Dankowski T., Schröder T., Möller S., Yu X., Ellinghaus D., Bär F., Fellermann K., Lehnert H., Schreiber S., Franke A., Sina C., Ibrahim S.M., König I.R. 2016. Male-specific association between MT-ND4 11719 A/G polymorphism and ulcerative colitis: A mitochondria-wide genetic association study. BMC Gastroenterol. 16 (1), 118.
Crakes K.R., Santos Rocha C., Grishina I., Hirao L.A., Napoli E., Gaulke C.A., Fenton A., Datta S., Arredondo J., Marco M.L., Sankaran-Walters S., Cortopassi G., Giulivi C., Dandekar S. 2019. PPARα-targeted mitochondrial bioenergetics mediate repair of intestinal barriers at the host-microbe intersection during SIV infection. Proc. Natl. Acad. Sci. U. S. A. 116 (49), 24819–24829.
Park W.H., Han Y.W., Kim S.H., Kim S.Z. 2007. An ROS generator, antimycin A, inhibits the growth of HeLa cells via apoptosis. J. Cell Biochem. 102 (1), 98–109.
Liu Y., Schubert D.R. 2009. The specificity of neuroprotection by antioxidants. J. Biomed. Sci. 16 (1), 98.
Janssen Duijghuijsen L.M., Grefte S., de Boer V.C.J., Zeper L., van Dartel D.A.M., van der Stelt I., Bekkenkamp-Grovenstein M., van Norren K., Wichers H.J., Keijer J. 2017. Mitochondrial ATP depletion disrupts Caco-2 monolayer integrity and internalizes claudin 7. Front. Physiol. 8, 794.
Smith S.A., Ogawa S.A., Chau L., Whelan K.A., Hamilton K.E., Chen J., Keilbaugh S., Fogt F., Bewtra M., Braun J., Xavier R.J., Clish C.B., Slaff B., Weljie A.M., Bushman F.D., Lewis J.D., Li H., Master S.R., Bennett M.J., Nakagawa H., Wu G.D. 2021. Mitochondrial dysfunction in inflammatory bowel disease alters intestinal epithelial metabolism of hepatic acylcarnitines. J. Clin. Invest. 131 (1), e133371.
Roediger W.E., Nance S. 1986. Metabolic induction of experimental ulcerative colitis by inhibition of fatty acid oxidation. Br. J. Exp. Pathol. 67 (6), 773–782.
Umar S. 2010. Intestinal stem cells. Curr. Gastroenterol. Rep. 12 (5), 340–348.
Chandel N.S., Jasper H., Ho T.T., Passegué E. 2016. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell. Biol. 18 (8), 823–832.
Rodríguez-Colman M.J., Schewe M., Meerlo M., Stigter E., Gerrits J., Pras-Raves M., Sacchetti A., Hornsveld M., Oost K.C., Snippert H.J., Verhoeven-Duif N., Fodde R., Burgering B.M. 2017. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature. 543 (7645), 424–427.
Zhang F., Pirooznia M., Xu H. 2020. Mitochondria regulate intestinal stem cell proliferation and epithelial homeostasis through FOXO. Mol. Biol. Cell. 31 (14), 1538–1549.
Bär F., Bochmann W., Widok A., von Medem K., Pagel R., Hirose M., Yu X., Kalies K., König P., Böhm R., Herdegen T., Reinicke A.T., Büning J., Lehnert H., Fellermann K., Ibrahim S., Sina C. 2013. Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology. 145 (5), 1055–1063.e3.
Wen Y.A., Xiong X., Scott T., Li A.T., Wang C., Weiss H.L., Tan L., Bradford E., Fan T.W.M., Chandel N.S., Barrett T.A., Gao T. 2019. The mitochondrial retrograde signaling regulates Wnt signaling to promote tumorigenesis in colon cancer. Cell Death Differ. 26 (10), 1955–1969.
Mihaylova M.M., Shaw R.J. 2011. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell. Biol. 13 (9), 1016–1023.
Chen L., Wang J., You Q., He S., Meng Q., Gao J., Wu X., Shen Y., Sun Y., Wu X., Xu Q. 2018. Activating AMPK to restore tight junction assembly in intestinal epithelium and to attenuate experimental colitis by metformin. Front. Pharmacol. 9, 761.
Chang K.W., Kuo C.Y. 2015. 6-Gingerol modulates proinflammatory responses in dextran sodium sulfate (DSS)-treated Caco-2 cells and experimental colitis in mice through adenosine monophosphate-activated protein kinase (AMPK) activation. Food Funct. 6 (10), 3334–3341.
Sun X., Yang Q., Rogers C.J., Du M., Zhu M.J. 2017. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 24 (5), 819–831.
Zorov D.B., Juhaszova M., Sollott S.J. 2014. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94 (3), 909–950.
Shanmugasundaram K., Nayak B.K., Friedrichs W.E., Kaushik D., Rodriguez R., Block K. 2017. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 8 (1), 997.
Forrester S.J., Kikuchi D.S., Hernandes M.S., Xu Q., Griendling K.K. 2018. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 122 (6), 877–902.
Butterfield D.A., Boyd-Kimball D. 2020. Mitochondrial oxidative and nitrosative stress and Alzheimer disease. Antioxidants (Basel). 9 (9), 818.
Dincer Y., Erzin Y., Himmetoglu S., Gunes K.N., Bal K., Akcay T. 2007. Oxidative DNA damage and antioxidant activity in patients with inflammatory bowel disease. Dig. Dis. Sci. 52 (7), 1636–1641.
Balmus I.M., Ciobica A., Trifan A., Stanciu C. 2016. The implications of oxidative stress and antioxidant therapies in inflammatory bowel disease: Clinical aspects and animal models. Saudi J. Gastroenterol. 22 (1), 3–17.
Kruidenier L., Kuiper I., Lamers C.B., Verspaget H.W. 2003. Intestinal oxidative damage in inflammatory bowel disease: Semi-quantification, localization, and association with mucosal antioxidants. J. Pathol. 201, 28–36.
Keshavarzian A., Banan A., Farhadi A., Komanduri S., Mutlu E., Zhang Y., Fields J.Z. 2003. Increases in free radicals and cytoskeletal protein oxidation and nitration in the colon of patients with inflammatory bowel disease. Gut. 52 (5), 720–728.
Yasukawa K., Hirago A., Yamada K., Tun X., Ohkuma K., Utsumi H. 2019. In vivo redox imaging of dextran sodium sulfate-induced colitis in mice using Overhauser-enhanced magnetic resonance imaging. Free Radic. Biol. Med. 136, 1–11.
Esworthy R.S., Steven Esworthy R., Aranda R., Martín M.G., Doroshow J.H., Binder S.W., Chu F.F. 2001. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281 (3), G848–G855.
Zhang H., Kuai X.Y., Yu P., Lin L., Shi R. 2012. Protective role of uncoupling protein-2 against dextran sodium sulfate-induced colitis. J. Gastroenterol. Hepatol. 27 (3), 603–608.
Rao R. 2008. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front. Biosci. 13, 7210–7226.
Gangwar R., Meena A.S., Shukla P.K., Nagaraja A.S., Dorniak P.L., Pallikuth S., Waters C.M., Sood A., Rao R. 2017. Calcium-mediated oxidative stress: A common mechanism in tight junction disruption by different types of cellular stress. Biochem. J. 474 (5), 731–749.
Chu F.F., Esworthy R.S., Doroshow J.H., Grasberger H., Donko A., Leto T.L., Gao Q., Shen B. 2017. Deficiency in Duox2 activity alleviates ileitis in GPx1- and GPx2-knockout mice without affecting apoptosis incidence in the crypt epithelium. Redox Biol. 11, 144–156.
Dashdorj A., Jyothi K.R., Lim S., Jo A., Nguyen M.N., Ha J., Yoon K.S., Kim H.J., Park J.H., Murphy M.P., Kim S.S. 2013. Mitochondria-targeted antioxidant Mi-toQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 11, 178.
Bauer C., Duewell P., Mayer C., Lehr H.A., Fitzgerald K.A., Dauer M., Tschopp J., Endres S., Latz E., Schnurr M. 2010. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 59 (9), 1192–1199.
Fedorov A.V., Chelombitko M.A., Chernyavskij D.A., Galkin I.I., Pletjushkina O.Y., Vasilieva T.V., Zinovkin R.A., Chernyak B.V. 2022. Mitochondria-targeted antioxidant SkQ1 prevents the development of experimental colitis in mice and impairment of the barrier function of the intestinal epithelium. Cells. 11 (21), 3441.
Gan H.T., Chen Y.Q., Ouyang Q. 2005. Sulfasalazine inhibits activation of nuclear factor-κB in patients with ulcerative colitis. J. Gastroenterol. Hepatol. 20 (7), 1016–1024.
Piechota-Polanczyk A., Fichna J. 2014. Review article: The role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn Schmiedebergs Arch. Pharmacol. 387 (7), 605–620.
Ji Y., Dai Z., Sun S., Ma X., Yang Y., Tso P., Wu G., Wu Z. 2018. Hydroxyproline attenuates dextran sulfate sodium-induced colitis in mice: Involvment of the NF-κB signaling and oxidative stress. Mol. Nutr. Food Res. 62 (21), e1800494.
He Y., Li Z., Xu T., Luo D., Chi Q., Zhang Y., Li S. 2022. Polystyrene nanoplastics deteriorate LPS-modulated duodenal permeability and inflammation in mice via ROS drived-NF-κB/NLRP3 pathway. Chemosphere. 307 (Pt. 1), 135662.
Lifei-Luo, Zhang J., Li X., Zhu Y., Wang Y., Liu D. 2023. Sericic acid ameliorates DSS-induced ulcerative colitis in mice by modulating the NF-κB and Nrf2 pathways. Curr. Mol. Pharmacol. 16 (7), 759‒770. https://doi.org/10.2174/1874467215666220928100319
Yan H., Wang H., Zhang X., Li X., Yu J. 2015. Ascorbic acid ameliorates oxidative stress and inflammation in dextran sulfate sodium-induced ulcerative colitis in mice. Int. J. Clin. Exp. Med. 8 (11), 20245–20253.
Wang Z., Li S., Cao Y., Tian X., Zeng R., Liao D.F., Cao D. 2016. Oxidative stress and carbonyl lesions in ulcerative colitis and associated colorectal cancer. Oxid. Med. Cell Longev. 2016, 9875298.
Shlapakova T.I., Kostin R.K., Tyagunova E.E. 2020. Reactive oxygen species: Participation in cellular processes and progression of pathology. Russ. J. Bioorg. Chem. 46 (5), 657–674.
Reutov V.P., Samosudova N.V., Sorokina E.G. 2019. A model of glutamate neurotoxicity and mechanisms of the development of the typical pathological process. Biophysics. 64 (2), 233–250.
Reutov V.P., Sorokina E.G. 2022. Causal relationship between physiological and pathological processes in the brain and in the gastrointestinal tract: The brain–intestine axis. Biophysics. 67 (6), 972–986.
McCafferty D.M. 2000. Peroxynitrite and inflammatory bowel disease. Gut. 46 (3), 436–439.
Chokshi N.K., Guner Y.S., Hunter C.J., Upperman J.S., Grishin A., Ford H.R. 2008. The role of nitric oxide in intestinal epithelial injury and restitution in neonatal necrotizing enterocolitis. Semin. Perinatol. 32 (2), 92–99.
Pavlick K.P., Laroux F.S., Fuseler J., Wolf R.E., Gray L., Hoffman J., Grisham M.B. 2002. Role of reactive metabolites of oxygen and nitrogen in inflammatory bowel disease. Free Radical Biol. Med. 33 (3), 311–322.
Abot A., Fried S., Cani P.D., Knauf C. 2022. Reactive oxygen species/reactive nitrogen species as messengers in the gut: Impact on physiology and metabolic disorders. Antioxid. Redox Signal. 37 (4‒6), 394–415.
Predonzani A., Calì B., Agnellini A.H., Molon B. 2015. Spotlights on immunological effects of reactive nitrogen species: When inflammation says nitric oxide. World J. Exp. Med. 5 (2), 64–76.
Guan G., Lan S. 2018. Implications of antioxidant systems in inflammatory bowel disease. Biomed. Res. Int. 2018, 1290179.
Theiss A.L., Idell R.D., Srinivasan S., Klapproth J.M., Jones D.P., Merlin D., Sitaraman S.V. 2007. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 21 (1), 197–206.
Jackson D.N., Panopoulos M., Neumann W.L., Turner K., Cantarel B.L., Thompson-Snipes L., Dassopoulos T., Feagins L.A., Souza R.F., Mills J.C., Blumberg R.S., Venuprasad K., Thompson W.E., Theiss A.L. 2020. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut. 69 (11), 1928–1938.
Zhu J., Wang K.Z.Q., Chu C.T. 2013. After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy. 9 (11), 1663–1676.
Brookes P.S., Yoon Y., Robotham J.L., Anders M.W., Sheu S.S. 2004. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287 (4), C817–C833.
Perry S.W., Norman J.P., Barbieri J., Brown E.B., Gelbard H.A. 2011. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques. 50 (2), 98–115.
Palikaras K., Tavernarakis N. 2014. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 56, 182–188.
Palikaras K., Lionaki E., Tavernarakis N. 2015. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 22 (9), 1399–1401.
Mancini N.L., Goudie L., Xu W., Sabouny R., Rajeev S., Wang A., Esquerre N., Al Rajabi A., Jayme T.S., van Tilburg Bernandes E., Nasser Y., Ferraz J.G.P., Shutt T., Shearer J., McKay D.M. 2020. Perturbed mitochondrial dynamics is a novel feature of colitis that can be targeted to lessen disease. Cell Mol. Gastroenterol. Hepatol. 10 (2), 287–307.
Mancini N.L., Rajeev S., Jayme T.S., Wang A., Keita Å.V., Workentine M.L., Hamed S., Söderholm J.D., Lopes F., Shutt T.E., Shearer J., McKay D.M. 2021. Crohn’s disease pathobiont adherent-invasive E. coli disrupts epithelial mitochondrial networks with implications for gut permeability. Cell Mol. Gastroenterol. Hepatol. 11 (2), 551‒571.
Nagai T., Abe A., Sasakawa C. 2005. Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: Critical role of the 16th leucine residue in EspF. J. Biol. Chem. 280 (4), 2998–3011.
Helle S.C.J., Feng Q., Aebersold M.J., Hirt L., Grüter R.R., Vahid A., Sirianni A., Mostowy S., Snedeker J.G., Šarić A., Idema T., Zambelli T., Kornmann B. 2017. Mechanical force induces mitochondrial fission. Elife. 6, 30292.
Singh S.B., Ornatowski W., Vergne I., Naylor J., Delgado M., Roberts E., Ponpuak M., Master S., Pilli M., White E., Komatsu M., Deretic V. 2010. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 12 (12), 1154–1165.
Liu B., Gulati A.S., Cantillana V., Henry S.C., Schmidt E.A., Daniell X., Grossniklaus E., Schoenborn A.A., Sartor R.B., Taylor G.A. 2013. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 305 (8), G573–G584.
Salem M., Ammitzboell M., Nys K., Seidelin J.B., Nielsen O.H. 2015. ATG16L1: A multifunctional susceptibility factor in Crohn disease. Autophagy. 11 (4), 585–594.
Ussakli C.H., Ebaee A., Binkley J., Brentnall T.A., Emond M.J., Rabinovitch P.S., Risques R.A. 2013. Mitochondria and tumor progression in ulcerative colitis. J. Natl. Cancer Inst. 105 (16), 1239–1248.
D’Errico I., Salvatore L., Murzilli S., Lo Sasso G., Latorre D., Martelli N., Egorova A.V., Polishuck R., Madeyski-Bengtson K., Lelliott C., Vidal-Puig A.J., Seibel P., Villani G., Moschetta A. 2011. Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) is a metabolic regulator of intestinal epithelial cell fate. Proc. Natl. Acad. Sci. U. S. A. 108 (16), 6603–6608.
ACKNOWLEDGMENTS
We are grateful to Dr. Sci. (Biol.) B.V. Chernyak (head of the Laboratory of Cell Bioenergetics, Belozersky Institute of Physicochemical Biology) for useful criticism.
Funding
This work was supported by the Russian Science Foundation (project no. 23-14-00061).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This work does not contain any studies involving animals or human participants performed by any of the authors.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
Additional information
Translated by T. Tkacheva
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations: AMPK, AMP-activated protein kinase; DNM1L, dynamin-1-like protein; IL, interleukin; IFN, interferon; MitoQ, mitoquinone (10-(6′-ubiquinonyl)-decyl-triphenylphosphonium); MitoTEMPO, mitochondria-targeted [2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl]triphenylphosphonium chloride; MPC, mitochondrial pyruvate carrier; OPA1, optic atrophy 1; OXPHOS, oxidative phosphorylation; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator 1-α; SkQ1, Skulachev’s quinone (10-(6'-plastoquinonyl)-decyl-triphenylphosphonium); TL1A, tumor necrosis factor family member TNF-like factor 1A; TNF, tumor necrosis factor; ZO, zonula occludens; RNS, reactive nitrogen species; ROS, reactive oxygen species; CD, Crohn’s disease; IBD, inflammatory bowel disease; DSS, dextran sulfate sodium; mtROS, mitochondrial reactive oxygen species; ETC, electron transport chain; UC, ulcerative colitis.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chernyavskij, D.A., Galkin, I.I., Pavlyuchenkova, A.N. et al. Role of Mitochondria in Intestinal Epithelial Barrier Dysfunction in Inflammatory Bowel Disease. Mol Biol 57, 1024–1037 (2023). https://doi.org/10.1134/S0026893323060043
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893323060043