
Abstract
Signal transducer and activator of transcription 3 (STAT3) is a well-known antitumor target. Exogenous ROS insult can lead to selective cytotoxicity against cancer cells. A combination of STAT3 inhibition and “oxidation therapy” may be a new strategy to address the multidrug-resistance issue due to their important roles in the survival and drug resistance of cancer cells. Here, a series of novel curcumin-BTP hybrids were designed and evaluated as STAT3 inhibitors with ROS production activity. Compound 6b exerted the best antitumor activity and selectivity for MCF-7 and MCF-7/DOX cells (IC50 = 0.52 μM and 0.40 μM, respectively), while its IC50 value for MCF-10A breast epithelial cells was 7.72 μM. Furthermore, compound 6b suppressed STAT3 phosphorylation, nuclear translocation and DNA-binding activity and the expression of STAT3 specific oncogenes. Increases in the level of IL-6-induced p-STAT3 were also inhibited by 6b without influencing IFN-γ-induced p-STAT1 expression. Additionally, 6b effectively promoted intracellular ROS accumulation, induced cancer cell apoptosis and cell cycle arrest, abolished the colony formation ability of breast cancer cells, and inhibited P-gp expression in MCF-7/DOX cells. Finally, 6b suppressed the growth of implanted human breast cancer in vivo. Our findings highlight that 6b may be a promising therapeutic agent for drug-sensitive and drug-resistant breast cancers.
Similar content being viewed by others

Introduction
Breast cancer is the second most fatal cancer in women. The prognosis is generally regarded as poor and the overall survival rate of all stages is less than 1% at 5 years1. The rapid emergence of drug resistance also limits the benefits of treatment of this disease2. Therefore, it is imperative to develop more effective treatments for breast cancer.
Signal transducer and activator of transcription 3 (STAT3) is a type of latent cytosolic transcription factor3,4,5,6,7,8. Persistent activation of the STAT3 signaling pathway has been documented in a wide range of solid cancers and some drug-resistant cancers in humans and is commonly associated with a worse prognosis2,9,10. Interference with the STAT3 signaling pathway in cancer cells has been shown to result in growth inhibition and the induction of apoptosis, making it an attractive target for cancer therapies11,12,13,14,15,16,17,18,19. Moreover, STAT3 inhibition is a viable treatment option for drug-resistant cancers through inhibiting STAT3-mediated MDR1 gene expression2.
Cancer cells are usually exposed to a moderate level of reactive oxygen species (ROS), primarily due to their active metabolism in response to oncogenic signals20. In fact, moderate oxidative stress is conducive to several important processes, such as proliferation, angiogenesis, and metastasis, in cancer cells21. Nevertheless, high levels of ROS irreversibly damage DNA and lipids and ultimately cause cancer cell apoptosis22. An exogenous ROS insult is tolerable to normal cells, but it may exceed the threshold that cancer cells can tolerate and thus lead to selective cytotoxicity against cancer cells. The pharmacological elevation of intracellular ROS has emerged as an effective strategy for selectively targeting cancer cells23,24, which was considered as “oxidation therapy”25,26,27.
In 2011, Raj, L. et al. reported that piperlongumine selectively kills cancer cells by targeting their stress response to ROS24. In 2014, Bharadwaj, U. et al. reported that piperlongumine is a direct STAT3 inhibitor with potent activity against breast cancer28. The inhibition of STAT3 and increase in ROS production are antitumor activities that jointly promote cancer cell apoptosis.
Curcumin is a bioactive component in Curcuma longa and the subject of extensive research due to its relative safety in large dosages. The complex chemistry of curcumin allows it to inhibit multiple oncogenic processes, including those associated with the JAK2/STAT3 pathway or the induction of ROS production29,30,31. To improve the potency of curcumin as a cancer therapeutic agent, many synthetic curcumin analogues have been developed as new JAK2/STAT3 inhibitors, including FLLL31, FLLL32 and FLL6217,18,19. These curcumin-based compounds improve the bioavailability and anticancer potential by stabilizing the central β-diketone linker.
The benzo[b]thiophene 1, 1-dioxide (BTP) moiety contributes to ROS production and is also a crucial pharmacophore of many STAT3 inhibitors (such as Stattic, HJC0123 and HJC0146). Inhibitors with this moiety significantly inhibit p-STAT3 levels, markedly enhance the ROS level and exhibit anti-proliferative activity in cancer cells6,32,33. Moreover, Stattic, a STAT3 inhibitor containing this moiety, circumvented the cisplatin resistance of an orthotopic xenograft of ovarian cancer in vivo. Nevertheless, because of their low activity or poor properties, none of these inhibitors has reached the market yet.
Given that a STAT3 inhibitor with ROS-promoting activity may be a new strategy for the treatment of solid tumors, we hypothesized that these new hybrids of curcumin with a BTP moiety may inhibit STAT3 activation and lead to ROS accumulation and ultimately induce cancer cell apoptosis. To further improve metabolic stability, half of the central linker was stabilized with piperazine, which has an equal length and is widely used in the market drugs (Fig. 1). In this study, twenty-two curcumin-BTP hybrids were designed, synthesized, and biologically evaluated as STAT3 inhibitors with ROS production activity. These new STAT3 inhibitors are promising candidates for breast cancer therapy, due to their drug-like properties and potent bioactivities in vitro and in vivo.

Design strategy of the new curcumin-BTP hybrids.
Results and Discussion
Chemistry
The synthetic routes for compounds 6a–v are shown in Scheme 1 in the supporting information. Compounds 2a and 2b were obtained by the reaction of commercially available 3-bromo-2-fluorobenzaldehyde (1a) and 4-bromo-2-fluorobenzaldehyde (1b), respectively, with methyl thioglycolate. Coupling of 2a and 2b with tert-butyl-1-piperazinecarboxylate in the presence of HATU resulted in compounds 3a and 3b, respectively, which were then reacted with 3-chlorine peroxide benzoic acid to produce compounds 4a and 4b, respectively. Compounds 5a and 5b were prepared from 4a and 4b, respectively, by deprotecting N-Boc protection with chlorine hydride gas in methanol and dichloromethane (1:1). The desired compounds (6a-6v) were generated by the coupling of 5a and 5b with different cinnamic acids using the same synthetic method used to generate compounds 3a and 3b. All the hybrids were confirmed by 1H NMR, IR, HRMS (ESI), and 13C NMR spectra. The 1H NMR spectrums of representative compounds were shown in the S1 of the Supporting Information.
In vitro cell growth inhibition activity
Given that compounds 6a–v were designed to target STAT3 in cancer cells, we first examined the levels of p-STAT3 expression in MCF-7, MCF-7/DOX and MCF-10A cells by western blot assays. The levels of p-STAT3 expression in MCF-7 and MCF-7/DOX cells were significantly higher than that in MCF-10A cells (Fig. 2). To determine the structure-activity relationships of the synthetic compounds, MCF-7 and MCF-7/DOX (doxorubicin) cells were treated with the designed compounds and the positive controls, DOX and curcumin, and then their proliferative activity was determined by the MTT assay. The IC50 values are summarized in Table 1. Notably, all the compounds exhibited stronger anti-proliferative activity than their mother compound, curcumin. Compound 6b was the most potent inhibitor of MCF-7 and MCF-7/DOX cell growth with IC50 values of 0.52 μM and 0.40 μM, respectively, which was a marked improvement the anti-proliferative activity compared to that of curcumin (IC50 = 37.7 μM and 32.7 μM, respectively). In general, the hybrids with a 5-Br substitute on the BTP ring had a slightly stronger activity than the hybrids with a 6-Br substitute. Compounds with electron-withdrawing substitutes such as chlorine (6h and 6s) and fluorine (6v) on the benzene ring of the hybrids usually showed less inhibition activity than those with electron-donating substitutes (6a, 6b, 6c, 6j and 6u) with exception of 6i. Compounds with a methoxy substitute in the para-position of the benzene ring (6a, 6b, 6l, 6j, 6m and 6u) exerted more potent activity, while replacement of the methoxy group with a hydroxyl group (6d, 6o and 6t) led to a marked decrease in activity.

Western blot analysis of p-STAT3 levels in MCF-7, MCF-7/DOX and MCF-10A cells.
Subsequently, the anti-proliferative efficacy of the most potent compound, 6b, was further investigated using the MTT assay. DOX had similar growth inhibition activity in both tumor and normal cells, while 6b exhibited a 7- to 21-fold increase in anti-proliferative selectivity for cancer cells than for normal MCF-10A and LO2 cells (IC50 = 7.7 μM and 8.3 μM, respectively), suggesting that 6b may have selective anti-proliferative activity against cancer cells (Table 2).
Molecular docking
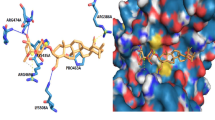
Given that these compounds were designed as STAT3 inhibitors, compound 6b was used to predict potential binding modes with STAT3 using docking assays. As shown in Fig. 3, 6b tightly bound to the SH2 domain of STAT3, indicating that 6b may be a STAT3-SH2 domain inhibitor. The docking mode of 6b perfectly overlapped with the binding mode of the native pTyr705-Leu706 peptide by forming three hydrogen bonding interactions with the Arg609, Lys626 and Gln635 residues. The predicted binding energy was −8.2 kcal/mol. In addition, the piperazine linker provided a suitable length and angle for the inhibitor to interact with the two binding pockets of STAT334. Together, 6b was inferred to be a direct STAT3 inhibitor.

The binding mode of 6b, native pTyr705-Leu706 peptide and STAT3 SH2 domain by AutoDock4.2.
Compound 6b inhibited STAT3 phosphorylation, nuclear translocation and DNA-binding activity in breast cancer cells
It was reported that targeting the STAT3-SH2 domain can effectively suppress the phosphorylation of STAT335. To determine the role of 6b as a STAT3 inhibitor, we investigated the effects of 6b on p-STAT3 (Tyr705) and p-STAT3 (Ser727) protein expression by western blot assays. Exposure of MCF-7 and MCF-7/DOX cells to 6b led to a dose-dependent inhibition of STAT3 phosphorylation at the pTyr705 residue but had little effect on STAT3 phosphorylation at the Ser727 residue or total STAT3 protein level (Fig. 4A).

(A) Western blot analysis of p-STAT3 (Tyr705), p-STAT3 (Ser727) and STAT3 in whole-cell lysates of equal total protein prepared from MCF-7 and MCF-7/DOX cells treated with compound 6b for 24 h. (B) EMSA analysis of nuclear extracts of equal total protein containing activated STAT3 from MCF-7 cells treated with compound 6b for 24 h after incubation with hSIE probe that binds STAT3. (C) MCF-7 cells were starved overnight followed by treatment with 6b or DMSO for 6 h. Cells were then treated with 50 ng/ml of IL-6 for 30 min. For immunofluorescent assay, cells were fixed and stained with anti-phospho-STAT3 (p-STAT3) and DAPI before subject to ImageXpress Micro Confocal analysis. Red: p-STAT3; blue: nuclei. (D) Western blot analysis of Bcl-2, Bax and Cyclin D1 in MCF-7 and MCF-7/DOX cells treated with compound 6b for 24 h.
Nuclear translocation of STAT3 plays a critical role in STAT3:STAT3/DNA complex formation and its target gene expression, so we further investigated the inhibitory effect of 6b on preventing STAT3 from translocating to the nucleus. As shown in Fig. 4B, p-STAT3 protein (red) was retained in both the cytoplasm and nucleus in MCF-7 cells not treated with 6b or IL-6. In MCF-7 cells treated with IL-6 at 50 ng/ml, an increased level of p-STAT3 protein (red) was observed in the nucleus. However, treatment with 6b led to a sharp decrease of the p-STAT3 level in the nucleus, suggesting that 6b suppressed STAT3 nuclear translocation.
As a STAT3 inhibitor, 6b is supposed to prevent dimeric STAT3 from binding to certain DNA sequences in the nucleus. Nuclear extracts of human breast MCF-7 cells treated with 6b were prepared and subjected to EMSA analysis using an hSIE probe. Compound 6b strongly inhibited STAT3 DNA-binding activity in a dose-dependent manner (Fig. 4C). Taken together, these results show that 6b effectively inhibited STAT3 activation.
Compound 6b regulated STAT3 target oncogenes in breast cancer cells
We had elucidated that 6b could effectively inhibit STAT3 phosphorylation, nuclear translocation and DNA-binding activity. To gain more insight into the antitumor activity and mechanistic effects of 6b on the STAT3 pathway, we examined its effect on STAT3 target genes such as Bcl-2 and Bax, which are related to proliferation, and Cyclin D1, which regulates the cell cycle. MCF-7 and MCF-7/DOX cells were treated with compound 6b and processed for western blot assays and qRT-PCR analysis. As shown in Fig. 4D, compound 6b inhibited the expression of Bcl-2 and Cyclin D1 and increased the expression of Bax. Similarly, Bcl-2 and Cyclin D1 gene levels were decreased in a dose-dependent manner and that of Bax was upregulated by 6b, while treatment with curcumin had little effect on these gene expression levels. The decreases in the gene expression level of Bcl-2 and at 1.0, 2.0, and 4.0 μM concentrations of 6b were 11.7%, 45.8% and 61.2%, respectively, in MCF-7 cells and 27.5%, 43.5% and 71.0%, respectively, in MCF-7/DOX cells. The decreases in the gene expression level of Cyclin D1 at 1.0, 2.0, and 4.0 μM concentrations of compound 6b were 16.0%, 28.4% and 45.5%, respectively, in MCF-7 cells and 18.5%, 51.0% and 75.0%, respectively, in MCF-7/DOX cells. Bax gene expression increased by 15.0%, 71.5% and 97.5% in MCF-7 cells and 14.0%, 76.5% and 125.0% in MCF-7/DOX cells when treated with 1.0, 2.0, and 4.0 μM concentrations of compound 6b, respectively (Fig. 5A).

(A) RT-PCR analysis of Bcl-2, Bax and Cyclin D1 in MCF-7 and MCF-7/DOX cells treated with compound 6b for 24 h with curcumin as reference. (B) Western blot analysis of P-gp levels in whole-cell lysates of equal total protein prepared from MCF-7, MCF-7/DOX cells and MCF-7/DOX cells treated with compound 6b for 24 h.
Compound 6b inhibited the expression of P-gp in MCF-7/DOX breast cancer cells
Previous studies have shown that overexpression of P-glycoprotein (P-gp) in cancer cells can lead to the export of anticancer drugs and cause consequent drug ineffectiveness, which is a leading factor for tumor multidrug-resistance (MDR) and inhibiting STAT3 activation could effectively decrease P-gp expression2,36. Thus, to further investigate the role of 6b in suppressing MCF-7/DOX cell proliferation, we examined the effect of 6b on P-gp in MCF-7/DOX cells. We determined the levels of P-gp expression in MCF-7 and MCF-7/DOX cells using western blot assays. As shown in Fig. 5B, high levels of P-gp were expressed in MCF-7/DOX cells while low levels of P-gp were detected in MCF-7 cells. Compound 6b decreased P-gp expression in MCF-7/DOX cells in a dose-dependent manner.
Compound 6b induced breast cancer cell apoptosis and inhibited breast cancer cell colony formation
How 6b regulated the apoptosis-related STAT3 target genes, Bcl-2 and Bax, has been elucidated. Next, we investigated the mechanism of cancer cell apoptosis induced by 6b. Compound 6b induced the cleavage of PARP, Caspase 9 and Caspase 3 in MCF-7 and MCF-7/DOX cells as detected by western blot assays. We further investigated the effects of compound 6b on inducing tumor cell apoptosis by flow cytometry. The results showed that 6b induced the apoptosis of MCF-7 and MCF-7/DOX cells in a dose-dependent manner. The increases in apoptosis rates at 0, 1, 2, and 4 μM 6b were 8.5%, 15.7%, 37.9% and 67.2%, respectively, in MCF-7 cells, and 9.7%, 18.5%, 50.1% and 66.4%, respectively, in MCF-7/DOX cells (Fig. 6A and B) compared to the small effect of the mother compound curcumin at 4 μM on cell apoptosis in MCF-7 and MCF-7/DOX cells (Fig. 6C). Similar to data from the MTT assay, there was little cleaved PARP and apoptosis induced by treatment with 6b at concentrations up to 4 μM in MCF-10A cells (Fig. 6D), suggesting that 6b is a selective antitumor agent.

Western blot analysis of Cleaved-PARP, Cleaved Caspase-3 and Cleaved Caspase-9 levels in whole-cell lysates. Annexin V-APC/7-AAD staining was carried out and the percentages of apoptotic cells were further determined using flow cytometry MCF-7 (A), MCF-7/DOX (B) and MCF-10A (D) cells were incubated with 6b at different concentrations for 24 h with curcumin (C) as reference. **p < 0.01, ***p < 0.001.
To further investigate the anti-proliferative activity of 6b on cancer cells, a colony survival assay was performed. As shown in Fig. 7, there was a significant reduction in clonogenic ability with 0.25 μM of compound 6b and almost a cessation of colony formation at 0.5 μM of compound 6b.

MCF-7 and MCF-7/DOX cells were treated with compound 6b at 0.25–1 μM for 24 h and cultured for 14 d until the colonies were visible. Crystal violet solution was used to stain the colonies for 4 h and imaged (A) and counted (B).
Compound 6b induced cell cycle arrest in breast cancer cells
We investigated the effect of 6b on the cell cycle in breast cancer cells by flow cytometry. As shown in Fig. 8A and B, there was a significant increase in cells in the Sub-G1 phase after treatment of MCF-7 and MCF-7/DOX cells with 6b which further suggested that 6b induced cancer cell apoptosis. In MCF-7/DOX cells, 6b also increased the percentage of cells in the G0/G1 phase, which was related to a decrease in Cyclin D1. In MCF-7 cells treated with 6b, the percentage of cells in S phase increased as well. Treatment with the mother compound, curcumin at 4 μM had little effect on the cycles of MCF-7 and MCF-7/DOX cells (Fig. 8C). These data suggest that 6b could induce cancer cell apoptosis and cell cycle arrest.

MCF-7 and MCF-7/DOX cells were incubated with 6b (A and B) and curcumin (C and D) at different concentrations for 24 h. Cells were harvested, washed with cold PBS, and fixed with precooled 70% ethanol incubated for a minimum of 4 h at 4 °C. The cells were then centrifuged, carefully aspirated from the supernatant, and then these cells were re-suspended in propidium iodide and RNase A solution. The suspension was incubated at 37 °C for 30 min prior to analysis by flow cytometry.
Compound 6b induced the generation of ROS in breast cancer cells
Curcumin and BTP derivatives induce the generation of reactive oxygen species, which would partially contribute to cell apoptosis20,37. We evaluated the effect of 6b on the ROS levels in tumor cells using flow cytometry. As expected, the ROS levels increased 2–8-fold at different concentrations of 6b in MCF-7 and MCF-7/DOX cells (Fig. 9A and B) while there was little effect of treatment with the mother compound curcumin at 4 μM on ROS generation in MCF-7 and MCF-7/DOX cells (Fig. 9C).

MCF-7 and MCF-7/DOX cells were incubated with 6b (A and B) and curcumin (C and D) at different concentrations for 24 h, and then cells were collected and stained by DCFH-DA and subjected to flow cytometry. ***p < 0.001, **p < 0.01.
Compound 6b significantly inhibited STAT3 phosphorylation induced by interleukin-6 (IL-6) over IFN-γ induced p-STAT1 without affecting the related kinases
The effect of 6b on IL-6-induced STAT3 phosphorylation and IFN-γ-induced STAT1 phosphorylation was also assessed. As shown in Fig. 10A, IL-6 significantly increased the phosphorylation levels of STAT3, which was decreased by 6b at low concentration. Treatment with 4 μM 6b almost completely abolished STAT3 phosphorylation induced by IL-6. Treatment with 6b had less of an effect on IFN-γ-induced STAT1 phosphorylation compared with its effect on IL-6-induced STAT3 phosphorylation (Fig. 10B). These data show that compound 6b is a selective STAT3 inhibitor.

MCF-7 cells were seeded in 6-well plates and allowed to adhere overnight. The following day, the cells were serum-starved. The cells were then left untreated or were treated with 6b (0.5–4 μM). After 6 h, the untreated and 6b treated cells were stimulated by IL-6 (50 ng/mL) (A) or IFN-γ (50ng/mL) (B). The cells were harvested after 30 min and analyzed by western blot. (C) Western blot analysis of p-Jak2, p-Src and p-Erk levels in whole-cell lysates of equal total protein prepared from MCF-7 and MCF-7/DOX cells treated with compound 6b for 24 h.
The activation of STAT3 phosphorylated at Tyr705 residue may be mediated by upstream kinases (such as Jak2 and Src). To further investigate the selective targeting of STAT3 by 6b, we evaluated the effect of 6b on p-Jak2, p-Src, p-Erk1/2 protein levels by western blot assays. Compound 6b had little effect on p-Src and p-Erk1/2 expression but inhibited the phosphorylation of Jak2 (Fig. 10C). Further analysis showed that the p-STAT3 level was significantly inhibited by 1.0 μM 6b in MCF-7 and MCF-7/DOX cells and that the inhibition occurred prior to the decrease of p-Jak2, indicating that inhibition of STAT3 was a Jak2-independent event. The data suggest that 6b mainly, or directly, suppresses p-STAT3 expression, independent of these related kinases.
Compound 6b exhibited anticancer activity in vivo
To evaluate the in vivo anticancer activity of 6b, BALB/c nude mice were inoculated subcutaneously with human MCF-7 breast cancer cells. After the establishment of solid tumors, the mice were randomly treated by intraperitoneal injection with the indicated doses of 6b or the vehicle (Cremophor-EL/DMSO/PBS (1:1:8)) daily for 21 consecutive days. Treatment with 10 mg/kg of 6b significantly reduced the volumes of the implanted human breast tumors (Fig. 11A and B). Importantly, the weight of the tumor tissues from mice treated with 6b was significantly reduced by 68.2% compared with the control (Fig. 11C), and there were no significant changes in the body weights of the mice (Fig. 11D). Immunoblotting analysis of lysates from the tumor tissues treated with 6b showed suppression of p-STAT3 compared with the control tumors (Fig. 11E). Immunohistochemical staining analysis for Ki-67 in tumor tissues obtained from the control and 6b-treated mice showed that 6b suppressed tumor cell proliferation (Fig. 11F). (Fig. 11G) Apoptosis analysis by the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay of tumor cells in mice treated with or without 6b showed that 6b induced tumor cell apoptosis. These results show that 6b has potent antitumor activity against the growth of implanted human breast tumors (MCF-7) with little toxicity.

(A) The growing curves of mice’s tumor size. (B) The treated and control mice before sacrificed. (C) The weight of tumor tissues of treated and control mice. (D) The growing curves of mice’s body weight. (E) Western blot analysis of p-STAT3 and STAT3 levels in tumor tissues prepared from control (C1, C2) or mice treated compound 6b (T1, T2). (F) Immunohistochemical staining for Ki-67 in tumour tissues obtained from the control and 6b-treated mice. (F) Apoptosis of tumor cells in mice treated with 6b, as determined by TUNEL assay.
Conclusions
The poor prognosis and the rapid emergence of drug resistance limits the benefits of breast cancer treatment. Over-activated STAT3 has been found in a large number of drug-sensitive and drug-resistant breast cancers, and STAT3 inhibition may be a viable treatment option for both drug-sensitive and drug-resistant breast cancers by regulating STAT3 target genes and inhibiting STAT3-mediated MDR1 gene expression2. Cancer cells are usually exposed to a moderate level of reactive oxygen species (ROS), primarily due to their active metabolism in response to oncogenic signals23. In fact, cancer cells take advantage of this moderate oxidative stress for several important processes such as proliferation, angiogenesis, and metastasis24. However, high levels of ROS irreversibly damage DNA and lipids and ultimately cause cancer cell apoptosis. While an exogenous ROS insult is tolerable to normal cells, it may exceed the threshold cancer cells can endure and lead to selective cytotoxicity against cancer cells27. In fact, there have been increasing efforts to increase the levels of ROS for what is termed “oxidation therapy,” specifically in cancer cells. A combination of STAT3 inhibition and a promotion of ROS activity may be a new strategy for the treatment of solid tumors24,28. Therefore, a new series of curcumin-BTP hybrids (6a-v) were designed, synthesized, and biologically evaluated as STAT3 inhibitors. According to our screening results, we found that 6b exerted potent and selective antitumor activity against MCF-7 and MCF-7/DOX cells with a weak effect on MCF-10A breast epithelial cells. This is because the p-STAT3 levels in MCF-7 and MCF-7/DOX cells are significantly higher than that in MCF-10A breast epithelial cells. Docking studies revealed that 6b could tightly bind to the SH2 domain of STAT3 suggesting that 6b may be a STAT3-SH2 domain inhibitor. Further biological evaluation showed that 6b inhibited persistent and IL-6-induced STAT3 phosphorylation, nuclear translocation, and DNA binding activity and regulated the expression of the STAT3 downstream genes, Bcl-2, Bax and Cyclin D1, with little effect on p-Src or p-Erk. Compound 6b also inhibited STAT3-mediated P-gp expression in MCF-7/DOX cells. Additionally, 6b promoted intracellular ROS production and accumulation, induced cancer cell cycle arrest and apoptosis, and abolished the colony formation activity of cancer cells. Furthermore, 6b significantly inhibited human implanted breast cancer in vivo without obvious toxicity. Together, our findings highlight that hybrid compounds similar to 6b that are novel, direct STAT3 inhibitors with ROS production activity are worth further investigation as potential therapeutics for breast cancer or drug-resistant breast cancer.
Materials and Methods
Chemistry
All reagent grade chemicals used were purchased from Sinopharm Chemical Reagent Co., Ltd. (China). Reaction progress was monitored using analytical thin layer chromatography (TLC) on precoated silica gel GF254 plates (Qingdao Haiyang Chemical Plant, Qingdao, China), and the spots were detected under UV light (254 nm). Melting points were measured using an XT-4 micro-melting point instrument and were uncorrected. IR (KBr-disc) spectra were recorded by a Bruker Tensor 27 spectrometer. 1H NMR and 13C NMR spectra were measured on a Bruker ACF-500 spectrometer at 25 °C and referenced to TMS. Chemical shifts were reported in ppm (δ) using the residual solvent line as an internal standard. Splitting patterns were designed as s, singlet; d, doublet; t, triplet; m, multiplet. Mass spectra were obtained on an MS Agilent 1100 Series LC/MSD Trap mass spectrometer (ESI-MS) and a Mariner ESI-TOF spectrometer (HRESI-MS). Column chromatography was performed on silica gel (90–150 μm; Qingdao Marine Chemical Inc). Intermediates 2a and 2b were prepared as previously described38.
5-bromobenzo[b]thiophene-2-carboxylic acid (2a)
Yield 96%, white solid; 1H NMR (500 MHz, DMSO) δ13.60 (s, 1H, COOH), 8.17 (s, 1H), 7.95 (d, J = 8.6 Hz, 1H), 7.85 (s, 1H), 7.56 (d, J = 8.6 Hz, 1H).
6-bromobenzo[b]thiophene-2-carboxylic acid (2b)
Yield 96%, white solid; 1H NMR (500 MHz, DMSO) δ 13.57 (s, 1H, COOH), 8.36 (s, 1H, Ar-H), 8.10 (s, 1H, Ar-H), 7.95 (d, J = 8.6 Hz, 1H, Ar-H), 7.61 (dd, J = 8.6, 1.6 Hz, 1H, Ar-H).
General procedures for the preparation of 3a and 3b
To a solution of 2a (1.0 g, 3.9 mmol) or 2b (1.0 g, 3.9 mmol) and HATU (1.78 g, 4.7 mmol) in the presence of trimethylamine (0.97 ml, 5.8 mmol), tert-butyl-1-piperazinecarbo-xylate (0.8 g, 4.3 mmol) was added and stirred overnight. After completion, dilute hydrochloric acid solution was added and extracted by dichloromethane, washed by saturated sodium carbonate solution three times. The combined organic layers were washed with brine and dried with sodium sulfate, filtered, and concentrated, loaded on silica gel, and purified by silica gel chromatography with petroleum/ethyl acetate as eluent to give pure 3a or 3b.
Tert-butyl-4-(5-bromobenzo[b]thiophene-2-carbonyl) piperazine-1-carboxylat ( 3a ) 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 1.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.68 (dd, J = 8.0, 1.5 Hz, 1H), 5.72–4.47 (m, 8 H) 1.46 (s, 9H).
Tert-butyl-4-(5-bromobenzo[b]thiophene-2-carbonyl) piperazine-1-carboxylat ( 3b ) 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 4.7 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 7.53–7.48 (m, 1H), 7.44 (s, 1H), 3.78–3.68 (m, 4H), 3.58–3.46 (m, 4H), 1.48 (s, 9H).
General procedures for the preparation of 4a and 4b
3-Chloroperbenzoic acid (1.27 g, 5.9 mmol, 80%) was added, in portions, to the stirred solution of compound 3a or 3b (1.0 g, 2.35 mmol) in dichloromethane (50 ml) over a 20–30 minute period. The mixture was heated to reflux for approximately 5 h and monitored by TLC. After completion, the mixture was cooled to room temperature and sodium hydrogen sulfite (20 ml) was added to it. The solution was stirred for 15 minutes and extracted with dichloromethane. The organic phase was washed with aqueous sodium bicarbonate. The organic phase was separated, washed with brine and dried with sodium sulfate, filtered, and the solvents were removed under reduced pressure, loaded onto a silica gel, and purified by silica gel chromatography with petroleum/ethyl acetate as an eluent to give pure 4a or 4b .
Tert-butyl-4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazine-1-carboxylate ( 4a ) 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 1.0 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.77 (dd, J = 8.0, 1.5 Hz, 1H), 5.72 (t, J = 5.9 Hz, 4H), 4.47 (dd, J = 5.8, 1.7 Hz, 4H) 1.46 (s, 9H).
Tert-butyl-4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazine-1-carboxylate ( 4b ) 1H NMR (500 MHz, CDCl3) δ 7.86 (s, 1H), 7.76–7.71 (m, 1H), 7.32–7.27 (m, 2H), 3.55–3.50 (m, 8H), 1.47 (s, 9H).
General procedures for the preparation of 5a and 5b
A solution of 4a or 4b in chlorine hydride gas in methanol and dichloromethane (1:1) (10 ml) was stirred for 48 h, after completion determined by TLC, the solvents were removed under reduced pressure to generate compound 5a or 5b.
(5-bromo-1,1-dioxidobenzo[b]thiophen-2-yl)(piperazin-1-yl)methanone hydrochloride ( 5a ) 1H NMR (500 MHz, DMSO) δ 7.98 (s, 1H), 7.92 (s, 2H), 7.84 (s, 1H), 3.83 (s, 4H), 3.16 (d, J = 3.6 Hz, 4H).
(5-bromo-1,1-dioxidobenzo[b]thiophen-2-yl)(piperazin-1-yl)methanone hydrochloride ( 5b ) 1H NMR (500 MHz, DMSO) δ 7.90 (s, 1H), 7.86–7.80 (m, 1H), 7.39–7.32 (m, 2H), 3.65–3.59 (m, 8H).
General procedures for the preparation of 6a-v
To a solution of 5a (0.1 g, 0.25 mmol) or 5b (0.1 g, 0.25 mmol) and HATU (0.096 g, 0.27 mmol) in the presence of trimethylamine (0.07 ml, 0.508 mmol), a different cinnamic acid (0.28 mmol) was added and the solution was stirred overnight. After completion, dilute hydrochloric acid solution was added and the resulting product was extracted with dichloromethane and washed with a saturated sodium carbonate solution three times. The combined organic layers were washed with brine and dried sodium sulfate, were filtered and concentrated, loaded onto a silica gel, and purified by silica gel chromatography with dichloromethane/ethyl acetate as an eluent to give the pure compounds 6a-v.
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one ( 6a ) Yield 94%, white solid; m.p. 244–246 °C; 3450, 2921, 1642, 1592, 1517, 1441, 1309, 1257, 1144, 1018, 993, 909, 810, 726, 616, 550 cm−1; 1H NMR (500 MHz, DMSO) δ 7.96 (s, 1H), 7.92 (s, 2H), 7.78 (s, 1H), 7.48 (d, J = 15.2 Hz, 1H), 7.36 (s, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.14 (d, J = 14.8 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 3.86–3.76 (m, 8H), 3.64 (s, 6H). 13C NMR (126 MHz, DMSO) δ 165.51 (s), 158.22 (s), 150.93 (s), 149.48 (s), 142.77 (s), 137.80 (s), 137.78 (s), 132.02 (s), 129.20 (s), 128.96 (s), 128.41 (s), 125.47 (s), 125.09 (s), 122.91 (s), 115.83 (s), 112.14 (s), 111.07 (s), 56.16 (s), 56.24 (s). MS (ESI) m/z 547.0 [M + H]+; HRMS (ESI) m/z 547.0532 [M + H]+ (calcd for 547.0533, C24H24BrN2O6S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-methoxyphenyl)prop-2-en-1-one ( 6b ) Yield 92%, white solid; m.p. 250–251 °C; 3448, 1644, 1603, 1512, 1431, 1314, 1225, 1176, 1156, 1064, 997, 967, 821, 745, 570 cm−1; 1H NMR (500 MHz, DMSO) δ 7.98 (s, 1H), 7.97–7.91 (m, 2H), 7.80 (s, 1H), 7.70 (d, J = 8.1 Hz, 2H), 7.51 (d, J = 15.3 Hz, 1H), 7.23–7.06 (m, 1H), 6.99 (d, J = 8.6 Hz, 2H), 3.81 (s, 5H), 3.66 (s, 6H). 13C NMR (126 MHz, DMSO) δ 158.94 (s), 151.92 (s), 136.82 (s), 130.02 (s), 129.69 (s), 127.16 (s), 126.46 (s), 125.49, 125.34, 123.43 (s), 119.12 (s), 111.66 (s), 110.60 (s), 54.47 (s). MS (ESI) m/z 517.0 [M + H]+; HRMS (ESI) m/z 517.0426 [M + H]+ (calcd for 517.0427, C23H22BrN2O5S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one ( 6c ) Yield 88%, white solid; m.p. 94–96 °C; 3448, 2936, 1643, 1505, 1426, 1312, 1266, 1154, 1124, 1062, 992, 823, 723, 577 cm−1; 1H NMR (500 MHz, DMSO) δ 7.98 (s, 1H), 7.97–7.90 (m, 2H), 7.81 (s, 1H), 7.50 (d, J = 15.3 Hz, 1H), 7.29–7.18 (m, 1H), 7.06 (d, J = 13.0 Hz, 2H), 3.94–3.78 (m, 8H), 3.78–3.58 (m, 9H). 13C NMR (126 MHz, DMSO) δ 164.75 (s), 162.21 (s), 157.54 (s), 152.99 (s), 142.25 (s), 138.97 (s), 134.54 (s), 134.20 (s), 131.53 (s), 130.81 (s), 130.53 (s), 129.78 (s), 127.58 (s), 123.09 (s), 116.94 (s), 105.76 (s), 60.02 (s), 56.05 (s), 38.16 (s), 35.68 (s). MS (ESI) m/z 577.0 [M + H]+; HRMS (ESI) m/z 577.0635 [M + H]+ (calcd for 577.0635, C25H26BrN2O7S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-hydroxy-3,5-dimethoxyphenyl)prop-2-en-1-one ( 6d ) Yield 76%, white solid; m.p. 125–127 °C; 3433, 1641, 1514, 1428, 1311, 1219, 1155, 1113, 993, 842, 724, 579 cm−1; 1H NMR (500 MHz, DMSO) δ 8.81 (s, 1H), 7.96 (s, 1H), 7.92 (s, 2H), 7.78 (s, 1H), 7.46 (d, J = 15.2 Hz, 1H), 7.16–7.04 (m, 1H), 7.02 (s, 2H), 3.81–3.65 (m, 14H). 13C NMR (126 MHz, DMSO) δ 165.19 (s), 157.66 (s), 148.09(s), 143.07 (s), 137.73 (s), 134.61 (s), 134.33 (s), 131.60 (s), 130.94 (s), 129.89 (s), 127.72 (s), 125.47 (s), 123.18 (s), 114.64 (s), 106.17 (s), 56.23 (s), 30.67 (s). MS (ESI) m/z 563.0 [M + H]+; HRMS (ESI) m/z 563.0484 [M + H]+ (calcd for 563.0482, C24H24BrN2O7S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-nitrophenyl)prop-2-en-1-one ( 6e ) Yield 86%, white solid; m.p. >250 °C; 3445, 1634, 1611, 1562, 1520, 1432, 1345, 1310, 1264, 1196, 1157, 1067, 992, 927, 841, 753, 576 cm−1; 1H NMR (500 MHz, DMSO) δ 8.25 (d, J = 8.6 Hz, 2H), 8.01 (d, J = 8.6 Hz, 2H), 7.96 (s, 1H), 7.94–7.88 (m, 2H), 7.78 (s, 1H), 7.62 (d, J = 15.4 Hz, 1H), 7.52 (d, J = 15.4 Hz, 1H), 3.86–3.75 (m, 8H). 13C NMR (126 MHz, DMSO) δ 165.13 (s), 157.62 (s), 148.06 (s), 143.03 (s), 137.71 (s), 134.59 (s), 134.29 (s), 131.58 (s), 130.90 (s), 129.86 (s), 127.68 (s), 125.44 (s), 123.16 (s), 114.61 (s), 106.16 (s), 56.20 (s), 30.64 (s). MS (ESI) m/z 532.0 [M + H]+; HRMS (ESI) m/z 532.0173[M + H]+ (calcd for 532.0172, C22H19BrN3O6S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(pyridin-2-yl)prop-2-en-1-one ( 6f ) Yield 65%, white solid; m.p. 241–242 °C; 3449, 3085, 2919, 1651, 1608, 1565, 1427, 1316, 1275, 1226, 1204, 1157, 1065, 994, 970, 938, 886, 817, 779, 746, 592, 569, 553, 453, 426 cm−1; 1H NMR (500 MHz, DMSO) δ 8.62 (d, J = 3.9 Hz, 1H), 7.96 (s, 1H), 7.94–7.89 (m, 2H), 7.86–7.83 (m, 1H), 7.78 (s, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.59–7.51 (m, 2H), 7.41–7.33 (m, 1H), 3.76–3.66 (m, 8H). 13C NMR (126 MHz, DMSO) δ 164.36 (s), 157.56 (s), 153.09 (s), 149.68 (s), 141.09 (s), 136.99 (s), 134.55 (s), 134.20 (s), 131.53 (s), 130.81 (s), 129.80 (s), 127.58 (s), 124.04 (s), 123.85 (s), 123.10 (s), 121.45 (s). MS (ESI) m/z 488.0 [M + H]+; HRMS (ESI) m/z 488.0275 [M + H]+ (calcd for 488.0274, C21H19BrN3O4S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-bromophenyl)prop-2-en-1-one ( 6g ) Yield 97%, white solid; m.p. >250 °C; 3450, 3079, 2916, 1644, 1601, 1564, 1486, 1429, 1314, 1266, 1226, 1205, 1156, 1067, 1044, 998, 967, 879, 813, 744, 589, 568, 552, 486, 452, 426 cm−1; 1H NMR (500 MHz, DMSO) δ 7.96 (s, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.78 (s, 1H), 7.70 (d, J = 7.3 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 15.4 Hz, 1H), 7.39–7.27 (m, 1H), 3.86–3.76 (m, 8H). 13C NMR (126 MHz, DMSO) δ 164.45 (s), 157.54 (s), 140.44 (s),136.48 (s), 134.53 (s), 134.31 (s), 132.25 (s), 131.61 (s), 131.25 (s), 131.42 (s), 130.83 (s), 129.92 (s), 129.78 (s), 127.87 (s), 127.57 (s), 125.30 (s), 123.09 (s), 122.74 (s), 118.94 (s). MS (ESI) m/z 566.9 [M + H]+; HRMS (ESI) m/z 566.9404 [M + H]+ (calcd for 566.9407, C22H19Br2N2O4S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-chlorophenyl)prop-2-en-1-one ( 6h ) Yield 95%, white solid; m.p. 225–227 °C; 3080, 2917, 1645, 1491, 1429, 1314, 1266, 1226, 1204, 1156, 1065, 998, 968, 880, 817, 744, 590, 569, 552, 490, 455, 423 cm−1; 1H NMR (500 MHz, DMSO) δ 7.96 (s, 1H), 7.94–7.88 (m, 2H), 7.77 (m, 3H), 7.49 (m, 3H), 7.32 (d, J = 14.9 Hz, 1H), 3.86–3.72 (m, 8H). 13C NMR (126 MHz, DMSO) δ 164.45 (s), 157.54 (s), 140.36 (s), 134.53 (s), 134.19 (s), 133.97 (s), 131.52 (s), 130.83 (s), 129.78 (s), 129.68 (s), 128.68 (s), 127.58 (s), 123.09 (s), 118.88 (s), 30.64 (s). MS (ESI) m/z 523.0 [M + H]+; HRMS (ESI) m/z 522.9009 [M + H]+ (calcd for 522.9911, C22H19BrClN2O4S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one ( 6i ) Yield 95%, white solid; m.p. 245–247 °C; 3443, 3084, 1603, 1431, 1329, 1268, 1228, 1205, 1168, 1067, 999, 968, 954, 827, 744, 590, 568, 453, 423 cm−1; 1H NMR (500 MHz, DMSO) δ 8.05–7.87 (m, 5H), 7.83–7.71 (m, 3H), 7.58 (d, J = 15.4 Hz, 1H), 7.46 (t, J = 12.2 Hz, 1H), 3.82–3.66 (m, 8H). 13C NMR (126 MHz, DMSO) δ 164.25 (s), 157.57 (s), 139.93 (s), 139.07 (s), 134.54 (s), 134.20 (s), 131.54 (s), 130.87 (s), 129.79 (s), 129.40 (s), 129.14 (s), 128.63 (d, J = 15.8 Hz), 127.58 (s), 125.48 (d, J = 3.8 Hz), 125.12 (s), 123.02 (d, J = 18.3 Hz), 121.05 (s). MS (ESI) m/z 555.0 [M + H]+; HRMS (ESI) m/z 555.0191 [M + H]+ (calcd for 555.0196, C23H19BrF3N2O4S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-hydroxy-3-methoxyphenyl)prop-2-en-1-one ( 6j ) Yield 64%, white solid; m.p.127–129 °C; 3397, 2923, 1641, 1514, 1432, 1311, 1155, 1063, 993, 842, 725, 557 cm−1; 1H NMR (500 MHz, DMSO) δ 9.43 (s, 1H), 7.96–7.95 (m, 2H), 7.93–7.93 (m, 2H), 7.78 (s, 1H), 7.45 (d, J = 15.2 Hz, 1H), 7.33 (s, 1 H), 7.10 (d, J = 8.1 Hz, 1H), 6.78 (d, J = 8.1 Hz, 1H), 3.87–3.58 (m, 11H). 13C NMR (126 MHz, DMSO) δ 165.06 (s), 162.21 (s), 157.54 (s), 148.55 (s), 147.80 (s), 142.56 (s), 134.55 (s), 134.20 (s), 131.53 (s), 130.79 (s), 129.78 (s), 127.58 (s), 126.55 (s), 123.09 (s), 122.53 (s), 115.42 (s), 114.22 (s), 111.28 (s), 55.77 (s), 38.16 (s), 35.68 (s). MS (ESI) m/z 533.0 [M + H]+; HRMS (ESI) m/z 533.0380 [M + H]+ (calcd for 533.0376, C23H22BrN2O6S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-fluorophenyl)prop-2-en-1-one ( 6k ) Yield 91%, white solid; m.p. >250 °C; 3445, 3083, 1602, 1509, 1430, 1314, 1269, 1221, 1157, 1065, 997, 967, 824, 744, 729, 590, 569, 552, 512, 422 cm−1; 1H NMR (500 MHz, DMSO) δ 7.96 (s, 1H), 7.92 (s, 2H), 7.85–7.74 (m, 3H), 7.52 (d, J = 15.4 Hz, 1H), 7.25 (t, J = 8.8 Hz, 3H), 3.72 (d, J = 80.2 Hz, 8H). 13C NMR (126 MHz, DMSO) δ 164.58 (s), 161.76 (s), 157.54 (s), 140.57 (s), 134.53 (s), 134.19 (s), 131.59 (d, J = 16.7 Hz), 130.83 (s), 130.18 (d, J = 8.4 Hz), 129.78 (s), 127.58 (s), 123.09 (s), 117.91 (s), 115.70 (s), 115.53 (s). MS (ESI) m/z 505.0 [M + H]+; HRMS (ESI) m/z 505.0225 [M + H]+ (calcd for 505.0227, C22H19BrFN2O4S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one ( 6l ) Yield 87%, white solid; m.p. 222–224 °C; 3422, 1641, 1600, 1511, 1438, 1308, 1260, 1218, 1156, 1073, 1030, 993, 967, 806, 607, 548 cm−1; 1H NMR (500 MHz, DMSO) δ 8.32 (s, 1H), 8.00 (dd, J = 8.0, 1.6 Hz, 1H), 7.87 (s, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 15.3 Hz, 1H), 7.38 (s, 1H), 7.24 (d, J = 8.1 Hz, 1H), 7.15 (d, J = 15.2 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 3.91–3.76 (m, 8 H), 3.68 (s, 6H). 13C NMR (126 MHz, DMSO) δ 164.93 (s), 157.64 (s), 150.36 (s), 148.91 (s), 142.20 (s), 137.23 (d, J = 5.6 Hz), 131.45 (s), 128.63 (s), 128.39 (s), 127.84 (s), 124.90 (s), 124.51 (s), 122.34 (s), 115.26 (s), 111.57 (s), 110.50 (s), 55.59 (d, J = 19.4 Hz). MS (ESI) m/z 547.0 [M + H]+; HRMS (ESI) m/z 547.0531 [M + H]+ (calcd for 547.0533, C24H24BrN2O6S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-methoxyphenyl)prop-2-en-1-one ( 6m ) Yield 89%, white solid; m.p. 220–222 °C; 3422, 1644, 1621, 1511, 1427, 1311, 1226, 1176, 1153, 1073, 1028, 996, 817, 745, 589, 545 cm−1; 1H NMR (500 MHz, DMSO) δ 8.33 (s, 1H), 8.00 (dd, J = 8.0, 1.5 Hz, 1H), 7.86 (s, 1H), 7.68 (dd, J = 14.3, 8.3 Hz, 3H), 7.51 (d, J = 15.3 Hz, 1H), 7.20–7.09 (m, 1H), 6.99 (d, J = 8.6 Hz, 2H), 3.82 (s, 5H), 3.67 (s, 6H). 13C NMR (126 MHz, DMSO) δ 164.91 (s), 160.45 (s), 157.65 (s), 141.68 (s), 137.20 (s), 131.46 (s), 129.63 (s), 128.64 (s), 128.39 (s), 127.63 (s), 124.90 (s), 124.52 (s), 115.25 (s), 114.15 (s), 55.21 (s), 38.16 (s). MS (ESI) m/z 517.0 [M + H]+; HRMS (ESI) m/z 517.0430 [M + H]+ (calcd for 517.0427, C23H22BrN2O5S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one ( 6n ) Yield 76%, white solid; m.p. 81–85 °C; 3449, 2928, 1643, 1505, 1425, 1314, 1252, 1225, 992, 825, 608, 549 cm−1; 1H NMR (500 MHz, DMSO) δ 8.34 (s, 1H), 8.00 (dd, J = 8.0, 1.7 Hz, 1H), 7.87 (s, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 15.3 Hz, 1H), 7.23 (d, J = 14.5 Hz, 1H), 7.08 (s, 2H), 3.84 (d, J = 5.2 Hz, 8H), 3.69 (d, J = 13.6 Hz, 9H). 13C NMR (126 MHz, DMSO) δ 164.74 (s), 157.65 (s), 152.99 (s), 142.27 (s), 138.97 (s), 137.21 (s), 131.46 (s), 130.54 (s), 128.63 (s), 128.38 (s), 124.90 (s), 124.52 (s), 116.93 (s), 105.75 (s), 60.02 (s), 56.05 (s), 38.16 (s). MS (ESI) m/z 577.0 [M + H]+; HRMS (ESI) m/z 577.0640 [M + H]+ (calcd for 577.0639, C25H26BrN2O7S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-hydroxy-3,5-dimethoxyphenyl)prop-2-en-1-one ( 6o ) Yield 61%, white solid; m.p. 112–114 °C; 3422, 2924, 2853, 1641, 1513, 1459, 1314, 1261, 1218, 1153, 1111, 992, 822, 548 cm−1; 1H NMR (500 MHz, DMSO) δ 8.83 (s, 1H), 8.34 (s, 1H), 8.00 (dd, J = 8.0, 1.3 Hz, 1H), 7.87 (s, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.48 (d, J = 15.2 Hz, 1H), 7.12 (d, J = 15.0 Hz, 1H), 7.04 (s, 2H), 3.91–3.78 (m, 8H), 3.67 (s, 6H). 13C NMR (126 MHz, DMSO) δ 165.00 (s), 157.64 (s), 147.98 (s), 142.93 (s), 137.65 (s), 137.21 (s), 131.44 (s), 128.63 (s), 128.39 (s), 125.37 (s), 124.90 (s), 124.52 (s), 114.53 (s), 106.12 (s), 56.13 (s), 38.16 (s). MS (ESI) m/z 563.0 [M + H]+; HRMS (ESI) m/z 563.0485 [M + H]+ (calcd for 563.0482, C24H24BrN2O7S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(2-hydroxy)prop-2-en-1-one ( 6p ) Yield 56%, white solid; m.p. >250 °C; 3444, 2924, 1641, 1433, 1311, 1218, 1153, 991, 841, 758, 550 cm−1; 1H NMR (500 MHz, DMSO) δ 10.04 (s, 1H), 8.34 (s, 1H), 8.00 (dd, J = 8.0, 1.4 Hz, 1H), 7.89–7.80 (m, 2H), 7.68 (dd, J = 17.9, 7.2 Hz, 2H), 7.27–7.15 (m, 2H), 6.91 (d, J = 8.1 Hz, 1H), 6.84 (t, J = 7.4 Hz, 1H), 3.78 (s, 2H), 3.67 (s, 6H). 13C NMR (126 MHz, DMSO) δ 171.83 (s), 165.17 (s), 157.65 (s), 156.14 (s), 151.02 (s), 137.33(s), 137.26 (s), 137.19 (s), 131.44 (s), 130.70 (s), 128.64 (s), 128.39 (s), 128.08 (s), 124.89 (s), 124.51 (s), 121.75 (s), 119.15 (s), 116.62 (s), 115.99 (s), 38.17 (s). MS (ESI) m/z 503.0 [M + H]+; HRMS (ESI) m/z 503.0269 [M + H]+ (calcd for 503.0271, C22H20BrN2O5S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(pyridin-2-yl)prop-2-en-1-one ( 6q ) Yield 67%, white solid; m.p. 97–99 °C; 3422, 2924, 2853, 1641, 1513, 1459, 1314, 1261, 1218, 1153, 1111, 991, 822, 548 cm−1; 1H NMR (500 MHz, DMSO) δ 8.62 (d, J = 3.5 Hz, 1H), 8.31 (s, 1H), 7.98 (dd, J = 8.0, 1.7 Hz, 1H), 7.89–7.81 (m, 2H), 7.74 (d, J = 7.7 Hz, 1H), 7.65 (d, J = 8.1 Hz, 1H), 7.52 (d, J = 14.8 Hz, 2H), 7.37 (dd, J = 7.1, 4.9 Hz, 1H), 3.77 (s, 2H), 3.67 (s, 6H). 13C NMR (126 MHz, DMSO) δ 164.37 (s), 149.68 (s), 149.10 (s), 137.00 (s), 136.05(s), 128.66 (s), 125.81 (s), 124.05 (s), 119.70 (s), 99.43 (s), 72.08 (s), 69.36 (s), 38.17 (s). MS (ESI) m/z 488.0 [M + H]+; HRMS (ESI) m/z 488.0276 [M + H]+ (calcd for 488.0274, C21H19BrN3O4S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-bromophenyl)prop-2-en-1-one ( 6r ) Yield 92%, white solid; m.p. 215–217 °C; 3425, 1646, 1603, 1433, 1316, 1154, 997, 815, 587, 549 cm−1; 1H NMR (500 MHz, DMSO) δ 8.34 (s, 1H), 8.00 (dd, J = 8.0, 1.3 Hz, 1H), 7.87 (s, 1H), 7.72 (d, J = 7.5 Hz, 3H), 7.65 (dd, J = 16.6, 8.1 Hz, 3H), 7.52 (d, J = 15.4 Hz, 1H), 7.36 (t, J = 11.2 Hz, 1H), 3.83 (s, 2H), 3.67 (s, 6H). 13C NMR (126 MHz, DMSO) δ 164.45 (s), 161.64 (s), 157.65 (s), 153.24 (s), 140.45 (s), 137.72 (s), 137.20 (s), 131.61 (s), 129.93 (s), 128.63 (s), 128.38 (s), 124.90 (s), 124.51 (s), 123.46 (s), 122.75 (s), 122.26 (s), 118.94 (s), 38.16 (s). MS (ESI) m/z 564.9 [M + H]+; HRMS (ESI) m/z 566.9404 [M + H]+ (calcd for 564.9427, C22H19Br2N2O4S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-chlorophenyl)prop-2-en-1-one ( 6s ) Yield 92%, white solid; m.p. 225–227 °C; 3421, 1645, 1602, 1491, 1432, 1315, 1222, 1153, 1074, 1153, 1074, 995, 817, 587, 548 cm−1; 1H NMR (500 MHz, DMSO) δ 8.34 (s, 1H), 8.00 (dd, J = 8.0, 1.7 Hz, 1H), 7.87 (s, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 1H), 7.54–7.51 (m, 3H), 7.34 (d, J = 15.1 Hz, 1H), 3.83 (s, 2H), 3.67 (s, 6H). 13C NMR (126 MHz, DMSO) δ 164.46 (s), 160.14 (s), 157.66 (s), 142.96 (s), 140.38 (s), 137.21 (s), 134.00 (s), 132.02 (s), 131.50 (s), 129.69 (s), 128.69 (s), 128.64 (s), 128.39 (s), 127.97 (s), 124.90 (s), 124.52 (s), 118.87 (s), 39.02 (s). MS (ESI) m/z 523.0 [M + H]+; HRMS (ESI) m/z 522.9014 [M + H]+ (calcd for 522.9911, C22H19BrClN2O4S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(3-hydroxy-4-methoxyphenyl)prop-2-en-1-one ( 6t ) Yield 64%, white solid; m.p.123–125 °C; 3397, 2923, 1641, 1514, 1432, 1311, 1155, 1063, 993, 842, 725, 557 cm−1; 1H NMR (500 MHz, DMSO) δ 9.43 (s, 1H), 7.96–7.95 (m, 2H), 7.92–7.91 (m, 2H), 7.78 (s, 1H), 7.46 (d, J = 15.2 Hz, 1H), 7.31 (s, 1H), 7.14 (d, J = 8.1 Hz, 1H), 6.78 (d, J = 8.1 Hz, 1H), 3.87–3.58 (m, 11H). 13C NMR (126 MHz, DMSO) δ 166.06 (s), 164.98 (s), 148.55 (s), 148.40 (s), 147.80 (s), 142.57 (s), 142.11 (s), 137.25 (s), 137.16 (s), 134.90 (s), 134.51 (s), 134.32 (s), 122.54 (s), 122.43 (s), 115.43 (s), 114.48 (s), 114.21 (s), 114.15 (s), 111.27 (s), 109.09 (s), 59.65 (s), 55.77 (s), 39.02 (s). MS (ESI) m/z 533.0 [M + H]+; HRMS (ESI) m/z 533.0377 [M + H]+ (calcd for 533.0376, C23H22BrN2O6S).
(E)-1-(4-(6-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-hydroxy-3-methoxyphenyl)prop-2-en-1-one ( 6u ) Yield 76%, white solid; m.p. 201–203 °C; 3423, 1638, 1578, 1511, 1432, 1318, 1261, 1227, 1130, 1026, 993, 547 cm−1; 1H NMR (500 MHz, DMSO) δ 9.06 (s, 1H), 8.34 (s, 1H), 8.00 (dd, J = 8.0, 1.4 Hz, 1H), 7.86 (s, 1H), 7.66 (d, J = 8.1 Hz, 1H), 7.41 (d, J = 15.2 Hz, 1H), 7.19 (s, 1H), 7.13 (d, J = 8.2 Hz, 1H), 7.05 (d, J = 15.2 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 3.83 (s, 5H), 3.66 (s, 6H). 13C NMR (126 MHz, DMSO) δ 164.92 (s), 157.64 (s), 149.27 (s), 146.51 (s), 142.13 (s), 137.22 (d, J = 6.2 Hz), 131.43 (s), 128.63 (s), 128.39 (s), 127.99 (s), 124.89 (s), 124.51 (s), 120.59 (s), 115.12 (s), 114.29 (s), 111.88 (s), 55.62 (s), 54.79 (s), 39.02 (s). MS (ESI) m/z 533.0 [M + H]+; HRMS (ESI) m/z 533.0381 [M + H]+ (calcd for 533.0376, C23H22BrN2O6S).
(E)-1-(4-(5-bromo-1,1-dioxidobenzo[b]thiophene-2-carbonyl)piperazin-1-yl)-3-(4-fluorophenyl)prop-2-en-1-one ( 6v ) Yield 92%, white solid; m.p. >250 °C; 3443, 1647, 1601, 1509, 1432, 1320, 1264, 1220, 1157, 1073, 992, 825, 548, 512 cm−1; 1H NMR (500 MHz, DMSO) δ 8.34 (s, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.87 (s, 1H), 7.83 (d, J = 7.8 Hz, 2H), 7.67 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 15.4 Hz, 1H), 7.27 (t, J = 8.5 Hz, 3H), 3.83 (s, 2H), 3.67 (s, 6H). 13C NMR (126 MHz, DMSO) δ 164.58 (s),162.01 (s), 157.66 (s), 148.59 (s), 140.58 (s), 137.75 (s), 137.20 (s), 135.51 (s), 131.48 (s), 130.18 (d, J = 8.4 Hz), 130.15 (s), 128.64 (s), 128.39 (s), 125.53 (s), 124.90 (s), 124.52 (s), 117.88 (s), 115.70 (s), 115.61 (d, J = 21.6 Hz). MS (ESI) m/z 505.0 [M + H]+; HRMS (ESI) m/z 505.0230 [M + H]+ (calcd for 505.0227, C22H19BrFN2O4S).
Docking
The computational docking program AutoDock4.2 was used to dock our designed small molecules to predict their binding modes and approximate binding free energies to the STAT3 SH2 dimerization sites. Briefly, compounds were docked using the Lamarckian Genetic Algorithm. The ligand and macromolecule were prepared using Schrödinger software. Gasteiger charges were assigned to the ligands by Auto Dock Tools. Then, AutoGrid maps were precomputed for all atom types in the ligand set. After 10 million energy evaluations were completed, the root-mean-square deviation threshold was set as 1.5 Å and all the resulting conformations of the ligands in the binding pocket of the macromolecule were clustered. Low energy clusters were identified and binding energies were evaluated.
Maintenance of cell line cultures and cell viability assays
All cell lines were purchased from Cell Bank of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Human breast carcinoma cells (MCF-7 and MCF-7/DOX), breast epithelial cells (MCF-10A) and normal human liver cells (LO2) were maintained in RPMI-1640 medium. All cells were supplemented with 12% fetal bovine serum containing 50 μg/ml penicillin and 50 μg/ml streptomycin. Cells were grown to 80% confluency in a tissue culture flask at 37 °C in a humidified atmosphere containing 5% CO2, and then were trypsinized with 1 × Trypsin-Versene and split. The MCF-7/DOX cell line we used was established from its parental cell line MCF-7 cell by gradually increasing the concentration of DOX to which the cells were exposed in a stepwise manner over a period of 8 months. A 5 nM to 100 nM range of DOX concentrations was added to MCF-7 cells, after which the cells were maintained in culture medium containing 100 nM DOX and displayed a 66.02-fold resistance to DOX compared with the corresponding parental, DOX-sensitive cells. The cells were incubated in drug-free medium for at least 1 week before use.
Cells (MCF-7, MCF-7/DOX, MCF-10A, LO2) were seeded in 96-well plates at a density of 3,000–6,000 cells per well. The cells were incubated overnight (16 h) in a humidified 5% CO2 incubator at 37 °C. After media removal, different concentrations of test compounds were added in triplicate to the plates in 200 μL of fresh media and were incubated with the cells at 37 °C for 48 h. The percentage of DMSO in the medium did not exceed 0.1%. Cell viability was evaluated using 3-(4, 5-dimethylthiazolyl)-2,5-diphenyltetrazoliumbromide (MTT). The absorbance was read by an ELISA reader (SpectraMax Plus384, Molecular Devices, Sunnyvale, CA) at a test wavelength of 570 nm and a reference wavelength of 630 nm. Cell viability was calculated using the following formula:

At and As denote the absorbance of the test substance and solvent control, respectively.
Western blot assays
MCF-7 or MCF-7/DOX cells were incubated with various concentrations of 6b for 24 h. After trypsinization, cells were treated with 1× RIPA lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA and protease inhibitors) (Amresco, Solon, USA) to extract the total proteins. An aliquot of proteins from the total cell lysates (30 to 60 μg/lane) was separated by sodium dodecyl sulfate (10%) polyacrylamide gel electrophoresis (SDS–PAGE, Bio-Rad Laboratories, Hercules, CA), wet-transferred to NC membrane (Bio-Rad Laboratories, Hercules, CA) and blotted with primary antibodies specific for STAT3, p-STAT3, p-Jak2, Jak2, p-Src, Src, Erk, P-Erk, PARP, cleaved PARP, Bcl-2, Bax, Cyclin D1, P-gp and GAPDH. Bound immuno-complexes were detected using ChemiDOC™ XRS + system (Bio-Rad Laboratories, Hercules, CA).
Quantitative real-time RT-PCR
Levels of mRNA expression were analyzed with the RT-PCR assay, with total RNA isolated from MCF-7 or MCF-7/DOX cells using an EASYspin Plus tissue/cell RNA extraction kit (Aidlab Biotechnologies Co. Ltd). RNA was quantified by measuring absorption at 260 nm and 1 μg of RNA was reverse transcribed to cDNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Basel, Switzerland). Thermal cycling conditions included 95 °C initial denaturation for 5 min, followed by 40 cycles of denaturation (10 s at 95 °C), annealing (15 s at 60 °C) and extension (15 s at 72 °C with a single fluorescence measurement), a melt curve program (60–95 °C with a 0.11 °C/s heat increase and continuous fluorescence measurement) and a cooling step to 40 °C. The Δ cycle threshold method was used for the calculation of relative differences in mRNA abundance with a LightCycler 480 (Roche Molecular Biochemicals, Mannheim, Germany). The data were normalized to the expression of GAPDH. The results were expressed as fold-changes. The RT-PCR primers that were used in this study are listed in the supporting information.
IL-6 induced STAT3 phosphorylation and IFN-γ induced STAT1 phosphorylation
MCF-7 or MCF-7/DOX cells were seeded in 6-well plates and allowed to adhere overnight. The following day, the cells were serum-starved. The cells were then left untreated or were treated with 6b (0.5–4 μM). After 6 h, the untreated and 6b-treated cells were stimulated with IL-6 (50 ng/ml). The cells were harvested after 30 min and analyzed by western blot.
Immunofluorescence staining
The basic protocol for the analysis of p-STAT3 subcellular location has been previously described39. Briefly, MCF-7 cells were starved overnight, followed by incubation with 6b for 6 h. Cells were then stimulated with 50 ng/ml IL-6 for 30 min before being harvested in Tris-buffered saline (TBS) containing 1 mM sodium orthovanadate. Cells were transferred to polylysine-coated slides by cytospin and air dried for 20 min at room temperature. After being fixed in 2% of paraformaldehyde for 10 min and permeabilized in 0.1% Triton X-100 for 5 min, cells were blocked in TBS containing 5% bovine serum albumin for 1 h at room temperature before being stained with p-STAT3 (Tyr705) overnight at 4 °C. Then, cells were gently washed and incubated with FITC–conjugated, goat, anti-rabbit IgG (Beyotime Biotechnology Ltd). Finally, slides were incubated with 5 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI, Beyotime) for 5 min. The subcellular location and relative abundance of p-STAT3 were immediately analyzed with an ImageXpress Micro Confocal analysis.
Electrophoretic mobility shift assay (EMSA) of DNA-binding activity
MCF-7 cells were cultured overnight and then treated with compound 6b in fresh regular growth medium for 24 h. The cells were harvested and nuclear extract preparations and EMSA analysis were carried out as previously described3. The 32P-labeled oligonucleotide, hSIE probe, was used to bind STAT3.
Flow cytometry analysis of apoptotic cells
MCF-7, MCF-7/DOX or MCF-10A cells, at a density of 2.5 × 105 cells per well, were cultured in regular growth medium in 6-well plates for 24 h and treated in duplicate with different concentrations of 6b or curcumin for 24 h. The cells were harvested, washed and stained with 5 μL of Annexin V-APC and 5 μL of 7-AAD at room temperature for 15 min. Cells were then analyzed by flow cytometry (488 nm excitation and 600 nm emission filters) using a BD FACSCalibur flow cytometer (Becton & Dickinson Company, Franklin Lakes, NJ).
Flow cytometry analysis of the cell cycle
MCF-7 or MCF-7/DOX cells were plated in 6-well culture plates (1.5 × 105 cells per well). After incubation overnight, various concentrations of 6b or curcumin were added and cells were incubated for 24 h. Cells were harvested, washed with cold PBS, and incubated with pre-cooled 70% ethanol for a minimum of 4 h at 4 °C to fix them. The cells were then centrifuged, carefully aspirated from the supernatant, and re-suspended in a propidium iodide (final concentration, 40 μg/ml) and RNase A (final concentration, 100 μg/ml) solution at a final cell density of 0.5 × 106 cells/ml. The suspension was incubated at 37 °C for 30 min prior to analysis by flow cytometry.
Detection of ROS using flow cytometry
MCF-7 or MCF-7/DOX cells were seeded at a density of 3 × 105 cells per well of a 6-well plate and treated with various concentrations of 6b (0, 1, 2, 4 μM) or 4 μM curcumin for 24 h. DCFH-DA was dissolved in serum-free medium and diluted to a final concentration of 10 μM. After treatment with 6b, the growth media was replaced with serum-free medium containing the probe. After incubation for 20 min at 37 °C, cells were washed with serum-free medium twice, digested by trypsin and resuspended in pre-warmed PBS buffer. The samples were then subjected to a flow cytometry assay using a BD FACSCalibur flow cytometer (Becton & Dickinson Company, Franklin Lakes, NJ).
Colony survival assay
MCF-7 or MCF-7/DOX cells were cultured a 6-well plate at a density of 800–1,000 cells/well with regular growth medium. Cells were treated for 24 h with the vehicle and 6b at 0.15–1 μM on the following day. Cells were allowed to grow for 10–14 d until colonies were visible. Crystal violet solution (Sigma, St. Louis, MO, USA) was used to stain the colonies for 4 hours and colonies were imaged.
In vivo studies
The working solution was prepared in a Cremophor and DMSO mixture (10% Cremophor, 10% DMSO and 80% 1 × PBS) to give a 1 mg/ml solution. Athymic BALB/c nude mice (15–18 g) were injected with human breast tumor MCF-7 cells (3 × 106 cells in a volume of 0.2 ml) into the subcutaneous tissue of the right auxiliary region of the mice. Three days after the tumor cell inoculation, the mice were randomly sorted into three groups with six mice per group. The tumor-bearing mice were either given a daily 10 mg/kg ip injection of compound 6b or the vehicle. The treatment was initiated when the tumor burden of the mice reached approximately 50 mm3. The tumor size was measured by caliper three times a week to document tumor growth and calculated by the formula: length × width × width/2, and the body weight was measured and recorded. Therapeutic efficacy was evaluated based on body weight loss, tumor growth inhibition [determined by using calipers and calculated by the formula: tumor inhibit rate (%) = (tumor volCon − tumor volTre)/tumor volCon] × 100. The body weight inhibitory ratio, BI (%) was calculated as BI (%) = (1 −MT/MC) × 100, where MT and MC were the average body weights of the treated and control groups, respectively. On the 22nd day, all mice were killed, and the tumors were segregated, weighed, and stored in −80 °C for later use. The levels of p-STAT3 in tumor tissues prepared from control or mice treated with 6b were analyzed by western blot assay. Apoptosis was evaluated via the TUNEL assay, and the Ki-67 assay was used to assess cancer cell proliferation.
The experimental mice were cared for and handled strictly according to the requirements of the Animal Ethics Committee of China Pharmaceutical University and the National Institutes of Health (NIH) standard guidelines for the Care and Use of Laboratory Animal. All experimental protocols were approved by the Animal Ethics Committee of China Pharmaceutical University.
Statistical analyses
All experiments were conducted more than three times. The results were analyzed using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA) to perform one-way ANOVA. The results are given as the mean ± SD. A p value less than 0.05 was considered statistically significant.
Additional Information
How to cite this article: Zhang, W. et al. Discovery of monocarbonyl curcumin-BTP hybrids as STAT3 inhibitors for drug-sensitive and drug-resistant breast cancer therapy. Sci. Rep. 7, 46352; doi: 10.1038/srep46352 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Lin, L. et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer research 70, 2445–2454 (2010).
Ji, T. et al. Abrogation of constitutive Stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer letters 341, 231–239 (2014).
Fletcher, S. et al. Antagonism of the Stat3-Stat3 protein dimer with salicylic acid based small molecules. ChemMedChem 6, 1459–1470 (2011).
Gabriella Miklossy et al. Hirsutinolide series inhibit Stat3 activity, alter GCN1, MAP1B, Hsp105, G6PD, Vimentin, TrxR1, and importin α-2 expression, and induce antitumor effects against human glioma. Journal of medicinal chemistry 19 (2015).
Huang, W. et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo . Oncogene 35, 783–792 (2016).
Chelsky, Z. L. et al. A Resveratrol Analogue Promotes ERKMAPK-Dependent Stat3 Serine and Tyrosine Phosphorylation Alterations and Antitumor Effects In Vitro against Human Tumor Cells. Molecular pharmacology 88, 524–533 (2015).
Lin, L. et al. A Novel Small Molecule, LLL12, Inhibits STAT3 Phosphorylation and Activities and Exhibits Potent Growth-Suppressive Activity in Human Cancer Cells. Neoplasia 12, 39–IN35 (2010).
Yuan, C. H. et al. Epigallocatechin gallate sensitizes cisplatin-resistant oral cancer CAR cell apoptosis and autophagy through stimulating AKT/STAT3 pathway and suppressing multidrug resistance 1 signaling. Environmental toxicology (2016).
Chiu, H. C. et al. Suppression of Stat3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochemical pharmacology 81, 1263–1270 (2011).
Lee, H. J. et al. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer cell 26, 207–221 (2014).
Hou, S. et al. Novel carbazole inhibits phospho-STAT3 through induction of protein-tyrosine phosphatase PTPN6. Journal of medicinal chemistry 57, 6342–6353 (2014).
Spitzner, M. et al. STAT3 inhibition sensitizes colorectal cancer to chemoradiotherapy in vitro and in vivo . International journal of cancer 134, 997–1007 (2014).
Yue, P. et al. Hydroxamic Acid and Benzoic Acid-Based STAT3 Inhibitors Suppress Human Glioma and Breast Cancer Phenotypes In Vitro and In Vivo . Cancer research 76, 652–663 (2016).
Zhang, M., Zhu, W. & Li, Y. Discovery of novel inhibitors of signal transducer and activator of transcription 3 (STAT3) signaling pathway by virtual screening. European journal of medicinal chemistry 62, 301–310 (2013).
Zhang, X. et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proceedings of the National Academy of Sciences of the United States of America 109, 9623–9628 (2012).
Jung, S. N. et al. Sugiol inhibits STAT3 activity via regulation of transketolase and ROS-mediated ERK activation in DU145 prostate carcinoma cells. Biochemical pharmacology 97, 38–50 (2015).
Wei. C. C. et al. Two small molecule compounds, LLL12 and FLLL32, exhibit potent inhibitory activity on STAT3 in human rhabdomyosarcoma cells. International journal of oncology 38 (2011).
Matthew A. Bill, J. R. F. et al. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Molecular Cancer 9 (2010).
Stacey L. Fossey, M. D. B. et al. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 11 (2011).
Waris, G. & Ahsan, H. Reactive oxygen species: role in the development of cancer and various chronic conditions. Journal of carcinogenesis 5, 14 (2006).
Wu, W. S. The signaling mechanism of ROS in tumor progression. Cancer metastasis reviews 25, 695–705 (2006).
Gorrini, C., Harris, I. S. & Mak, T. W. Modulation of oxidative stress as an anticancer strategy. Nature reviews. Drug discovery 12, 931–947 (2013).
Fang, J., Seki, T. & Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Advanced drug delivery reviews 61, 290–302 (2009).
Raj, L. et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475, 231–234 (2011).
Lee, S. et al. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell research 21, 817–834 (2011).
Mingxin Zuo, C. L. & Jiayuh Lin, Milind Javle . LLL12, a novel small inhibitor targeting STAT3 for hepatocellular carcinoma therapy. Oncotarget 13, 10940–10949 (2015).
Song, H., Wang, R., Wang, S. & Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America 102, 4700–4705 (2005).
Bharadwaj, U. et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene 34, 1341–1353 (2015).
Ai, Y. et al. Discovery of New Monocarbonyl Ligustrazine-Curcumin Hybrids for Intervention of Drug-Sensitive and Drug-Resistant Lung Cancer. Journal of medicinal chemistry 59, 1747–1760 (2016).
Chen, Q.-H. Curcumin-Based Anti-Prostate Cancer Agents. Anti-Cancer Agents in Medicinal Chemistry 15 (2015).
Prakobwong, S. et al. Curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signaling pathways. Carcinogenesis 32, 1372–1380 (2011).
Chen, H. et al. Discovery of potent anticancer agent HJC0416, an orally bioavailable small molecule inhibitor of signal transducer and activator of transcription 3 (STAT3). European journal of medicinal chemistry 82, 195–203 (2014).
Chen, H. et al. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. European journal of medicinal chemistry 62, 498–507 (2013).
Kraskouskaya, D., Duodu, E., Arpin, C. C. & Gunning, P. T. Progress towards the development of SH2 domain inhibitors. Chemical Society reviews 42, 3337–3370 (2013).
Yu, W., Xiao, H., Lin, J. & Li, C. Discovery of novel STAT3 small molecule inhibitors via in silico site-directed fragment-based drug design. Journal of medicinal chemistry 56, 4402–4412 (2013).
Yun, M. et al. Cinnamaldehyde derivative (CB-PIC) sensitizes chemo-resistant cancer cells to drug-induced apoptosis via suppression of MDR1 and its upstream STAT3 and AKT signalling. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 35, 1821–1830 (2015).
Alonso, M. M. et al. New benzo(b)thiophenesulphonamide 1,1-dioxide derivatives induce a reactive oxygen species-mediated process of apoptosis in tumour cells. Oncogene 22, 3759–3769 (2003).
Zhang, W. D. et al. Antagonizing STAT3 activation with benzo[b] thiophene 1, 1-dioxide based small molecules. European journal of medicinal chemistry 125, 538–550 (2016).
Zubin, Z. et al. A novel small molecule agent displays potent anti-myeloma activity by inhibiting the JAK2-STAT3 signaling pathway. Oncotarget 7, 9296–9308 (2016).
Acknowledgements
This research work was supported by National Natural Science Foundation of China (Grant No. 81402791 and No. 81673298), Natural Science Foundation of the Jiangsu Higher Education Institutions of China (Grant No. BK20140670), Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R63), Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Contributions
W.-D. Zhang carried out the experimental work. J.-P. Guo, S.-S. Li, T. Ma, D.-Q. Xu, C. Han, F.-Y. Liu, W.-Y. Yu and L.-Y. Kong provided oversight. W.-D. Zhang, W.-Y. Yu and L.-Y. Kong conceived the experiments and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, W., Guo, J., Li, S. et al. Discovery of monocarbonyl curcumin-BTP hybrids as STAT3 inhibitors for drug-sensitive and drug-resistant breast cancer therapy. Sci Rep 7, 46352 (2017). https://doi.org/10.1038/srep46352
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46352
- Springer Nature Limited