
Abstract
The interaction between the bone marrow microenvironment and malignant hematopoietic cells can result in the protection of leukemia cells from chemotherapy in both myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). We, herein, characterized the changes in cytokine expression and the function of mesenchymal stromal cells (MSC) in patients with MDS, AML with myelodysplasia-related changes (MRC), a well-recognized clinical subtype of secondary AML, and de novo AML. We observed a significant inhibitory effect of MDS-MSC on T lymphocyte proliferation and no significant differences in any of the cytokines tested. AML-MSC inhibited T-cell proliferation only at a very low MSC/T cell ratio. When compared to the control, AML-MRCderived MSC presented a significant increase in IL6 expression, whereas de novo AML MSC presented a significant increase in the expression levels of VEGFA, CXCL12, RPGE2, IDO, IL1β, IL6 and IL32, followed by a decrease in IL10 expression. Furthermore, data indicate that IL-32 regulates stromal cell proliferation, has a chemotactic potential and participates in stromal cell crosstalk with leukemia cells, which could result in chemoresistance. Our results suggest that the differences between AML-MRC and de novo AML also extend into the leukemic stem cell niche and that IL-32 can participate in the regulation of the bone marrow cytokine milieu.
Similar content being viewed by others
Introduction
Myelodysplastic syndromes (MDS) are heterogeneous clonal haematopoietic stem cell (HSC) disorders that incur an increased risk of evolution to acute myeloid leukemia (AML)1, a well-recognized clinical subtype of secondary AML with myelodysplasia-related changes (AML-MRC)2. The biological and prognostic differences between de novo and secondary AML have been extensively documented, such as the worse outcome of younger patients with secondary AML, compared with de novo AML3.
HSC self-renewal, differentiation and proliferation are regulated in local tissue microenvironments called niches. One of the main cellular components of the HSC niche are the mesenchymal stromal cells (MSC), which are important regulators of haematopoiesis, as well as of the immune system4,5. It is rational to assume that MSC, derived from patients with hematological malignancies, harbor some partial defects, either primary or secondary, due to their exposure to altered marrow components. Extensive data have already shown interactions between leukemic cells and their microenvironment, supporting the idea that defects in the HSC microenvironment may play a role either in MDS or in AML development6,7,8,9. For instance, interactions between MSC from the leukemic stem cell niche and malignant cells are critical components of resistance to many chemotherapy agents10,11,12.
One of the hallmarks of malignancy13, inflammation, has been recognized as an important factor in the pathogenesis of MDS and AML, and involves different molecular and cellular signaling pathways1,14,15,16. Thus, the continuous inflammatory state provided by the HSC leukemic niche can contribute to the initiation and progression of diseases. Interleukin (IL)-32 is a proinflammatory cytokine, expressed as several isoforms17,18, that is thought to contribute to the pathogenesis of infection19,20,21, autoimmune diseases21 and cancer22,23. IL-32 induces inflammatory cytokines such as TNF-α, IL-1β, IL-6, and chemokines through the NF-κB and p38 MAPK signaling pathways17. Previous data support a role for IL-32 in the pathophysiology of clonal myeloid diseases24.
In this study, we characterized cytokine expression changes and the function of MSC from patients with MDS, AML-MRC and de novo AML, in comparison to healthy control (HC) MSC. Moreover, we studied the ability of IL-32 to promote cell proliferation, chemotaxis of leukocytes and chemoprotection towards cytarabine (AraC) in the microenvironment.
Results
Expansion and characterization of MSC
MSC were cultured to confluence until the fourth passage. All 8 samples obtained from HC were successfully cultured, while only 71% of the samples obtained from MDS (22 of 31), 70% from AML-MRC (7 of 10) and 71% from de novo AML (12 of 17) were able to proliferate. The mean time to reach 80% confluency of samples obtained from MDS and AML-MRC were similar to those of HC (15 ± 6.2; 12.6 ± 6.1; 13.5 ± 2.4 days, respectively, p > 0.05).
De novo AML cells reached 80% confluency in 21.2 ± 8.2 days, which represents a significantly slower growth than that of HC and AML-MRC samples (p < 0.05). Confirming mesenchymal origin, all patient-derived MSC presented a typical profile, standardized by the International Society for Cellular Therapy25, and very similar to that of HC-MSC (Supplementary Table 2). Low levels of CD34 and CD31 were sometimes detected in the AML-MRC group, either due to leukemia cell or endothelial cell contamination26 or due to macrophage contamination27.
To assess the capacity and efficiency for self-renewal of the MSC, a colony-forming unit-fibroblast (CFU-F) assay was performed in some of our patient-derived cells (5 samples of MDS and 2 samples of AML). The MSC derived from MDS patients showed a median of 23.2 CFU-F (range: 16–37) per 103 cells. The samples from MSC-derived AML presented 28 and 8 CFU-F per 103 cells. These results indicate the self-renewal capacity of our MSC derived from patient bone marrow.
Immunomodulative capability
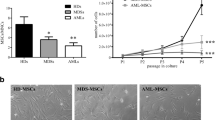
The immunomodulatory properties of MSC have been well characterized28, including suppression of T cell-mediated immunity29. Herein, the ability of patient-derived MSC to inhibit T cell proliferation was analyzed by mixing different ratios of MSC and CFSE-labeled CD3+ cells, in the presence of PHA. We observed a significant inhibitory effect of MDS-MSC on CD3+ cell proliferation up to the ratio of 1:100 (p < 0.05), in a dose-dependent manner, similar to that of HC-MSC (Fig. 1). This result remained significant when MDS patients were classified into subgroups, according to the WHO 2008 classification (data not shown). On the other hand, in contrast to HC-MSC, which presented normal inhibitory properties, AML-derived MSC were able to significantly inhibit T-cell proliferation at lower ratios. AML-MRC derived-MSC only inhibited proliferation at a ratio of 1:2 (p < 0.001), and the de novo AML MSC inhibited up to a ratio of 1:10 (p < 0.01) (Fig. 1). Further studies are needed to address the key aspects of the reduction of AML-MSC-mediated immunosuppression.

T cell proliferation assays were performed using CFSE-labeled CD3+ T cells activated with PHA and cocultured with MSC (white and shades of gray columns) from healthy donors, myelodysplastic syndromes (MDS), acute myeloid leukemia with myelodysplasia-related changes (AML-MRC) and de novo acute myeloid leukemia (de novo AML) patients, or without MSC (positive control; black column) for 4 days at MSC:T cell ratios of 1:2, 1:5, 1:10, 1:50 and 1:100 as shown in the figure. Cell proliferation was determined by flow cytometry after gating the lymphocyte population on the forward and side scatter plot and measuring the percentage of CFSE positive T cells. Results are shown as mean ± SEM and the number of samples in each group is shown in the figure. ANOVA, Bonferroni’s post-tests; *p < 0.05, **p < 0.01, ***p < 0.001.
Cytokine profile in MSC
We next characterized the mRNA expression of cytokines and other molecules in patient-derived MSC and compared these to the HC samples. No significant difference was detected in the MDS-derived MSC group (Fig. 2). AML-MRC-derived MSC showed a significant increase in IL6 expression (p = 0.02). However, de novo AML MSC presented a significant increase in expression levels of vascular endothelial growth factor A (VEGFA), stromal cell-derived factor 1 (CXCL12), receptor of prostaglandin E2 (RPGE2), indoleamine 2,3-dioxygenase (IDO), IL6 and IL32 (all p < 0.05), followed by a decrease in IL10 expression (p = 0.009) when compared to the HC group. We also observed a significantly increased expression of VEGFA, CXCL12, RPGE2 and IL32 in de novo AML, when compared with AML-MRC-derived MSC (p < 0.05). There were no differences in the expressions of TGFβ1 and IL1β.

qPCR analyses of mRNA expression of VEGFA (A), CXCL12 (B), TGFβ1 (C), RPGE2 (D), IDO (E), IL10 (F), IL1β (G), IL6 (H) and IL32 (I) in bone marrow MSC obtained from HC, MDS, AML-MRC and de novo AML patients. The “y” axis represents the relative mRNA expression. Horizontal lines indicate medians. The number of samples in each group and p values are indicated in the graph. Mann Whitney test.
We also analyzed the expression of the four best characterized isoforms of IL32 in our cohort. De novo AML MSC presented a significant increase in the expression levels of IL32γ(p = 0.01), the IL-32 isoform with the highest biological activity30. There were no differences in the expressions of the IL32α, β and δ transcripts (Supplemental Figure S1).
Silencing of IL-32 by miRNA and HS5 cell proliferation
Several studies have demonstrated that IL-32 plays a role in the inflammatory microenvironment and that there is a network between IL-32 and other cytokines such as IL-1β, IL-6, IL-10 and VEGF17,19,21,22,31,32,33. To investigate the relation between IL-32 and MSC, we used HS5, a cell line with a stromal phenotype and with the ability to secret several cytokines, including IL-6 and IL-1, and to support hematopoiesis34. Since inflammation triggers MSC activity35, we also performed our experiments upon stimulation with IFN-γ and TNF-α. HS5 cells were stably transduced with two lentiviral constructs encoding miRNA targeting IL32 (miIL32#1 and miIL32#2) or with miControl. After polyclonal cell selection with blasticidin, the efficiency of IL-32 silencing was analyzed by Western blotting. A significant reduction in IL-32 protein levels was observed in both constructions of miIL32 HS5, compared with miControl cells under regular and inflammatory conditions (Fig. 3A and B).

Western blotting analyses of IL-32 expression for HS5 cells in the presence or not of TNF-α and IFN-γ (10 ng/mL) as indicated. The levels of IL-32 relative to actin were quantified. In the figures are reported the cropped gels/blots. All gels were run in the same experimental conditions (see material and methods for details). (Full-length blots are reported in Supplementary Figure S2). Results are shown relative to miControl cells, as mean ± SEM of three independent experiments. ANOVA, Bonferroni’s post-tests (**p < 0.01; ***p < 0.001). (C) Cell viability was determined by MTT assay after 48 hours of incubation of miIL32 (#1 and #2) and normalized by the corresponding miControl cells. Results are shown as mean ± SEM of four independent experiments; The MTT assay was performed in the presence or not of IFN-γ and TNF-α (10 ng/mL) as indicated. ANOVA, Bonferroni’s post-tests (*p < 0.05, **p < 0.01); (D) Ki-67 mean of fluorescence intensity (M.F.I.) was determined by flow cytrometry after incubation of miIL32 for 48 h and normalized by the corresponding miControl cells. The dotted line represents the mean of miControl cells. Results are shown as mean ± SEM of four independent experiments; **p < 0.01, Student t test.
IL-32 is reported to have hematopoietic growth factor properties36, while IL-32 silencing results in a reduction in endothelial cell proliferation37. To determine whether IL-32 silencing affects stromal cell proliferation and/or viability, MTT assays were performed. Unexpectedly, viability of miIL32 HS5 cells was significantly increased (by ~40%) when compared with miControl cells, with or without pro-inflammatory stimulation (p < 0.05; Fig. 3C). Ki-67 analysis revealed that IL-32 silencing significantly increased cell proliferation (Fig. 3D).
Chemotactic activity of IL-32 on PBMCs
Cell migration is essential for the induction of an effective immune response. To test whether IL-32 has any effect on PBMC recruitment, transwell chemotaxis of PBMC was performed in coculture with miControl or miIL32 HS5 cells. The number and type of PBMC that migrated through the membrane were analyzed. There was a significant decrease in CD45+ cells migration, which mainly reflects the significant decrease in CD4+ migration, with or without proinflammatory stimulation (p < 0.05; Fig. 4).

miIL32 and miControl HS5 cells were seeded in the lower chamber of transwell plates with or without the addition of IFN-γ and TNF-α for 24 h before adding PBMCs, which were allowed to migrate for 5 h. Data represent mean ± SEM (4 independent experiments). ANOVA, Bonferroni’s post-tests (**p < 0.01; ***p < 0.001).
Similar effects on cell function were observed after IL-32 silencing with both the miIL32 sequences, implying that these effects are the result of RNAi-mediated silencing of the IL-32 gene rather than off-target effects. Since miIL32#2 showed a slightly better efficiency, we chose to carry out the subsequent experiments with this sequence.
Chemoprotection to AraC conferred by miIL32 HS5 cells
We firstly evaluated whether IL-32 silencing could modify the chemoprotection to AraC-induced apoptosis, the most effective drug for the treatment of AML38, conferred by HS5 to U937 cells39. As observed in Fig. 5A, IL-32 silencing does not modify the resistance of U937 cells to AraC when they are in contact with HS5 (p < 0.001).

U937 cells were cultured in the presence or absence of miControl or miIL32 HS5 cells in two distinct conditions: (A) direct contact or (B and C) noncontact (using a tranwell system). Cells were allowed to grow for 96 h and AraC was added (1 μM) for the last 18 h of experiment. (C) TNF-α was added (10 ng/mL) at the same time as AraC. Apoptosis was assessed by flow cytometry analysis of Annexin-V-APC/PI–stained cells. Data represent means ± SEM (6 independent experiments). ANOVA, Bonferroni’s post-tests (*p < 0.05; **p < 0.01; ***p < 0.001). (D) Dot plots are representative of one experiment.
Next, we tested whether the supernatant of miIL32 HS5 cells could change the chemosensitivity of U937 cells to AraC, using a transwell system. In these conditions, we observed that HS5 cells still protect U937 from AraC cytotoxicity. However, noncontact with miIL32 HS5 was significantly more effective than the miControl in inhibiting AraC-induced apoptosis of U937 cells (p < 0.05; Fig. 5B and D). Interestingly, this effect was reverted by the addition of TNF-α (Fig. 5C and D).
Modulation of cytokines, chemokines, MAPK and NF-κB signaling by IL-32
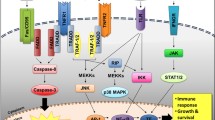
We also measured, in serum-free supernatants from cell culture of miControl and miIL32, the levels of cytokines and chemokines, in order to assess whether IL-32 regulates their expression in the stromal cell. The major cytokines downmodulated by IL-32 are demonstrated in Fig. 6A and B.

(A,B) The fold-change in cytokine concentration in miIL32 HS5 cell supernatant compared to miControl cell values is plotted, in the presence or not of IFN-γ and TNF-α (10 ng/mL) as indicated. Results are shown as mean ± SEM of 3 independent experiments. (C) HS5 cells transduced either with miIL32#1 or miIL32#2 and miControl. Western blot for IL-32, p-NF-κB, p-IKKβ, and p-IKKα; (D) p-JNK and p-p38 MAPK. (E) Activation status of NF-κB signaling in AML-MSC patients; western blot for p-NF-κB in total protein of MSC cells. Gels were run under the same experimental conditions while images of western blots displayed in cropped format. Membranes were reprobed with antibodies against actin or total protein. Full-length blots/gels are presented in Supplementary Figure S3.
The ability of IL-32 to activate the p38 mitogen-activated protein kinase (MAPK) and nuclear factor-kappa B (NF-κB) pathways has been previously reported17,40,41. Herein, we sought to examine the effect of IL-32 on MAPK and NF-κB signaling in our stromal cell line model. We observed that IL-32 depletion (Fig. 6C) resulted in a decrease in phosphorylation of NF-κB, IKKα, IKKβ, c-Jun N-terminal kinase (JNK) and p38 MAPK (Fig. 6C and D). Furthermore, we analyzed the phosphorylation levels of NF-κB in samples from patient-derived MSC and HC-derived MSC. In this small set of patient-derived cells, NF-κB phosphorylation levels were higher in the AML-derived MSC, especially in de novo AML samples (Fig. 6E). This finding is worthy of further research.
Discussion
In this report, we characterized and compared MSC derived from patients with MDS, AML-MRC and de novo AML with these from healthy controls. Although MSC from patients exhibited a typical antigen expression profile, as already described by others42, we observed a reduced immunosuppressive ability of AML-MSC when compared to the control. There are few studies about the immunosuppressive ability of AML-derived MSC43, although studies about MDS-MSC immunosuppressive function have been controversial43,44,45,46. We found that MDS-MSC were capable of satisfactorily inhibiting T cell proliferation in vitro induced by PHA, even when we classified our cohort as low or high-risk MDS, according to the WHO 2008 classification2. Supporting this finding, we observed no significant difference in any cytokine or molecule studied in the MDS-MSC. In agreement with Klaus et al.44, our data suggest that MDS-derived MSC are not the main factor responsible for the aberrant T cell response, sometimes observed in MDS patients.
We also demonstrated a distinct cytokine profile between AML-MRC and de novo AML-derived MSC, suggesting that the difference between these two AML subtypes also extends into the leukemic stem cell niche. AML-MRC is characterized by the persistence of the malignancy, alterations in the hematopoietic niche caused by prior MDS treatment, a higher frequency of molecular mutations and cytogenetic abnormalities47. We observed that, despite the impaired immunosuppressive ability of the AML-MRC-derived MSC, their cytokine profile is very similar to that of the control group. Conversely, de novo AML-derived MSC presented a significant increase in their expressions of VEGF and IL6, both of which are secreted by the leukemic blast in order to promote their survival and proliferation48,49,50,51. Recently, Kim et al.52 demonstrated that leukemia stem cells from de novo AML patients can induce extensive alterations in the mesenchymal niche, resulting in an altered expression of crosstalk molecules, including CXCL12. Together with the lower IL10 expression53,54, and the higher expression of IDO55,56,57, CXCL1258,59,60 and RPGE261,62,63 observed in our de novo AML cohort, we suggest that at least at the beginning of this disease, prior to any treatment, MSC provides a permissive niche for leukemogenesis rather than normal hematopoiesis. Marcondes et al.22 showed an increase in IL32 expression in the bone marrow stromal cells from 13 patients with MDS. Nevertheless, in our cohort of 22 MDS patients we could not see any statistical difference between the MDS and HC groups. These differences may due to either a lack of standardized culture for bone marrow MSC or the heterogeneity of MDS patients. Furthermore, in de novo AML-derived MSC, we observed a significantly higher expression level of IL32, which reflects the expression of the most active isoform, IL32γ. It has been shown that the over expression of IL-32γ in an animal model resulted in the inhibition of the cell proliferation of melanoma and colon tumors64. Our results suggest that the higher expression of IL32γ in de novo AML-derived MSC could contribute to the slower-growing cells generated from these patients samples, as observed in our study. Accordingly, our in vitro results show that IL-32 inhibition resulted in a significantly increased stromal cell proliferation, with our without proinflammatory stimulation. IL-32 may have pleiotropic effects on cell proliferation, as indicated by many other cellular functions65,66,67,68,69,70.
Son et al. demonstrated that endogenously-secreted IL-32γ may regulate a variety of immune responses via CCL5 expression from dendritic cells, including chemotaxis of activated T cells71. We observed that silencing of IL-32 resulted in lower concentrations of CCL5 in the supernatant of HS5 cells, which may explain the significantly decreased migration of CD4+ cells when cocultured with HS5 miIL32 cells. Additionally, many other important inflammatory molecules, including VEGF, TNF-α, IFN-γ, and IL-6 were reduced in the supernatant of HS5 miIL32 cells.
Our study on AraC-mediated cytotoxicity has shown that coculture of HS5 miControl or miIL32 with U937 cells in vitro resulted in the same levels of enhanced protection compared to U937 without HS5 cells. However, the coculture of HS5 miIL32 with U937 cells using transwell assays resulted in a significant protection from AraC-induced apoptosis when compared to the coculture with HS5 miControl. It has been described that HS5 cells can secrete a soluble factor(s) that protect U937 from AraC induced apoptosis72. Herein, we showed that IL-32 has a role in stromal cell crosstalk with leukemia cells, and that TNF-α must be part of this network, at least in vitro, since the protection from AraC-induced apoptosis conferred by HS5 miIL32 was reversed upon the addition of TNF-α. In a recent studies, it was shown that IL-32α suppressed colon cancer development by promoting the death signaling of TNFR173, and that IL-32γ can induce TFN-α production in differentiated THP1 cell line74.
Proliferation, leukocyte chemotaxis and regulation of the apoptotic threshold depend on appropriate signals through a favorable cytokine milieu for their homing to the bone marrow75,76,77,78. Our results suggest that IL-32 participates in the regulation of the bone marrow cytokine milieu, at least in part, through MAPK and NF-κB signaling.
In conclusion, we have shown a distinct function and cytokine profiles in AML-MRC and de novo AML-derived MSC, including the expression of IL-32. We also demonstrate that IL-32 takes part in the chemoresistance induced by MSC.
Materials and Methods
Primary samples
For isolation and expansion of MSC, bone marrow cells were obtained from 8 healthy donors (healthy control; HC) and 22 MDS and 19 AML patients. For the T cell proliferation and chemotaxis assay, peripheral blood mononuclear cells (PBMC) from HC (n = 26) were obtained by Ficoll-Hypaque gradient separation. All HC and patients provided their informed written consent, and all experiments were performed in accordance with the ethical and care guidelines and were approved by the local ethics committee of the University of Campinas and was adherent to the Declaration of Helsinki. Patients were untreated at the time of sample collection. Patients’ characteristics are described in Supplementary Table 1. MDS patients were classified according to the WHO 2008 classification2. Acute promyelocytic leukaemia [APL or AML with t(15;17)(q22;q12)] was excluded from the de novo AML group.
Cell lines and chemical reagents
The human bone marrow stromal cell line, HS5, and the human acute myeloid leukemia cell line, U93779, were obtained from the American Type Culture Collection (Manassas, VA, USA). 293FT and HT1080 cells were acquired in 2012 from Invitrogen (Carlsbad, CA, USA). Cells were cultured in appropriated medium, according to the manufacturer’s instruction, containing 10% fetal bovine serum (FBS) and glutamine with penicillin/streptomycin and amphotericin B, and maintained at 37 °C, 5% CO2. Recombinant human TNF-α and IFN-γ were purchased from PeproTech (Rocky Hill, NJ, USA). Cytarabine (AraC) was obtained from Intas Pharmaceuticals (Ahmedabad, India) and prepared as a 10 mM stock solution.
Isolation and expansion of MSC from bone marrow
Mononuclear cells were isolated from bone marrow by density gradient centrifugation and seeded at a density of 106cells/cm2. After 3–4 days of adhesion, non-adherent cells were removed. Cells were cultured at 37 °C, 5% CO2 in DMEM medium containing 1% penicillin/streptomycin, 1% L-glutamine and 10% fetal bovine serum, as described previously80. After achieving 80% confluence, the cells were removed with trypsin and plated again at a concentration of 4 × 103 cells/cm2, for further passages. We performed all analyses at the fourth passage.
Quantitative PCR (q-PCR)
Total RNA was purified using the TRIzol Reagent (Invitrogen). The reverse transcription reaction was performed using RevertAid First Strand cDNA synthesis kit (MBI Fermentas, Amherst, NY, USA). Expression of mRNAs was detected by qPCR with the Maxima SYBR Green qPCR master mix (MBI Fermentas) using the ABI 7500 Sequence Detection System (PE Applied Biosystems, Foster City, CA, USA). HPRT was used as an endogenous control (sequences of all genes at Supplementary Table 3). The relative quantification value was calculated using the equation, 2−ΔΔCT 81.
Flow cytometry
Immunophenotyping of MSC was evaluated by flow cytometry (FACSCalibur, Becton-Dickinson, San Jose, CA, USA). Fluorochrome conjugated monoclonal antibodies, anti-CD45-FITC or -PercP, CD34-APC, CD73-PE, CD31-FITC27, HLADR-FITC, CD90-PeCy5, and CD105PE. The U937 leukemia cell line was also stained for the detection of apoptotic cells (Annexin-V-APC positive/propidium iodide (PI) negative populations) after coculture with HS5 cells. Analyses were performed using the FACSDiva software (version 4.0.1, Becton-Dickinson) or FlowJo software (Treestar, Inc., San Carlos, CA, USA).
Fibroblastic colony-forming unit (CFU-F) assay
CFU-F assays were performed by plating 1 × 103 cells/well in 6-well plates. The medium was changed on day 7. After 15 days of culture, adherent cells were washed twice with phosphate-buffered saline, and stained with 1% crystal-violet in methanol. CFU-F colonies were macroscopically enumerated and clusters of more than 50 cells were considered as colonies.
T cell proliferation assay
CD3+ cells were purified from isolated PBMC, by magnetic separation of bead-bound cells (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Purity of CD3+ cells was 90–95%, as assessed by flow cytometry. CFSE-labeled CD3+ cells were resuspended and added to wells (105 cells/well) containing MSC at MSC/T cells ratios of 1:2, 1:5, 1:10, 1:50 and 1:100 in the presence of PHA (2,5 μg/mL). Four days later the CFSE fluorescence intensity was analyzed by flow cytometry (Becton-Dickinson).
Preparation of lentiviral vectors
Lentiviral vectors, expressing microRNA (miRNA) and targeting human IL-32 or LacZ were prepared using the BLOCK-iT Pol II miR RNAi Expression Vector with EmGFP System (Invitrogen), following the manufacturer’s instructions. The target sequence used to silence LacZ was 5′-AAATCGCTGATTTGTGTAGTC-3′. Two distinct sequences to silence IL-32 were used; sequence 1 (5′-AGAGGGCTACCTGGAGACAGT-3′) and sequence 2 (5′-GAGACAGTGGCGGCTTATTAT-3′). Viral concentrations were determined by in vitro transduction in HT1080 cells and using blasticidin selection.
Transduction of lentivirus
HS5 cells were transduced with lentivirus-mediated miRNA targeting LacZ (named miControl) or lentivirus-mediated miRNA targeting IL-32; miIL32#1 for sequence 1 and miIL32#2 for sequence 2. Briefly, HS5 cell lines were seeded onto six-well plates at 5 × 104cells/well, grown overnight, and transduced with lentiviral vectors at a multiplicity of infection equal to 1 in a minimal volume of medium containing 6 mg/mL of polybrene (Sigma-Aldrich, St. Louis, MO, USA). The transduced cells were selected for 15 days using blasticidin (10 μg/mL) before functional analyses.
Western blotting
Western blot analysis was performed as described previously82. Anti-IL-32 (sc-134446) and actin (sc-1616) antibodies were obtained from Santa Cruz Biotechnology. Anti-phospho-SAPK/JNK (Thr183/Tyr185; #9251), SAPK/JNK (9252), phospho-p38 MAPK (Thr180/Tyr182; 9211), p38 MAPK (8690), phospho-NF-κB p65 (Ser468; 3039), NF-κB p65 (4764), phospho-IKKα/β (Ser176/180; 2697), IKKα (2682) and IKKβ (8943) were obtained from Cell Signaling. Quantification of band intensity was performed by UN-SCAN-IT (Silk Scientific, Orem, UT, USA).
Cell viability of HS5 cells
Cell viability was measured by methylthiazoletetrazolium (MTT) assay. After 16 h of serum starvation, cells were stimulated to reenter the cell cycle and to proliferate using DMEM supplemented with 10% FBS. Serum starvation was not toxic to the cells (evaluated by Trypan blue; data not shown). A total of 9 × 103 cells per well were plated in 96-well plates in DMEM 10% FBS in the absence or presence of proinflammatory cytokines (10 ng/mL TNF-α and 10 ng/mL IFN-γ) for 48 h. In brief, 10 μL of a 5 mg/mL solution of MTT was added to the wells and incubated at 37 °C for 4 h. The reaction was stopped using 100 μL of 0.1 N HCl in anhydrous isopropanol. Cell viability was evaluated by measuring the absorbance at 570 nm, using an automated plate reader. All conditions were tested in six replicates.
Cell proliferation by Ki-67 staining
After 16 h of serum starvation, cells were stimulated to reenter the cell cycle and to proliferate using DMEM supplemented with 10% FBS, exposed or not to the proinflammatory environment (10 ng/mL TNF-α and 10 ng/mL IFN-γ). Ki-67 staining was performed following the manufacturer’s instructions (Ki-67 APC clone B56; Becton-Dickinson) and the mean fluorescence intensity (M.F.I.) was obtained by flow cytometry (FACSCalibur, Becton Dickinson). An IgG isotype was used as a negative control for each condition. Ten thousand events were acquired for each sample.
Chemotaxis assay
We performed a coculture of miControl or miIL32 HS5 cells and PBMC using a transwell culture system (8 μM-pore size membrane, Corning, NY, USA). HS5 cells were seeded in the lower chamber in DMEM/0.3% BSA exposed or not to an inflammatory environment (10 ng/mL TNF-α and 10 ng/mL IFN-γ). After 24 h, PBMC were seeded in the upper chamber (105cells/well). After 5 h, migrated cells were analyzed; cells from the lower chamber were trypsinized, stained with anti-CD45, anti-CD14, anti-CD4, and anti-CD8 and analyzed by flow cytometry. Results were calculated as the percentage of the 100% migration value (input).
Chemosensitivity assay to AraC
U937 cells were cocultured with miControl or miIL32 HS5 cells in two distinct conditions: direct contact or noncontact (using a 0.4 μm porous transwell insert that allows passage of soluble growth factors). Cells were allowed to grow for 96 h and AraC (1 μM) with or without TNF-α (10 ng/mL) was added in the last 18 h of experiment. Cells were collected and submitted to Annexin-V-APC/PI double staining for flow cytometry. U937 cells were distinguished from miControl or miIL32 HS5 cells by a gate in GFP-positive cells. The analysis was performed using FACSDiva software (version 4.0.1, Becton-Dickinson).
Bio-Plex human cytokine quantification assay
Culture supernatants from miControl and miIL32 HS5 cells, exposed or not to the proinflammatory environment (10 ng/mL TNF-α and 10 ng/mL IFN-γ), were assayed for several cytokines, chemokines and growth factors using a Bio-Plex human cytokine 27-plex and 21-plex panel assay (Bio-Rad, Hercules, CA, USA). Samples were tested according to the manufacturer’s instruction. Data were collected and analyzed using a Bio-Rad BioPlex 200 instrument equipped with Bio-Plex Manager software version 6.0 (Bio-Rad Laboratory, Hercules, CA, USA).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). For comparisons, Mann-Whitney or Student t tests were used for measured factors with 2 levels; ANOVA followed by post-hoc Bonferroni was used for measured factors with 3 or more levels. A p < 0.05 was considered as statistically significant.
Additional Information
How to cite this article: Lopes, M. R. et al. De novo AML exhibits greater microenvironment dysregulation compared to AML with myelodysplasia-related changes. Sci. Rep. 7, 40707; doi: 10.1038/srep40707 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Rankin, E. B., Narla, A., Park, J. K., Lin, S. & Sakamoto, K. M. Biology of the bone marrow microenvironment and myelodysplastic syndromes. Mol Genet Metab 116, 24–28 (2015).
Swerdlow, S. H., Campo, E., Harris, N. L., Jaffe, E. S., Pileri, S. A., Stein, H., Thiele, J. & Vardiman, J. W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (IARC Press, 2008).
Granfeldt Ostgard, L. S. et al. Epidemiology and Clinical Significance of Secondary and Therapy-Related Acute Myeloid Leukemia: A National Population-Based Cohort Study. J Clin Oncol 33, 3641–3649 (2015).
Ellis, S. L. & Nilsson, S. K. The location and cellular composition of the hemopoietic stem cell niche. Cytotherapy 14, 135–143 (2012).
Boulais, P. E. & Frenette, P. S. Making sense of hematopoietic stem cell niches. Blood 125, 2621–2629 (2015).
Blau, O. et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 118, 5583–5592 (2011).
Kastrinaki, M. C. et al. Mesenchymal stem cells in immune-mediated bone marrow failure syndromes. Clin Dev Immunol 2013, 265608 (2013).
Medyouf, H. et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 14, 824–837 (2014).
Binato, R., de Almeida Oliveira, N. C., Du Rocher, B. & Abdelhay, E. The molecular signature of AML mesenchymal stromal cells reveals candidate genes related to the leukemogenic process. Cancer Lett 369, 134–143 (2015).
Iwamoto, S., Mihara, K., Downing, J. R., Pui, C. H. & Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest 117, 1049–1057 (2007).
Kurtova, A. V. et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 114, 4441–4450 (2009).
Jacamo, R. et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood 123, 2691–2702 (2014).
Colotta, F., Allavena, P., Sica, A., Garlanda, C. & Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081 (2009).
Yang, L., Qian, Y., Eksioglu, E., Epling-Burnette, P. K. & Wei, S. The inflammatory microenvironment in MDS. Cell Mol Life Sci 72, 1959–1966 (2015).
Allampallam, K. et al. Biological significance of proliferation, apoptosis, cytokines, and monocyte/macrophage cells in bone marrow biopsies of 145 patients with myelodysplastic syndrome. Int J Hematol 75, 289–297 (2002).
Aggarwal, B. B. & Gehlot, P. Inflammation and cancer: how friendly is the relationship for cancer patients? Curr Opin Pharmacol 9, 351–369 (2009).
Kim, S. H., Han, S. Y., Azam, T., Yoon, D. Y. & Dinarello, C. A. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity 22, 131–142 (2005).
Kang, J. W. et al. Interaction network mapping among IL-32 isoforms. Biochimie 101, 248–251 (2014).
Nold, M. F. et al. Endogenous IL-32 controls cytokine and HIV-1 production. J Immunol 181, 557–565 (2008).
Li, W. et al. Activation of interleukin-32 pro-inflammatory pathway in response to influenza A virus infection. PLoS One 3, e1985 (2008).
Joosten, L. A. et al. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci USA 103, 3298–3303 (2006).
Marcondes, A. M. et al. Dysregulation of IL-32 in myelodysplastic syndrome and chronic myelomonocytic leukemia modulates apoptosis and impairs NK function. Proc Natl Acad Sci USA 105, 2865–2870 (2008).
Cheon, S. et al. Overexpression of IL-32alpha increases natural killer cell-mediated killing through up-regulation of Fas and UL16-binding protein 2 (ULBP2) expression in human chronic myeloid leukemia cells. J Biol Chem 286, 12049–12055 (2011).
Marcondes, A. M., Ramakrishnan, A. & Deeg, H. J. Myeloid Malignancies and the Marrow Microenvironment: Some Recent Studies in Patients with MDS. Curr Cancer Ther Rev 5, 310–314 (2009).
Dominici, M. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317 (2006).
Lin, G., Finger, E. & Gutierrez-Ramos, J. C. Expression of CD34 in endothelial cells, hematopoietic progenitors and nervous cells in fetal and adult mouse tissues. European journal of immunology 25, 1508–1516 (1995).
Newman, P. J. The biology of PECAM-1. The Journal of clinical investigation 99, 3–8 (1997).
Bartholomew, A. et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo . Exp Hematol 30, 42–48 (2002).
Crop, M. J. et al. Donor-derived mesenchymal stem cells suppress alloreactivity of kidney transplant patients. Transplantation 87, 896–906 (2009).
Choi, J. D. et al. Identification of the most active interleukin-32 isoform. Immunology 126, 535–542 (2009).
Bai, X. et al. IL-32 is a host protective cytokine against Mycobacterium tuberculosis in differentiated THP-1 human macrophages. J Immunol 184, 3830–3840 (2010).
Netea, M. G. et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1beta and IL-6 production through a caspase 1-dependent mechanism. Proc Natl Acad Sci USA 102, 16309–16314 (2005).
Kang, J. W. et al. Interleukin-32delta interacts with IL-32beta and inhibits IL-32beta-mediated IL-10 production. FEBS Lett 587, 3776–3781 (2013).
Roecklein, B. A. & Torok-Storb, B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood 85, 997–1005 (1995).
Madrigal, M., Rao, K. S. & Riordan, N. H. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J Transl Med 12, 260 (2014).
Moldenhauer, A. et al. Interleukin 32 promotes hematopoietic progenitor expansion and attenuates bone marrow cytotoxicity. Eur J Immunol 41, 1774–1786 (2011).
Nold-Petry, C. A. et al. IL-32 promotes angiogenesis. J Immunol 192, 589–602 (2014).
Kanno, S. et al. Characterization of resistance to cytosine arabinoside (Ara-C) in NALM-6 human B leukemia cells. Clin Chim Acta 377, 144–149 (2007).
Garrido, S. M., Appelbaum, F. R., Willman, C. L. & Banker, D. E. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp Hematol 29, 448–457 (2001).
Yousif, N. G., Al-Amran, F. G., Hadi, N., Lee, J. & Adrienne, J. Expression of IL-32 modulates NF-kappaB and p38 MAP kinase pathways in human esophageal cancer. Cytokine 61, 223–227 (2013).
Zeng, Q. et al. Interleukin-32 contributes to invasion and metastasis of primary lung adenocarcinoma via NF-kappaB induced matrix metalloproteinases 2 and 9 expression. Cytokine 65, 24–32 (2014).
Chandran, P. et al. Mesenchymal stromal cells from patients with acute myeloid leukemia have altered capacity to expand differentiated hematopoietic progenitors. Leuk Res 39, 486–493 (2015).
Zhi-Gang, Z., Wei-Ming, L., Zhi-Chao, C., Yong, Y. & Ping, Z. Immunosuppressive properties of mesenchymal stem cells derived from bone marrow of patient with hematological malignant diseases. Leuk Lymphoma 49, 2187–2195 (2008).
Klaus, M. et al. Reserves, functional, immunoregulatory, and cytogenetic properties of bone marrow mesenchymal stem cells in patients with myelodysplastic syndromes. Stem Cells Dev 19, 1043–1054 (2010).
Zhao, Z. et al. The different immunoregulatory functions of mesenchymal stem cells in patients with low-risk or high-risk myelodysplastic syndromes. PLoS One 7, e45675 (2012).
Han, Q. et al. Impairment in immuno-modulatory function of Flk1(+)CD31(−)CD34(−) MSCs from MDS-RA patients. Leuk Res 31, 1469–1478 (2007).
Vardiman, J. & Reichard, K. Acute Myeloid Leukemia With Myelodysplasia-Related Changes. Am J Clin Pathol 144, 29–43 (2015).
Bellamy, W. T. et al. Vascular endothelial cell growth factor is an autocrine promoter of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood 97, 1427–1434 (2001).
Kampen, K. R., Ter Elst, A. & de Bont, E. S. Vascular endothelial growth factor signaling in acute myeloid leukemia. Cell Mol Life Sci 70, 1307–1317 (2013).
Wierenga, A. T., Schuringa, J. J., Eggen, B. J., Kruijer, W. & Vellenga, E. Downregulation of IL-6-induced STAT3 tyrosine phosphorylation by TGF-beta1 is mediated by caspase-dependent and -independent processes. Leukemia 16, 675–682 (2002).
Dokter, W. H., Tuyt, L., Sierdsema, S. J., Esselink, M. T. & Vellenga, E. The spontaneous expression of interleukin-1 beta and interleukin-6 is associated with spontaneous expression of AP-1 and NF-kappa B transcription factor in acute myeloblastic leukemia cells. Leukemia 9, 425–432 (1995).
Kim, J. A. et al. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res 75, 2222–2231 (2015).
Sanchez-Correa, B. et al. Cytokine profiles in acute myeloid leukemia patients at diagnosis: survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine 61, 885–891 (2013).
Westermann, F. et al. Interleukin 10 inhibits cytokine production of human AML cells. Ann Oncol 7, 397–404 (1996).
Curti, A. et al. Acute myeloid leukemia cells constitutively express the immunoregulatory enzyme indoleamine 2,3-dioxygenase. Leukemia 21, 353–355 (2007).
Curti, A. et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25− into CD25+ T regulatory cells. Blood 109, 2871–2877 (2007).
Prendergast, G. C. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene 27, 3889–3900 (2008).
Kryczek, I., Wei, S., Keller, E., Liu, R. & Zou, W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol 292, C987–995 (2007).
Bianchi, G., Borgonovo, G., Pistoia, V. & Raffaghello, L. Immunosuppressive cells and tumour microenvironment: focus on mesenchymal stem cells and myeloid derived suppressor cells. Histol Histopathol 26, 941–951 (2011).
Burger, J. A. & Kipps, T. J. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 107, 1761–1767 (2006).
Burr, S. P., Dazzi, F. & Garden, O. A. Mesenchymal stromal cells and regulatory T cells: the Yin and Yang of peripheral tolerance? Immunol Cell Biol 91, 12–18 (2013).
Malissein, E. et al. PGE(2) receptor subtype functionality on immature forms of human leukemic blasts. Leuk Res 30, 1309–1313 (2006).
Nataraj, C. et al. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest 108, 1229–1235 (2001).
Oh, J. H. et al. IL-32gamma inhibits cancer cell growth through inactivation of NF-kappaB and STAT3 signals. Oncogene 30, 3345–3359 (2011).
Heinhuis, B. et al. Tumour necrosis factor alpha-driven IL-32 expression in rheumatoid arthritis synovial tissue amplifies an inflammatory cascade. Ann Rheum Dis 70, 660–667 (2011).
Heinhuis, B. et al. Inflammation-dependent secretion and splicing of IL-32{gamma} in rheumatoid arthritis. Proc Natl Acad Sci USA 108, 4962–4967 (2011).
Kang, J. W. et al. A proinflammatory cytokine interleukin-32beta promotes the production of an anti-inflammatory cytokine interleukin-10. Immunology 128, e532–540 (2009).
Meyer, N. et al. IL-32 is expressed by human primary keratinocytes and modulates keratinocyte apoptosis in atopic dermatitis. J Allergy Clin Immunol 125, 858–865, e810 (2010).
Kim, Y. G. et al. Effect of interleukin-32gamma on differentiation of osteoclasts from CD14+ monocytes. Arthritis Rheum 62, 515–523 (2010).
Netea, M. G. et al. Interleukin-32 induces the differentiation of monocytes into macrophage-like cells. Proc Natl Acad Sci USA 105, 3515–3520 (2008).
Son, M. H., Jung, M. Y., Choi, S., Cho, D. & Kim, T. S. IL-32gamma induces chemotaxis of activated T cells via dendritic cell-derived CCL5. Biochem Biophys Res Commun 450, 30–35 (2014).
Macanas-Pirard, P. et al. Bone marrow stromal cells modulate mouse ENT1 activity and protect leukemia cells from cytarabine induced apoptosis. PLoS One 7, e37203 (2012).
Yun, H. M. et al. IL-32alpha suppresses colorectal cancer development via TNFR1-mediated death signaling. Oncotarget 6, 9061–9072 (2015).
Kim, M. S. et al. IL-32theta gene expression in acute myeloid leukemia suppresses TNF-alpha production. Oncotarget 6, 40747–40761 (2015).
Sercan Alp, O. et al. Memory CD8(+) T cells colocalize with IL-7(+) stromal cells in bone marrow and rest in terms of proliferation and transcription. Eur J Immunol 45, 975–987 (2015).
Di Rosa, F. T-lymphocyte interaction with stromal, bone and hematopoietic cells in the bone marrow. Immunol Cell Biol 87, 20–29 (2009).
Van Etten, R. A. Aberrant cytokine signaling in leukemia. Oncogene 26, 6738–6749 (2007).
Blalock, W. L. et al. Signal transduction, cell cycle regulatory, and anti-apoptotic pathways regulated by IL-3 in hematopoietic cells: possible sites for intervention with anti-neoplastic drugs. Leukemia 13, 1109–1166 (1999).
Dickson, G. J. et al. HOXA/PBX3 knockdown impairs growth and sensitizes cytogenetically normal acute myeloid leukemia cells to chemotherapy. Haematologica 98, 1216–1225 (2013).
Manzini, B. M. et al. Useful properties of undifferentiated mesenchymal stromal cells and adipose tissue as the source in liver-regenerative therapy studied in an animal model of severe acute fulminant hepatitis. Cytotherapy 17, 1052–1065 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Traina, F., Carvalheira, J. B., Saad, M. J., Costa, F. F. & Saad, S. T. BCR-ABL binds to IRS-1 and IRS-1 phosphorylation is inhibited by imatinib in K562 cells. FEBS Lett 535, 17–22 (2003).
Acknowledgements
The authors would like to thank Dr. Nicola Conran for English review. We thank the staff of the Life Sciences Core Facility (LaCTAD) from University of Campinas, for cell biology analysis. We would like to thank Cleide Moreira Silva for assistance with statistical analysis. This work received financial support from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). The Hematology and Transfusion Medicine Center of UNICAMP forms part of the National Institute of Blood, Brazil (INCT do Sangue–CNPq/MCT).
Author information
Authors and Affiliations
Contributions
M.R.L. Conception and design, data analysis and interpretation and manuscript writing. J.K.N.P. Conception and design, data analysis and interpretation. P.M.C. Provision of patients and data analysis and interpretation and manuscript writing. J.A.M.N. Conception and design, data analysis and interpretation and manuscript writing. F.T. Provision of patients and data analysis and interpretation and manuscript writing. S.T.O.S. Conception and design, provision of patients and manuscript writing. P.F. Conception and design, data analysis and interpretation, manuscript writing and final approval of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lopes, M., Pereira, J., de Melo Campos, P. et al. De novo AML exhibits greater microenvironment dysregulation compared to AML with myelodysplasia-related changes. Sci Rep 7, 40707 (2017). https://doi.org/10.1038/srep40707
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40707
- Springer Nature Limited
This article is cited by
-
Mesenchymal stromal cell senescence in haematological malignancies
Cancer and Metastasis Reviews (2023)
-
Catch me if you can: how AML and its niche escape immunotherapy
Leukemia (2022)
-
Most Variable Genes and Transcription Factors in Acute Lymphoblastic Leukemia Patients
Interdisciplinary Sciences: Computational Life Sciences (2019)