
Abstract
The review summarizes the results of our own studies and published data on the biological markers of psychiatric disorders, with special emphasis on the activity of platelet monoamine oxidase. Pharmacotherapy studies in patients with the mixed anxiety-depressive disorder and first episode of schizophrenia have shown that the activity of platelet monoamine oxidase could serve as a potential biomarker of the efficacy of therapeutic interventions in these diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Biological markers are biochemical, physiological, or anatomical indicators specific for a certain state or a disease. The most important criterion for the use of biomarkers as diagnostic tools in medicine is their correlation with particular diseases.
Understanding molecular processes of psychiatric pathologies is a fundamental requirement for the development of diagnostic, differential diagnostic, and prognostic tools, as well as for the efficient therapy. Biomarkers isolated from easily available body fluids (cerebrospinal fluid, blood serum or plasma, urine, saliva) can help in the identification of disease subtypes, prognosis and monitoring of the patient’s response to therapy, and evaluation of the degree of treatment compliance. Moreover, biomarkers provide an opportunity to identify the targets of pharmaceutical preparations, which is important for the development of not only new drugs with desired properties, but of new therapeutic approaches as well. Identification of biological markers can promote detailed understanding of disease pathogenesis and pathophysiological mechanisms [1, 2].
There are several classifications of biomarkers. One of them, which is the most practical in our opinion, was developed by Lopresti et al. [3]. The authors classified biomarkers in the following groups:
-
diagnostic biomarkers used to confirm the presence or absence of the disease;
-
therapeutic biomarkers that can be used for selection of the optimal treatment for a particular patient;
-
biomarkers for the evaluation of therapeutic intervention efficacy;
-
prognostic biomarkers to estimate the likely disease outcomes.
Understanding neurochemical process involved in the emergence of mental disorders is crucial for elucidation of the underlying pathophysiological mechanisms, identification of disrupted metabolic processes, and development of pharmaceuticals with new properties and targets. Such information is obtained in biochemical (in broad terms) studies. In recent years, genetic studies have significantly contributed to the understanding of pathogenetic mechanisms of mental diseases, as these studies can reveal particular disruptions at the genomic, chromosomal, and gene levels.
Hence, pathogenetic and pathophysiological mechanisms of mental disorders could be investigated by combining genetic and biochemical studies. Biomarkers are used in multiple areas of clinical practice, e.g., in the diagnostics and treatment of cardiovascular diseases, kidney damage, and others. However, no biomarkers for the application in psychiatry have been found so far [4, 5].
In this regard, biological psychiatry can significantly benefit from the systemic approach [2, 6, 7]. The data obtained with the use of this approach (unlike the reductionist one) indicate a very complex nature of mental disorders. These multifactorial diseases are characterized by the interaction of genetic and environmental factors cause both functional and structural changes at the neuronal level. Hence, the diagnostics of mental disorders should be conducted based on the assessment of all components of these pathological states [2]. Clinical manifestations of mental disorders are usually a result of the combined effects of different genes and numerous epigenetic mechanisms [8].
Presently, scientists are gaining more insight into the complexity of mental disorders and inadequacy of existing diagnostic, prognostic, and therapeutic approaches (e.g., the absence of biomarkers for particular diseases) The complex nature of mental disorders is associated with a high etiological heterogeneity that involves numerous multifactorial (environmental and genetic) effects [9, 10].
Studying mental disorders has numerous limitations related, in particular, to the unavailability of brain tissues for testing. The symptoms of mental disorders often overlap, which hinders disease diagnostics and prevents identification of specific diagnostic biomarkers by modern imaging techniques, such as functional magnetic resonance imaging (fMRI), positron-emission tomography (PET), single photon emission computed tomography (SPECT), and others [9].
Modern methods of molecular biology can facilitate accurate diagnostics of mental diseases, which, in turn, can promote the development of targeted personalized therapy. Studying the effects of a single gene or a single protein is insufficient for understanding mental disorders [2, 7]. Such reductionist approaches fail to provide a comprehensive picture of the structure and dynamics of such complex biological system as mammalian (including, human) organism.
The reductionist approach limits our understanding of the compensatory and adaptive responses of biological system to diverse external and, possibly, internal factors. The systemic approach is more instrumental in evaluating the multifactorial aspects of mental disorders and elucidating pathogenetic and pathophysiological mechanisms of these diseases.
According to the published data, depression mostly affects five biological systems (neurotransmitter, neuroendocrine, neurotrophic, metabolic, and cytokine). At the same time, these systems are the main sources of potential biomarkers [11]. The studies on the activity of enzymes of neurotransmitter metabolism in patients with mental disorders are scarce.
The objective of this review was to discuss the role of monoamine oxidase (MAO), one of main enzymes of monoamine metabolism, as a potential biomarker of mental disorders.
MONOAMINE OXIDASE
MAO is one of the enzymes that are of great interest to psychiatrists. This enzyme is a key component in the inactivation of biologically active monoamines – neurotransmitters and neuromodulators.
The major pathways of the synthesis and degradation (metabolism) of important biologically active monoamines (shown in bold and italic) are presented below:
Tyrosine → L‑DOPA (L‑dihydroxyphenylalanine) → dopamine → homovanillic acid
Tyrosine → L‑DOPA → dopamine → norepinephrine → vanillylmandelic acid
Tryptophan → 5‑hydroxytriptophan → serotonin (5‑hydroxytryptamine) → 5‑hydroxyindolylacetic acid
MAO catalyzes oxidative deamination of aromatic amines in the mitochondria. MAO substrates include biogenic monoamines, e.g., indolealkylamines (serotonin and tryptamine) and catecholamines (dopamine and noradrenaline), trace amines (beta-phenylethylamine, tyramine, and octopamine), dietary monoamines, and a number of other compounds. MAO-catalyzed reactions can also yield highly reactive and toxic compounds, such as aldehydes, ammonia, and hydrogen peroxide:
Monoamine (M–NH2) → aldehyde + hydrogen peroxide + ammonia
The two types of MAO that have been characterized are MAO‑A and MAO‑B. Even before their molecular characterization, it had been found that these two isoforms differ in the substrate specificity and sensitivity to inhibitors. Thus, MAO‑A exhibits a higher affinity to serotonin and noradrenaline and is irreversibly inhibited by clorgyline, whereas MAO‑B has a higher affinity to phenylethylamine and benzylamine and is selectively inhibited by deprenyl. Both MAO isoforms deaminate dopamine with equal efficiency and have similar molecular weights: 59.7 and 58.0 kDa for MAO-A and MAO-B, respectively [12].
The identity of the primary structures of MAO‑A and MAO‑B is 70%, both enzymes contain the Ser-Gly-Gly-Cys-Tyr fragment that binds the flavin cofactor via a thioester covalent bond with the cysteine residue [13].
The existence of two isoforms with different molecular structure was confirmed by cloning the corresponding genes. Both MAOA and MAOB genes are located on the X chromosome (locus Xp11.23) and have the same exon-intron organization that includes 15 exons. Taken together, these data suggest that these two genes have originated from a common ancestral gene via duplication [14]. The detailed consideration of the problems of enzymatic catalysis and analysis of results of genetic studies were beyond the scope of our review; these issues have been discussed in detail in a number of publications [12, 15-18].
The differences between the MAO isoforms have been confirmed by the observations that MAO‑A and MAO‑B are activated and repressed by different transcription factors [18]. Furthermore, both isoforms are expressed in almost all peripheral organs and tissues; however, MAO‑A has been detected predominantly in the fibroblasts and placenta, while MAO‑B is the only isoform found in the thrombocytes and lymphocytes [12]. It is also known that the serotoninergic neurons of the raphe nuclei contain mostly MAO‑B, while the noradrenergic neurons of the locus coeruleus predominately express MAO‑A [12].
In the human brain, MAO‑B is more abundant than MAO‑A starting from adolescence till old age; however, fetal brain expresses more MAO-A than MAO-B [19]. The total distribution volume of MAO‑A (MAO‑A Vt) can be examined in the human brain in vivo by the positron emission tomography (PET) using [11C]‑harmine [20-22]. The content of MAO‑A has been assessed in different brain areas, such as the prefrontal cortex, anterior and posterior cingulate cortex, caudate nucleus, basal ganglia, thalamus, frontotemporal lobe, midbrain, hippocampus, and structures adjacent to the hippocampus. Based on the obtained results, it was suggested that the density (content) of MAO‑A in the brain tissues positively correlates with the MAO‑A activity [19, 21].
The levels of MAO‑B were found to be significantly higher than of MAO‑A in all investigated brain areas, with the MAO‑B to MAO‑A ratio varying from 2.6 (occipital lobe) to 17.8 (corpus callosum) [19].
Tong et al. [19] suggested that the enzymatic activity and concentration positively correlate with each other for both enzymes [19]. However, this statement has caused some doubts, because the activity of MAO cannot be directly measured in the human brain in vivo, and protein content does not always correspond the activity of the enzyme. Although this correlation seems reasonable under normal conditions (in a healthy individual), the direct dependence of the MAO activity on the enzyme content under pathological conditions is arguable, as mentioned above. To support our point of view, we can refer to our own studies of albumin conformation which demonstrated that although the content of albumin in healthy individuals and patients with the first-episode of schizophrenia (FES) was the same, the antioxidant activity of this protein in the patients with schizophrenia was significantly lower [23].
Although direct evaluation of the MAO activity in human brain is impossible, this issue can be addressed using the peripheral model of the central nervous system suggested in biological psychiatry [24]. This model is based on the assumption that the functioning of enzymes, receptors, and other biochemical components of blood cells to a large degree reflects the state of biochemical systems in the brain. In particular, platelets with detected MAO activity were found to be similar to brain synaptosomes in the active transport of biogenic amines, formation of amine reserve forms, functions of reserve granules, and activity of monoamine receptors [25].
MAO was first detected in platelets in 1964 [26].
Platelets and neurons have different embryonic origins. Platelets are formed from megakaryocytes originating in the mesoderm, while neurons form from the ectoderm [27]. Despite different embryonic origin, both platelets and neurons exhibit the same responses in many neurological and mental disorders, such as Alzheimer’s diseases, Parkinson’s diseases, various forms of schizophrenia, and depression [25]. For example, platelets express large amounts of APP (amyloid precursor protein); the highest concentration of this protein was found in the brain and in platelets [28]. It was suggested that changes observed in the brain in Alzheimer’s diseases are reflected in platelets [29].
According to Pearse [30], both platelets and neurons are components of the APUD (amine precursor uptake and decarboxylation) system. Platelets, as fragments of megakaryocytes, lack the nuclei; however, they contain a large number of mitochondria, dense granules (organelles containing small molecules, such as ADP and others), and α‑granules (reservoirs of serotonin and secretory proteins). Moreover, both platelets and neurons have a system for the active serotonin transport, binding sites for the neurotransmitters and drugs, dense bodies (serotonin-storing granules), and mitochondria containing MAO-B [25, 29]. Platelets express exclusively MAO‑B, which has the same amino acid sequence as the brain enzyme [31, 32].
MENTAL DISORDERS
Depression is one of the most common mental diseases and causes of disability [33, 34]. It places the most severe clinical, emotional, socioeconomic burden on a person and society as a whole. Approximately 300 million people suffer from depression, with 5 to 17% of the human population experiencing depression at least once in a lifetime. In 1999, the World Bank predicted that by 2020, the number of people suffering for depression would increase, and depression would become the second major cause of disability after cardiovascular diseases [35]. According to the analysis conducted by the World Health Organization in 2012, depression will be the leading cause of disability by 2030 [36]. In reality, depression has become the leading cause of disability in Europe already by the end of the 2020s [37].
Depression is also a risk factor in many somatic diseases [34]. It aggravates the disease course and aggravates the risk of complications and premature death [38]. Depression presents a serious socio-medical problem that will become more urgent in the coming years. Hence, comprehensive studies of depression and its pathogenetic mechanisms are currently one of the major tasks of medical science. Special emphasis in these studies is placed on prognosis and evaluation of the efficacy of therapeutic interventions, due to their high importance.
Treatment of patients suffering from depression and anxiety, i.e., diseases with closely related pathogenetic mechanisms, requires choosing between the thymoleptic (antidepressant) and anxiolytic (anti-anxiety) preparations [38].
Introduction of anti-depressants selectively acting on the serotoninergic system (serotonin reuptake inhibitors and activators) into clinical practice could significantly improve the efficacy of therapy and the patient’s quality of life. However, this requires further investigations of the neurochemical mechanisms underlying the effects of these preparations, as well as clarification of indications for their use and studies of the clinical and metabolic picture in the process of treatment.
In our studies of the mixed anxiety-depressive disorder, the condition of the patients was evaluated according to the ICD-10 (International Statistical Classification of Diseases, 10th revision) as a single episode of the major depressive disorder (F32.1) and in the context of recurrent depressive disorder (F33.1). The main criterion for the patient inclusion in this study was the presence of the anxiety component in the depressive disorder.
The use of a combination of biochemical and neurochemical parameters developed at the Moscow Research Institute of Psychiatry [39, 40] allowed us to identify significant disruptions in the metabolism of monoaminergic neurotransmitters in the patients.
According to the Hamilton Depression Rating Scale (HDRS), the average score of the patients before the therapy was 21.8, which corresponded to severe depression. The average anxiety score according to the Hamilton Anxiety Rating Scale (HARS) was 18.4, which corresponded to a moderate severity anxiety [41].
The activity of MAO in the platelets of patients before the therapy was significantly (by 94%) higher than in the control group (16.91 vs. 8.68 nmol benzaldehyde/mg protein per hour, respectively). It should be mentioned that the blood plasma contains semicarbazide-sensitive amine oxidase (SSAO) that can interfere with the activity of platelet MAO-B; hence, particular attention was paid to the process of platelet isolation [39, 40].
The cortisol content in the blood serum of patients was more than 10 times higher than the upper limit of the normal cortisol level (306 vs. 30.2 µg/dl, respectively).
Th antidepressant therapy with tianeptine significantly improved the state of the patients: the average HDRS score after 14-day therapy was 18.0; the average HARS score was 13.1. The treatment caused statistically significant 10% decrease in the average MAO activity in platelets in comparison with the enzyme activity before the treatment on the background of improved clinical signs [40]. In 2017, our results were confirmed in the study by Zeb et al. [42]. The authors established that the platelet MAO activity in the patients with depression prior to the treatment was on average 25% higher than in the control group. The two-week treatment with the antidepressant fluoxetine caused a reliable decrease in the MAO activity (by 20%).
Our data on the increased MAO activity in the platelets of patients with depression were also confirmed by the Meyer group [43, 44]. To determine the density (content) of MAO-B in the brain of patients suffering from depression, the researchers employed the high-resolution PET using radioactively labeled MAO ligand [11C]SL25.1188 ([(S)-5-methoxymethyl-3-[6-(4,4,4-trifluorobutoxy)-benzo[d]isoxazol-3-yl]-oxazolidin-2-[11C]one]) [43]. It was found that the MAO‑B Vt in depression was increased by 26% on average in almost all cerebral cortex regions in comparison with the control group. The differences between the patients and the control group were most pronounced in the cortical regions proximal to the ventrolateral area of the prefrontal cortex and in the thalamus. The authors suggested that the longer a patient suffered from depression, the higher was the density (content) of MAO-B in the brain cortex (ventrolateral, dorsolateral, orbitofrontal, temporal, and occipital areas, inner parietal lobe) and the thalamus [44].
Hence, our study [40] and works by the Meyer group [44] demonstrated a direct association between the MAO (MAO-B) activity in platelets and density of the MAO‑B protein in the brain of patients with depression. Summarizing the results of the above-mentioned studies, it can be stated that the platelets indeed are the “window into the brain” [24].
The situation with MAO‑A studies is different. The content of MAO‑A was also evaluated based on the protein density index. Human brains were examined in vivo by PET using [11C]‑harmine as a MAO‑A ligand [20-22]. [11C]‑harmine displays both high affinity (Ki = 2 nM) and selectivity toward MAO-A. It was established that the MAO‑A Vt in the patients with depression was increased by 34% on average in all brain areas [19, 21]. The authors suggested that the density (content) of MAO-A in the brain tissue positively correlated with the MAO‑A activity [19, 21].
It must be mentioned that the lack of studies and, correspondingly, the data on the dynamics of MAO-A density during pharmacotherapy is an essential drawback of these works.
What is the mechanism responsible for the increase in the MAO-A protein content in depression? Under normal conditions, the CDCA7L protein (cell division cycle-associated 7-like protein, also called protein R1) represses transcription of the MAOA gene by interacting with the binding site of the transcription factor Sp1 (simian virus 40 promoter factor 1) in the gene promoter. This process regulates MAO‑A biosynthesis and maintains its balance with other cellular components [45]. Another situation is observed in depression. Johnson et al. [46] studied the post-mortem brains of patients with depression to assess the content of R1 and MAO‑A proteins and the catalytic activity of MAO‑A in patients not subjected to therapy (untreated group) and patients treated with antidepressants (treated group). The authors demonstrated that the assessment of the MAO‑A catalytic activity in the post-mortem human brain was specific and concluded that:
-
the content of R1 was significantly lower in both untreated and treated patients (by 37.5 and 30.3%, respectively) vs. the control. This suggested that the antidepressant therapy did not affect the content of the R1 protein;
-
expression of MAO‑A in the prefrontal cortex was significantly higher in the untreated (by 40% on average) and treated (by 39% on average) patients vs. the control;
-
the catalytic activity of MAO‑A in the prefrontal cortex was higher in both treated (by 19%) and untreated (by 24.5%) patients in comparison with the control [46].
Interestingly enough, the observed increase in the MAO‑A enzymatic activity (24%), was significantly less pronounced than the increase in the MAO-A protein content (40%). This observation is in agreement with our previous suggestion that the content (density) of the protein does not directly reflect its enzymatic activity.
Hence, the post-mortem examination of the brains of patients with depression demonstrated that the level of the R1 protein in the prefrontal cortex was significantly lower in comparison with the control, which was likely the reason for the increase in the MAO-A content and, respectively, its enzymatic activity [46].
The in vivo studies of MAO‑A conducted by the Meyer group [21, 46] using PET with [11C]‑harmine in patients suffering with depression and in the post-mortem brain samples are very informative and interesting; however, they are of fundamental nature and lack clinical relevance, as they cannot be applied in a regular psychiatric practice. Such clinical applications require the use of peripheral parameters that would reflect the level and activity of MAO-A in the brain. Unfortunately, no such parameters have been identified in the blood so far. Nevertheless, the studies conducted by the Meyer group could be of interest to pharmacologists and chemists in their search for specific MAO-A inhibitors.
Impaired MAO activity can disturb the metabolic processes in the brain by [47]:
-
disrupting the metabolism of monoamine neurotransmitters and neuromodulators;
-
shifting the balance between the major monoamine neurotransmitters (dopamine, noradrenaline, and serotonin);
-
disrupting the mitochondrial membrane structure;
-
activating free-radical reactions (via hydrogen peroxide);
-
increasing of the level of toxic compounds (aldehydes, ammonia).
Excessive expression of MAO‑B accompanied by the activation of free-radical reactions is manifested as the disruption of mitochondrial functions. The levels of pyruvate dehydrogenase, succinate dehydrogenase, and mitochondrial aconitase decrease and the activity of the mitochondrial complex 1 is inhibited due to the formation of dopaminochrome from dopamine [48]. It should be mentioned that the decreased activity of the mitochondrial complex 1 in the prefrontal cortex has been observed in patients with depression [49].
In 2001, Kasper [50] suggested that depression and anxiety are controlled not by the absolute levels of serotonin and norepinephrine, but by the balance between the activities of the serotoninergic and noradrenergic systems. This implies that the regulation of the metabolism of these monoamines in the patients with depression and anxiety is disrupted. The ‘balance’ hypothesis is in agreement with the fact that the raphe nuclei (serotoninergic system center in the brain) and locus coeruleus (noradrenergic system center) are interrelated and interdependent structures [51].
We believe that the ‘balance’ hypothesis suggested by Kasper is based on the concepts proposed and experimentally proven by Gromova in the middle of 1970s. According to Gromova [51], the serotoninergic and noradrenergic systems interact reciprocally, i.e., activation of one system leads to the inhibition of the other and vice versa. The balance between the activities of these systems to a large extent defines the emotional reactivity. Serotonin and norepinephrine play specific roles in the pathogenetic mechanisms of anxiety and depression due to their modulating functions. The activity of these monoaminergic systems is controlled by other neurotransmitter systems, such as the GABAergic system [52]. Moreover, any of these neurotransmitters can initiate a cascade of processes that cause anxiety in patients and gradually form the depressive state [53]. Disruptions in the regulation of the serotoninergic system (its inhibition) facilitate the hyperactivation of the noradrenergic system, which might exacerbate the depressive symptoms [50].
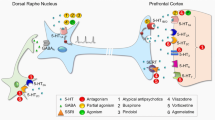
Studying biochemical mechanisms involved in the disruption of the balance between the serotoninergic and noradrenergic systems is of both theoretical and practical interest. Analysis of data of clinical/biochemical studies and experimental works conducted with the models of depression [54], together with our own data, allowed us to suggest a plausible neurochemical mechanism underlying these disruptions in the mixed anxiety-depressive disorder [55, 56] (figure).

As mentioned above, mixed anxiety-depressive disorder is accompanied by the increase in the MAO activity in the platelets and upregulation of the cortisol secretion. We have proposed a hypothesis on the possible mechanism of glucocorticoid involvement in the MAO activation and suppression of the functional activity of serotoninergic and noradrenergic systems in the brain [40, 55]. The decrease of the functional activity of the serotoninergic and noradrenergic systems in the mixed anxiety-depressive disorder eliminates the control of the monoaminergic system over glucocorticoid secretion, as manifested by the activation of cortisol synthesis (figure). Increased cortisol levels associated with depression [40, 55] activate tryptophan pyrrolase, thus redirecting the metabolism of tryptophan (serotonin precursor) to other metabolic pathways (e.g., kynurenine pathway) [57], which, in turn, causes reduction in the serotonin biosynthesis [58]. On the other hand, upregulated cortisol secretion promotes the activity of tyrosine transaminase, which could decrease the fraction of tyrosine used for the biosynthesis of catecholamines and reduce the level of these compounds, in particular, norepinephrine [59].
MAO contributes significantly to the serotonin and norepinephrine content in the brain. As mentioned above, the activity of platelet MAO reflects the activity of the brain enzyme [32]. It is possible that high cortisol levels can promote MAO activity. As we showed earlier, the deaminating activity of MAO in patients with the mixed anxiety-depressive disorder was on average 2 times higher than in the control. It must be mentioned that MAO deaminates serotonin and noradrenaline at different rates [16], which could disrupt the balance between these compounds in the mixed anxiety-depressive disorder.
What are the mechanisms associated with the MAO activity increase in depression?
It has been established that disturbances of metabolism observed in different forms of depression are associated with the increased secretion of glucocorticoids [11, 40, 44, 45, 55, 57]. In turn, upregulated expression of MAO in depression is accompanied by the increased secretion of glucocorticoids [44, 45, 55], which activate MAOB transcription via the following mechanisms:
-
glucocorticoids bind to the glucocorticoid response element 4 (GRE4) in the MAOB promoter and activate gene transcription [44];
-
increased MAO activity could be associated with the decrease in the activity of the transcription factors EAPP (E2F-associated phosphoprotein) and R1 (also designated as RAM2/CDCA7L/JPO2). Under normal conditions, EAPP and R1 inhibit MAOB transcription by interacting with the Sp1-binding sites in the center of the MAOB promoter. The binding of glucocorticoids to GRE4 with simultaneous decrease in the binding activity of EAPP and R1 allows endogenous Sp1 to promote MAOB transcription [45, 60];
-
glucocorticoids stimulate expression of TIEG2 (transforming growth factor-β-inducible early gene 2) that interacts with the proximal region of the Sp1-binding site, thus upregulating MAOB transcription [61].
It was shown that the activity of R1 (inhibitor of MAOB transcription in the cell culture) in the prefrontal cortex of patients with depression is decreased, while the activity of TIEG2 (activator of MAOB transcription in vitro) is increased [45, 60]. We believe that the increase in the MAO activity could be associated with changes in the protein conformation. This hypothesis is based on the results of our studies demonstrating that the conformation of the blood serum albumin in patients suffering from the mixed anxiety-depressive disorder [62] and melancholic depression [63] was significantly disturbed.
Therefore, activation of the hypothalamic-pituitary-adrenal axis and excessive secretion of cortisol are some of the most significant impairments occurring in depression. Analysis of the published data had brought our attention to the necessity of the introduction of drugs decreasing glucocorticoid secretion to the complex antidepressant therapy [56, 64].
According to the results of presented studies, MAO activity in platelets could serve as a potential biomarker of the efficacy of antidepressant pharmacotherapy.
First episode of schizophrenia. Schizophrenia is a severe mental disorder manifested by the pathological brain functioning and changes in behavior. Approximately 1% of the human population suffers from this disease. The cardinal signs of schizophrenia include positive (hallucinations, delusions) and negative (apathy, abulia, emotional and social withdrawal) symptoms, as well as cognitive impairments and disrupted social functioning [65]. The disease is characterized by impaired development and structure of the nervous system and abnormal behavior. It is assumed that such pathological changes are associated with disruptions in the functioning of the genetic apparatus and pathological effects of different environmental factors that facilitate the oxidative stress development and cause damage of the structural elements in the brain [66].
At present, the etiology of mental disorders (schizophrenia, depression) remains unknown. Numerous studies have been focused on the identification of genes and polymorphisms associated with the development of mental disorders. However, single-gene mutations cannot explain the mental disease phenotype and no monogenic forms of schizophrenia or depression have been described so far. It is believed that these diseases result from complex interactions between genetic, epigenetic, and environmental factors [8].
Special attention is paid to early diagnostics and timely help to patients with FES. Early interventions can accelerate disease remission and lessen social inadequacy, as well as mitigate neurocognitive deficit and improve the quality of such patients [65]. Hence, comprehensive investigation of pathophysiological mechanisms of FES is a priority. The pathogenetic mechanisms of FES are still poorly understood despite intensive research in this area [67, 68].
An alternative to genetic studies is the search for disease biomarkers. However, only few studies of biochemical processes in the patients with FES have been conducted, and their results are often contradictory.
Based on the known fact that the monoamine metabolism is disrupted in patients with schizophrenia, we decided to investigate the activity of MAO, as the key enzyme of monoamine metabolism, in patients with FES. We were able to find only one relevant study [69], which showed that the MAO activity in platelets of patients with FES was the same as in the healthy volunteers. According to another study, MAO activity in the platelets decreased in the patients with chronic schizophrenia [70]. On the contrary, our study of FES patients revealed almost a 2-fold increase in the activity of this enzyme [71]. Analysis of the data on the monoamine metabolism allows to propose the reason for such increase in the platelet MAO activity in the FES patients prior to the treatment. Based on the data of PET and SPECT studies, Laruelle and Abi-Dargham [72] suggested hyperactivation of dopaminergic neurotransmission in patients with FES, resulting in the increased release of dopamine into the synaptic cleft. Moreover, the binding activity of the dopamine transporter responsible for the dopamine reuptake from the synaptic cleft to the presynaptic endings was decreased in the patients with FES [73], which also promoted excessive dopamine accumulation in the synaptic cleft. It can be presumed that the observed increase in the MAO activity in FES patients is a compensatory mechanism aimed to decrease the concentration of dopamine in the synaptic cleft. This concept is in agreement with the data in the study [74], that reported significantly increased levels of homovanillic acid (product of MAO-catalyzed dopamine deamination) in patients with FES in comparison with the control.
Monoamine neurotransmitters secreted into the synaptic cleft are captured by the MAO-containing astrocytes following their interaction with the respective receptors [6]. Chemical inactivation of monoamines (deamination) occurs in the astrocytes. The increase in the MAO activity aimed to reduce the elevated dopamine levels has both positive and negative consequences. In particular, platelets of patients with FES exhibit significantly lower serotonin content [75] resulting from either decrease in the synthesis of this neurotransmitter or activation of serotonin deamination by MAO. Hence, an increase in the MAO activity can disrupt the balance between monoamine neurotransmitters. On the other hand, the shift of this balance caused by changes in the glutamatergic system activity cannot be entirely ruled out [76].
The consequences of the increased MAO activity could be evaluated from several points of view. Firstly, such increase indicates disruptions in the monoamine metabolism. Considering that MAO displays different affinity to dopamine and serotonin [16], deamination of these compounds occurs at different rates. It can be assumed that not only the absolute concentrations of these neurotransmitters change in the patients with FES, but the balance between them is disturbed as well. Secondly, MAO is an integral component of the outer mitochondrial membrane. Disruption of the activity of this enzyme in FES patients could be accompanied by the damage to the membrane structures and emergence of toxic products in the blood. Thirdly, the increase in the MAO activity results in the elevated production of hydrogen peroxide. Hydrogen peroxide formed in the MAO-catalyzed deamination reaction is the main source of free radicals in the brain [77, 78]. Hence, the increase in the MAO activity in patients with FES might activate free-radical reactions and lipid peroxidation. Fourthly, the products of the monoamine deamination reaction catalyzed by MAO include aldehyde and ammonia. The increase in the MAO activity leads to the increased production of these toxic compounds, contributing to the endotoxicosis [47].
As mentioned above, we investigated MAO activity in patients with FES [71]. Regression and factorial analysis of the obtained data revealed a positive correlation between the MAO activity and disease severity according to the positive and negative syndrome scale (PANSS). Hence, it is expected that the activity of MAO increases with the increase in the disease severity. Based on these data, we can state with sufficient certainty that MAO is a specific component of the pathogenetic mechanisms of the first episode of schizophrenia.
The average PANSS score was 81.4 before the antipsychotic therapy with risperidone (3-6 mg per day for 45 days) and decreased to 58 after the treatment. This was accompanied by a significant decrease in the platelet MAO activity (p = 0.04) in comparison with the levels detected prior to the treatment. Nevertheless, even after the treatment, the activity of MAO was significantly higher than in the control group, indicating that the therapy failed to normalize the MAO activity and metabolism as a whole [79]. In line with these observations, Ivanova et al. [80] reported that the antipsychotic treatment of patients with chronic schizophrenia noticeably improved the clinical signs, but the metabolic processes, e.g., those involving glucose-6-phosphate dehydrogenase (one of most important enzymes), remained significantly disrupted.
Statistical analysis revealed a positive correlation between the MAO activity in platelets and PANSS score both before and after the antipsychotic therapy [79]. Our results suggest that the platelet MAO activity could serve as a potential biomarker of the pharmacotherapy efficacy in FES patients. Based on the results, the following conclusions can be made:
-
1.
The fact that changes in the MAO activity in FES and mixed anxiety-depressive disorder are very similar is of particular interest. Although the reason for this similarity is currently unknow, it can be presumed that it involves genetic factors, most likely, complex interactions of numerous genes. As a result, similar metabolic disruptions in an organism can cause different sets of symptoms. Systemic approach could help in understanding these processes [2]. Changes in the neurotransmission and monoaminergic system in schizophrenia and depression are mostly associated with dopamine and serotonin, respectively. One can presume that MAO predominately deaminates dopamine in schizophrenia and serotonin and, partially, norepinephrine – in depression.
-
2.
Although pharmacotherapy significantly improves the clinical signs in patients with the first episode of schizophrenia, the metabolic processes remain disrupted.
CONCLUSIONS
It can be concluded based on the results of our studies that the platelet MAO could serve as a potential biomarker of the efficacy of pharmacotherapy of mental disorders such as first episode psychosis and depression.
Abbreviations
- FES:
-
first-episode of schizophrenia
- MAO:
-
monoamine oxidase
- R1:
-
factor inhibiting MAOB gene transcription
- Sp1:
-
transcription factor Sp1 (simian virus 40 promoter factor 1)
References
Scarr, E., Millan, M., Bahn, S., Bertolino, L., Turck, C., et al. (2015) Biomarkers for psychiatry: the journey from fantasy to fact, a report of the 2013 CINP think tank, Int. J. Neuropsychopharmacol., 18, 1-9, https://doi.org/10.1093/ijnp/pyv042.
Uzbekov, M. G., Gurovich, I. Ya., and Ivanova, S. A. (2016) Potential biomarkers of mental disorders from the standpoint of systems biology, Soc. Clin. Psychiatry, 26, 98-109.
Lopresti, A. L., Maker, G. L., Hood, S. D., and Drummond, P. D. (2014) A review of peripheral biomarkers in major depression: the potential of inflammatory and oxidative stress biomarkers, Prog. Neuropsychopharmacol. Biol. Psychiatry, 48, 102-111, https://doi.org/10.1016/j.pnpbp.2013.09.017.
Kim, H., Blumberger, D., Downar, J., and Daskalakis, Z. (2020) Systematic review of biological markers of therapeutic repetitive transcranial magnetic stimulation in neurological and psychiatric disorders, Clin. Neurophysiol., 132, 429-448, https://doi.org/10.1016/j.clinph.2020.11.025.
Zafar, T. (2020) Potential biomarkers of emotional stress induced neurodegeneration, eNeurologica Sci., 21, 100292-100297, https://doi.org/10.1016/j.ensci.2020.100292.
Uzbekov, M. G. (2009) Antidepressant action of tianeptine is connected with acceleration of serotonin turnover in the synapse: a hypothesis, Neuropsychopharmacol. Hung., 10, 85-89.
Alawieh, A., Zaraket, F., Li, J., Mondello, S., Nokkari, A., and Razafsha, M. (2012) Systems biology, bioinformatics and biomarkers in neuropsychiatry, Front. Neurosci., 6, 187-193, https://doi.org/10.3389/fnins.2012.00187.
Cacabelos, R., Hashimoto, R., and Takeda, M. (2011) Pharmacogenomics of antipsychotics efficacy for schizophrenia, Psychiatry Clin. Neurosci., 65, 3-19, https://doi.org/10.1111/j.1440-1819.2010.02168.x.
Linden, D. E. (2012) The challenges and promise of neuroimaging in psychiatry, Neuron, 73, 8-22, https://doi.org/10.1016/j.neuron.2011.12.014.
Stober, G., Ben-Shachar, D., Cardon, M., Falkai, P., Fonteh, A. N., et al. (2009) Schizophrenia: From the brain to peripheral markers – a consensus paper of the WFSBP Task Force on biological markers, World J. Biol. Psychiatry, 10, 127-155, https://doi.org/10.1080/15622970902898980.
Nobis, A., Zalewski, D., and Waszkiewicz, N. (2020) Peripheral markers of depression. Review, J. Clin. Med., 9, 3793-3847, https://doi.org/10.3390/jcm9123793.
Shih, J. C., Chen, K., and Ridd, M. J. (1999) Monoamine oxidase: from genes to behavior, Annu. Rev. Neurosci., 22, 197-217, https://doi.org/10.1146/annurev.neuro.22.1.197.
Bach, A. W. J., Lan, N. C., Johnson, D. L., Abell, C. W., Bemkenek, M. E., et al. (1988) cDNA cloning of human liver monoamine oxidase A and B: molecular basis of differences in enzymatic properties, Proc. Natl. Acad. Sci. USA, 85, 4934-4938.
Lan, N. C., Heinzmann, C., Gal, A., Klisak, I., Orth, U., et al. (1989) Human monoamine oxidase A and B genes map to Xp 11.23 and are deleted in a patient with Norrie disease, Genomics, 4, 552-559.
Gorkin, V. Z. (1981) Aminoxidases and their importance in medicine, Meditsina, Moscow.
Medvedev, A. E., and Gorkin, V. Z. (1992) Biogenic amines and monoamine oxidases in the regulation of activities of membrane-bound mitochondrial enzymes, Biogenic Amines, 8, 323-338.
Gorkin, V. Z., and Medvedev, A. E. (1994) Monooxidase, Proteins and Peptides (Ivanov, V. T., and Lipkin, V. M., eds) vol. 1, Nauka, Moscow.
Shih, J. C. (2018) Monoamine Oxidase isoenzymes: genes, functions and targets for behavior and cancer therapy, J. Neural Transm. (Vienna), 125, 1553-1566, https://doi.org/10.1007/s00702-018-1927-8.
Tong, J., Meyer, J., Furukawa, Y., Boileau, I., Chang, L.-J., et al. (2013) Distribution of monoamine oxidase proteins in human brain: implications for brain imaging studies, J. Cereb. Blood Flow Metab., 33, 863-871, https://doi.org/10.1038/jcbfm.2013.19.
Bergstrom, M., Westerberg, G., and Langstrom, B. (1997) 11C-harmine as a tracer for monoamine oxidase A (MAO-A): in vitro and in vivo studies, Nucl. Med. Biol., 24, 287-293.
Meyer, J. H., Ginovart, N., Boovariwala, A., Sagrati, S., Hussey, D., et al. (2006) Elevated monoamine oxidase a levels in the brain: an explanation for the monoamine imbalance of major depression, Arch. Gen. Psychiatry, 63, 1209-1216, https://doi.org/10.1001/archpsyc.63.11.1209.
Sacher, J., Rekkas, P., Wilson, A., Houle, S., Romano, S., et al. (2015) Relationship of monoamine oxidase-a distribution volume to postpartum depression and postpartum crying, Neuropsychopharmacology, 40, 429-435, https://doi.org/10.1038/npp.2014.190.
Brilliantova, V. V., Smolina, N. V., Syreishchikova, T. I., Uzbekov, M.G., and Dobretsov, G. E. (2018) The state of albumin thiol groups in patients with the first episode of schizophrenia, Naurochem. J., 35, 96-100, https://doi.org/10.7868/S102781331801003X.
Stahl, S. M. (1985) Peripheral models for the study of neurotransmitter receptors in man, Psychopharmacol. Bull., 21, 663-671.
Asor, E., and Ben-Shachar, D. (2012) Platelets: a possible glance into brain biological processes in schizophrenia, World J. Psychiatry, 22, 124-133, https://doi.org/10.5498/wjp.v2.i6.124.
Paasonen, M. K., Solatunturi, E., and Kivalo, E. (1964) Monoamine oxidase activity of blood platelets and their ability to store 5-hydroxytryptamine in some mental deficiencies, Psychopharmacologia, 6, 120-124.
George, J. (2000) Platelets, Lancet, 355, 1531-1539.
Bush, A. I., Martins, R. N., Rumble, B., Moir, R., Fuller, S., et al. (1990) The amyloid precursor protein of Alzheimer’s disease is released by human platelets, J. Biol. Chem., 265, 15977-15983.
Veitinger, M., Varga, B., Guterres, S. B., and Zellner, M. (2014) Platelets, a reliable source for peripheral Alzheimer’s disease biomarkers? Acta Neuropathol. Commun., 2, 65-80, https://doi.org/10.1186/2051-5960-2-65.
Pearse, A. G. (1977) The diffuse neuroendocrine system and the apud concept: related “endocrine” peptides in brain, intestine, pituitary, placenta, and anuran cutaneous glands, Med. Biol., 55, 115-125.
Youdim, M. B. (1988) Platelet monoamine oxidase B: use and misuse, Experientia, 44, 137-141.
Chen, K., Wu, H. F., and Shih, J. C. (1993) The deduced amino acid sequences of human platelet and frontal cortex monoamine oxidase B are identical, J. Neurochem., 61, 187-190.
Goodvin, F. K., and Jamison, K. R. (2007) Maniac Depressive Illness: Bipolar Disorders and Recurrent Depression, 2nd Edn., Oxford University Press, N.Y.
Kessler, R. C., and Ustün, T. B. (2008) The WHO World Mental Health Surveys: Global Perspectives on the Epidemiology of Mental Disorders, Cambridge University Press, N.Y.
Kupfer, D. (1999) Depression: a major contributor to world-wide disease burden, Int. Med. News, 99, 1-2.
Health Assembly (2012) Global Burden of Mental Disorders and the Need for a Comprehensive, Coordinated Response from Health and Social Sectors at the Country Level, Report by The Secretariat World Health Organization, Geneva, Switzerland.
Spinney, L. (2009) European brain policy forum: depression and the european society, Eur. Psychiatry, 24, 550-551, https://doi.org/10.1016/j.eurpsy.2009.04.001.
Krasnov, V. N. (2012) Problems of modern diagnostics of depression, S. S. Korsakov J. Neurol. Psychiatry, 112, 3-11.
Uzbekov, M. G., Misionzhnik, E. Y., Shmukler, A. B., Gurovich, I. Ya., Gryzunov, Yu. A., et al. (2009) Disruption of monoamine oxidase activity and indices of endogenous intoxication in patients with the first episode of schizophrenia, S. S. Korsakov J. Neurol. Psychiatry, 109, 48-52.
Uzbekov, M. G., Misionzhnik, E. Y., Maximova, N. M., and Vertogradova, O. P. (2006) Biochemical profile in patients with anxious depression under the treatment with serotonergic antidepressants with different mechanisms of action, Hum. Psychopharmacol. Clin. Exp., 21, 109-115, https://doi.org/10.1002/hup.749.
Hamilton, M. (1988) In Handbook of Anxiety Disorders (Last, C., and Hersen, M., eds.) Pergamon Press, Oxford, pp. 143-155.
Zeb, F., Naqvi, S., Rahman, R., and Farooq, A. D. (2017) Depressive symptoms, monoamines levels, MAO-B activity and effect of treatment in a subset of depressed individuals from government sector hospital at Karachi, Pak. J. Pharm. Sci., 30, 1509-1519.
Rusjan, P., Wilson, A., Miler, L., Fan, I., Mizrahi, R., et al. (2014) Kinetic modeling of the monoamine oxidase B radioligand [11C]SL25.1188 in human brain with high-resolution positron emission tomography, J. Cereb. Blood Flow Metab., 34, 883-889, https://doi.org/10.1038/jcbfm.2014.34.
Moriguchi, S., Wilson, A., Miler, L., Rusjan, P., Vasdev, N., et al. (2019) Monoamine oxidase B total distribution volume in the prefrontal cortex of major depressive disorder. An [11C]SL25.1188 positron emission tomography study, JAMA Psychiatry, 76, 634-641, https://doi.org/10.1001/jamapsychiatry.2019.0044.
Higuchi, Y., Soga, T., and Parhar, I. (2017) Regulatory pathways of monoamine oxidase A during social stress, Front. Neurosci., 11, 604-615, https://doi.org/10.3389/fnins.2017.00604.
Johnson, S., Stockmeier, C. A., Meyer, J. H., Austin, M. C., Albert, P. R., and Wang, J. (2011) The reduction of R1, a novel repressor protein for monoamine oxidase A, in major depressive disorder, Neuropsychopharmacology, 36, 2139-2148, https://doi.org/10.1038/npp.2011.105.
Uzbekov, M. G. (2019) Endogenous intoxication and its role in the pathogenetic mechanisms of mental disorders, Soc. Clin. Psychiatry, 29, 14-20.
Mallajosyula, J. K., Chinta, S. J., Rajagopalan, S., Nicholls, D. G., and Andersen, J. K. (2009) Metabolic control analysis in a cellular model of elevated MAO-B: relevance to Parkinson’s disease, Neurotox. Res., 16, 186-193, https://doi.org/10.1007/s12640-009-9032-2.
Andreazza, A. C., Shao, L., Wang, J. F., and Young, L. T. (2010) Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder, Arch. Gen. Psychiatry, 67, 360-368, https://doi.org/10.1001/archgenpsychiatry.2010.22.
Kasper, S. (2001) Depression and anxiety – separate or continuum, World J. Biol. Psychiatry, 2, 162-163, https://doi.org/10.3109/15622970109026804.
Gromova, E. A. (1979) In Catecholaminergic Neurons (Turpaeva, T. M., and Budantsev, A. Yu., eds.) Nauka, Moscow, pp. 97-105.
Petty, F., Davis, L. L., Kabel, D., and Kramer, G. L. (1996) Serotonin dysfunction disorders: a behavioral neurochemistry perspective, J. Clin. Psychiatry, 5 (Suppl 8), 11-16.
Paul, S. M. (1988) Anxiety and depression: a common neurobiological substrate? J. Clin. Psychiatry, 49, 13-16.
Hoffman, K. L. (2013) Role of murine models in psychiatric illness drug discovery: a dimensional view, Expert Opin. Drug Discov., 8, 865-877, https://doi.org/10.1517/17460441.2013.797959.
Uzbekov, M. G., and Maximova, N. M. (2016) Monoamine-hormone interactions in the pathogenesis of anxious depression, Neurosci. Behav. Physiol., 46, 673-676, https://doi.org/10.1007/s11055-016-0295-9.
Uzbekov, M. G., and Maximova, N. M. (2018) Some neurobiological aspects of the pathogenesis of anxiety depression and antiglucocorticoid pharmacotherapy, Ross. Psikh. Zhurn., 2, 31-39.
Lapin, I. P. (2004) Stress – Anxiety – Depression – Alcoholism – Epilepsy (Neurokynurenine mechanisms and new approaches to treatment), DEAN, St.-Petersburg.
Curzon, P. (1969) Tryptophan pyrrolase – a biochemical factor in depressive illness, Br. J. Psychiatry, 115, 1367-1374.
Nemeth, S. (1978) The effect of stress or glucose feeding on hepatic tyrosine aminotransferase activity and liver and plasma tyrosine levels of intact and adrenalectomized rats, Horm. Metab. Res., 10, 144-147.
Chen, K., Ou, X. M., Wu, J. B., and Shih, J. C. (2011) Transcription factor E2F-associated phosphoprotein (EAPP), RAM2/CDCA7L/JPO2 (R1), and simian virus 40 promoter factor 1 (Sp1) cooperatively regulate glucocorticoid activation of monoamine oxidase B, Mol. Pharmacol., 79, 308-317, https://doi.org/10.1124/mol.110.067439.
Ou, X. M., Chen, K., and Shih, J. C. (2004) Dual functions of transcription factors, transforming growth factor-β-inducible early gene (TIEG)2 and Sp3, are mediated by CACCC element and Sp1 sites of human monoamine oxidase (MAO) B gene, J. Biol. Chem., 279, 21021-21028, https://doi.org/10.1074/jbc.M312638200.
Uzbekov, M., Syrejshchikova, T., Smolina, N., Maximova, N., Shikhov, S., and Brilliantova, V. (2019) Antidepressant Therapy has restored albumin conformation in anxious depression, Biomed. J. Sci. Tech. Res., 24, 16103-16105, https://doi.org/10.26717/BJSTR.2019.21.003647.
Uzbekov, M. G., Syrejshchikova, T. I., Smolina, N. V., Brilliantova, V. V., Dobretsov, G. E., and Shikhov, S. N. (2018) Serum albumin conformation in patients with melancholic depression under antidepressant therapy, Biomed. J. Sci. Tech. Res., 7, 1-2, https://doi.org/10.26717/BJSTR.2018.07.001540.
Lenze, E., Hershey, T., Newcomer, J., Karp, J., Blumberger, D., et al. (2014) Antiglucocorticoid therapy for older adults with anxiety and cooccurring cognitive dysfunction: results from a pilot study with mifepristone, Int. J. Geriatr. Psychiatry, 29, 962-969, https://doi.org/10.1002/gps.4085.
Gurovich, I. Ya., and Neufeldt, O. G. (2007) Modern Development Trends and New Forms of Mental Health Care, Medpraktika-M, Moscow.
Uzbekov, M. G. (2017) Biological Markers of Schizophrenia: Search and Clinical Application (Bokhan, N. A. and Ivanova, S. A., eds.) SB RAS, Novosibirsk, pp. 67-89.
Gurovich, I. Ya., Shmukler, A. B. (2010) First Psychotic Episode (Problems and Mental Health Care) Medpraktika-M, Moscow.
Albert, N., and Weibell, M. (2019) The outcome of early intervention in first episode psychosis, Int. Rev. Psychiatry, 31, 413-424, https://doi.org/10.1080/09540261.2019.1643703.
Spivak, B., Kosower, N., Zipzer, Y., Shreiber-Schul, N., Apter, A., et al. (1994) Platelet monoamine oxidase activity in neuroleptic-naïve schizophrenic patients: lack of influence of chronic perphenazine treatment, Clin. Neuropharmacol., 17, 83-88.
Ertugrul, A., Ucar, G., Basar, K., Demir, B., Yabanoglu, S., and Ulug, B. (2007) Influence of clozapine on platelet serotonin, monoamine oxidase and plasma serotonin level, Psychiatry Res., 149, 49-57, https://doi.org/10.1016/j.psychres.2005.12.009.
Uzbekov, M. G., Misionzhnik, E. Yu., Gurovich, I. Y., and Shmukler, A. B. (2013) Aspects of metabolic changes in first-episode drug-naïve schizophrenic patients, Acta Neuropsychiatrica, 25, 268-274, https://doi.org/10.1017/neu.2013.1.
Laruelle, M., and Abi-Dargham, A. (1999) Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies, Psychopharmacology, 13, 358-371.
Mateos, J. J., Lomena, F., Parellada, E., Font, M., Fernandez, E., et al. (2006) Striatal dopamine transporter density decrease in the first episode schizophrenic patients treated with risperidone, Rev. Esp. Med. Nucl., 25, 159-165, https://doi.org/10.1157/13088411.
Yoshimura, R., Ueda, N., Shinkai, K., and Nakamura, J. (2003) Plasma levels of homovanillic acid and the response to risperidone in the first episode untreated acute schizophrenia, Int. Clin. Psychopharmacol., 18, 107-11, https://doi.org/10.1097/00004850-200303000-00008.
Marcinko, D., Pivac, N., Martinac, M., Jakovljevic, M., Mihaljevic-Peles, A., and Muck-Seler, D. (2007) Platelet serotonin and serum cholesterol concentrations in suicidal and non-suicidal male patients with a first episode of psychosis, Psychiatry Res., 150, 105-108.
Tan, H. Y., Callicott, J. H., and Weinberger, D. R. (2007) Dysfunctional and compensatory prefrontal cortical systems, genes and the pathogenesis of schizophrenia, Cereb. Cortex, 17 (Suppl 1), 171-181, https://doi.org/10.1093/cercor/bhm069.
Beckman, K. B., and Ames, B. N. (1998) The free radical theory of aging matures, Physiol. Rev., 78, 547-581, https://doi.org/10.1152/physrev.1998.78.2.547.
Uzbekov, M. G. (2014) Lipid peroxidation and antioxidant systems in mental illness. Message I, Soc. Clin. Psychiatry, 24, 97-103.
Uzbekov, M., Brilliantova, V., Shikhov, S., Smolina, N., Syrejshchikova, T., et al. (2019) First episode of schizophrenia: potential biomarkers of pharmacotherapy efficacy, Eur. Neuropsychopharmacol., 29, S96-S97, https://doi.org/10.1016/j.euroneuro.2018.11.1081.
Ivanova, S. A., Smirnova, L. P., Shchigoreva, Yu. G., Boyko, A. S., Semke, A. V., et al. (2014) Activity of glucose-6-phosphate dehydrogenase and catalase in erythrocytes of patients with schizophrenia in the dynamics of pharmacotherapy with traditional antipsychotic drugs, Neurochem. J., 31, 79-83, https://doi.org/10.1134/S1819712414010061.
Acknowledgments
The author is grateful to all colleagues that participated in our studies: I. Ya. Gurovich, A. B. Shmukler, S. A. Ivanova, S. N. Shikhov, E. Yu. Misionzhnik, N. V. Smolina, N. M. Maximova, V. V. Brilliantova, and E. B. Skokina.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares no conflict of interests in financial or any other sphere. All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or with comparable ethical standards.
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Uzbekov, M.G. Monoamine Oxidase as a Potential Biomarker of the Efficacy of Treatment of Mental Disorders. Biochemistry Moscow 86, 773–783 (2021). https://doi.org/10.1134/S0006297921060146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297921060146