
Abstract
Phytoplankton blooms are a worldwide problem and can greatly affect ecological processes in aquatic systems, but its impacts on the functional potential of microbial communities are limited. In this study, a high-throughput microarray-based technology (GeoChip) was used to profile the functional potential of free-living microbes from the Xiamen Sea Area in response to a 2011 Akashiwo sanguinea bloom. The bloom altered the overall community functional structure. Genes that were significantly (p < 0.05) increased during the bloom included carbon degradation genes and genes involved in nitrogen (N) and/or phosphorus (P) limitation stress. Such significantly changed genes were well explained by chosen environmental factors (COD, nitrite-N, nitrate-N, dissolved inorganic phosphorus, chlorophyll-a and algal density). Overall results suggested that this bloom might enhance the microbial converting of nitrate to N2 and ammonia nitrogen, decrease P removal from seawater, activate the glyoxylate cycle, and reduce infection activity of bacteriophage. This study presents new information on the relationship of algae to other microbes in aquatic systems, and provides new insights into our understanding of ecological impacts of phytoplankton blooms.
Similar content being viewed by others

Introduction
Phytoplankton are the most important primary producers in marine systems and can accumulate to bloom proportions from eutrophication, especially in coastal waters. Blooms dramatically change seawater systems through their efficient autotrophic processes and the exudation of dissolved organic matter1, followed by changes in the bacterial2,3,4,5, archaeal6, viral7,8 and other microbial communities.
In the phytoplankton bloom area, bacterial abundance increases greatly4,9, especially species that consume organic matter released by the algae. Virioplankton also increase following increases in bacterial abundance, particularly during peaks in bloom activity. Through these changes to the microbial communities, blooms would eventually affect biogeochemical cycling of elements such as C, N, P and Fe in the marine system10,11,12.
Studies of the influence of phytoplankton blooms on marine ecosystems have primarily focused on the changes of other microbial communities as revealed by 16S rDNA clone libraries13, polymerase chain reaction-denaturing gradient gel electrophoresis14,15, flow cytometry16, terminal restriction fragment length polymorphism13, fluorescence in situ hybridization17,18, metatranscriptomics19, and high-throughput sequencing20,21. These methods mainly focus on the microbial community instead of their functional potential, except the metatranscriptomics which could be used to investigate the real time function fluctuation based on RNA. These studies confirmed increased bacterial abundance during blooms and subsequent increases in bacteriophage abundance. And there are some interesting findings based on metatranscriptomic analysis. Viral transcription was much higher in the post-bloom environment than during bloom, and bacterioplankton within the bloom had greater upregulation of genes for organic acid utilization and increasing cell surface adhesiveness19. Compared to when in monocultures, the dominant dinoflagellate expresses toxin biosynthesis related genes at a higher level in the presence of competitors, predators and prey22. It seems that viruses might have less activity and bacteria have enhanced substrate utilization levels during bloom.
The influence of phytoplankton bloom on the biochemical process in the water system is limited, except the sulfur cycle. Phytoplankton in ocean plays an important role on global sulfur cycle by producing massive quantities of dimethylsulfoniopropionate (DMSP)23 and promoting specific bacteria species for DMSP turnover, e.g. Roseobacter24. What’s more, our former study25 (based on Illumina sequencing of 16S rDNA) showed the bacterial diversity decreased, species evenness increased, the bacterial community structure significantly changed, and many bacterial species were stimulated in this bloom, e.g. SAR86 and SAR116 clades and the AEGEAN-169 marine group. Despite these findings, there is still a lot to understand in regards to the functional changes in the microbial community during a bloom.
To obtain a more comprehensive understanding of changes in community function, functional gene arrays (FGAs), microarrays that contain functional gene probes, can be used to simultaneously detect key genes involved in multiple microbial functional processes. The most comprehensive FGAs are the GeoChip arrays. GeoChip 4.2 contains 107,950 probes for genes involved in cycling of C, N, S, P and metals, virulence and antibiotic resistance, the biodegradation of environmental contaminants, stress responses26, fungi, pathogens and soil beneficial genes. GeoChips have been successfully used to analyze microbial communities in several marine environments including marine sediments27, marine basalt28, deep sea hydrothermal vents29, the Gulf of Mexico during the 2010 oil spill30, mangroves31, and coral mucous32.
In this study, we used the GeoChip to functionally profile free-living microbial communities in response to an A. sanguinea bloom. Specifically, we aimed to address the following questions: (i) does the bloom significantly change the microbial community functional potential? (ii) what functional processes are influenced by the bloom? and (iii) are there specific correlations between the A. sanguine population and the functional potential of the associated microbial communities? Our results showed that this bloom significantly altered overall community functional structure and promoted organic carbon degradation, ammonium accumulation and decreased P removal; there were statistical correlations between A. sanguine abundance and the functional potential of the microbial community. This study provides new insight into our understanding of the ecological impacts of phytoplankton blooms on microbial communities.
Experimental Procedures
Study sites and sample collection
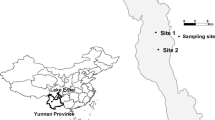
Samples were collected along the Xiamen coast at sites within (A1; N 24°35′53.40′′, E 118°9′29.67′′) or near (H1; N 24°36′56.31′′, E 118°9′15.92′′) the A. sanguinea bloom (Fig. 1), during the summer of 2011. Samples were collected each day from 31 July to 5 August as described previously25. Briefly, three replicate water samples (20 l) were collected from each site and filtered through 5-μm diameter pore-size filters (Millipore, US) to remove A. sanguinea and other large algae. The total free-living bacteria in the filtrate were then collected with 0.22-μm filters (Millipore, US) and stored at −70 °C until analysis.

Location of sampling sites.
This map was created based on ArcGIS (Version 10, http://www.esri.com/software/arcgis/arcgis-for-desktop) by CYY.
DNA extraction, amplification and labeling
For RNA and proteins, sampling procedure (e.g., filtration) could easily induce their expression change33. Experiments based on DNA are more stable than that of RNA and proteins, so DNA analysis was focused on in this study. High molecular weight DNA was extracted from filters using a previously described phenol–chloroform–isoamyl alcohol (25:24:1) extraction method4 and resuspended in sterile water. DNA quality and quantity was assessed using a spectrophotometer (ND-1000, Nanodrop Inc., DE, U.S.) and a PicoGreen assay34, respectively. An aliquot of DNA (25 ng) from each sample was amplified using a TempliPhi kit (GE Healthcare, Piscataway, NJ, U.S.) with a modified buffer35, and the amplified DNA (~1 μg) from each sample was labeled and then purified for hybridization36,37. Control samples from 1 August did not contain enough DNA to perform hybridization, so those samples were removed.
GeoChip hybridization and data processing
GeoChip 4.2 was used in this study and sample preparation and hybridization were performed as previously described31. After hybridization, the arrays were scanned using an MS200 Microarray Scanner (NimbleGen) and images were processed as previously described31,38,39. Spots were scored as positive if the signal-to-noise ratio (SNR) was ≥2.0 and the CV of the background was <0.8. The probes that appeared in only one of the three replicates were removed as noise.
Hybridization data is available at the Institute for Environmental Genomics, University of Oklahoma (http://ieg2.ou.edu/NimbleGen/batch_upload.cgi) and the data was processed as previously described7. The relative abundance of each probe signal was then multiplied by the mean of the sums of the original signal intensity for all probes in each sample. All statistical analyses were performed in R40. Dissimilarity tests between the bloom and control areas were based on the Bray-Curtis dissimilarity index using analysis of similarities, and the Bray-Curtis dissimilarities among samples using Adonis, ANOSIM and MRPP36, and multi-response permutation procedures27. Monte Carlo permutation was used to test statistical significance and a significance level of p < 0.05 was adopted for all comparisons. Detrended correspondence analysis (DCA) was used to compare the different functional gene communities, and canonical correlation analysis (CCA) was used to link microbial communities to environmental variables. Variation partitioning analysis (VPA) was conducted to examine the contribution of environmental factors in influencing functional gene in the CCA analysis. R statistics were performed running the Vegan (v. 1.15-1)41 and agricolae package. Hierarchical clustering was performed with CLUSTER 3.0 with uncentered correlations and the complete linkage for both genes and samples; trees were visualized with Java Treeview.
Results
The A1 and H1 were bloom and control site, respectively (Fig. 1). The water was distinctly dark in A1 because of the high density of A. sanguinea, especially on sampling day 2 and 5. H1 was beside A1 and there was no bloom phenomenon during sampling in this site. The measurement of water parameters (e.g. phytoplankton species identification, cell counts, concentration of chlorophyll a, dissolved inorganic phosphorus, nitrate, and so on) were determined as previously described25. Based on the algal density dynamics (Table S1), 31 July through 4 August were during the bloom (B); peak algal densities occurred on 1 and 4 August (bloom peaks; BP1 and BP2); and 5 August was after the bloom (AB).
A total of 14171 probes for 575 genes were detected across all samples, including: 1470 probes (39 genes) for C cycling, 1108 probes (17 genes) for N cycling, 196 probes (3 genes) for P cycling, 492 probes (11 genes) for S cycling, 2578 probes (45 genes) for stress response, 85 probes (32 genes) for bacteria phage and 594 probes (13 genes) for virulence (585, 6 and 3 probes from bacteria, fungi and archaea, respectively).
Total overall functional diversity and structure of free-living microbial communities
The number and relative abundance of functional genes detected were used to calculate the Shannon–Weiner index (H’) and the Simpson Evenness index (E). For most days, neither the difference in the overall microbial functional diversity (Fig. 2a) nor the evenness (Fig. 2b) between the bloom and control areas was significant (p > 0.05). However, on BP2, the functional gene diversity in the bloom area was significantly (p < 0.05) higher than that in the control area, while evenness (Fig. 2) was significantly (p < 0.05) lower than in the control area. The greatest percentage of unique genes (18.1%) was found in the day 1 bloom samples. Days 3, 4 and 5 had 10.2, 14.8 and 16.5% unique genes (data not shown).

(a) Shannon diversity and (b) Simpson evenness of total functional genes for bloom and control samples on day 1, 3, 4 and 5. Day 5 was bloom peak 2 (BP2). *p < 0.05; the error bars represent standard errors.
Dissimilarity tests (Table 1) showed there were significant (p < 0.01) differences in the functional gene structures of the bloom and control areas based on MRPP analysis on days 1, 3, 4, and 5. And the difference for the total was greater since the p values were all less than 0.05 based on Adonis, Anosim and MRPP analyses. So, the differences were more pronounced between bloom and control areas than over time. The DCA ordination plot (Fig. 3) shows bloom samples grouped in the middle, while the control samples were scattered around the bloom samples, suggesting that the functional gene composition was more similar or stable within the bloom.

Detrended correspondence analysis of functional genes.
Significantly changed functional genes in response to bloom
Based on the functional genes detected, functional potential of microbial communities within bloom samples were significantly (p < 0.05) different from control samples (Table 1), comparison was then calculated by T-test among all samples (data not shown). The abundance of 288 genes (1260 probes) were significantly (p < 0.05) changed during the bloom. Hierarchical cluster analysis of these 288 genes indicated that samples were well separated by site condition (Fig. 4a).

Hierarchical cluster analysis of functional genes from probes with significantly different abundance between bloom and control samples (a). Red indicates signal intensities above background (black), while green indicates signal intensities below background. Brighter red or green coloring indicates higher or lower signal intensities. Two major groups were observed. (b) The sum of signal intensities for genes from bloom and control sites in groups 1 and 2, (c) and the number of genes detected from each gene category in groups 1 and 2.
Based on the functional gene cluster results, two large groups were observed (Fig. 4a). The two groups represented genes with higher abundance either within the bloom (referred to as the Bloom Group, 179 genes) or the control site (referred to as the Control Group, 109 genes) (Fig. 4a,b). There were more functional genes with high abundance in bloom samples. The groups mainly contained organic remediation, carbon cycling, fungi function and stress related genes (Fig. 4c), and the gene numbers of these categories were much higher in the Bloom Group than in Control Group. The most abundant genes were mainly involved in organic carbon degradation: 17 (bloom) and 9 (control) from carbon degradation by bacteria, 13 (bloom) and 7 (control) genes from carbon degradation of fungi, 38 (bloom) and 16 (control) genes from aromatic carbon degradation. Gene abundance changes followed similar patterns for both bloom and control samples in Control Group, while these changes varied for bloom and control samples in Bloom Group (Fig. 4b).
There were more N cycling related genes in Bloom Group (11 genes) than Control Group (2 genes), and the 11 genes were involved in denitrification (nosZ, nirS, nirK and narG), assimilatory N reduction (nasA, nir and nirA), dissimilatory N reduction (napA and nrfA), nitrification (amoA) and nitrogen fixation (nifH); the 2 genes were involved in ammonification (gdh and ureC). The signal intensity changing pattern of those genes was shown in the simplified N cycling diagram (Fig. S1). This suggests the bloom community may have an accelerated N cycle stimulating the utilization of nitrate and nitrite, e.g., ammonification, N fixation and denitrification processes. Bloom Group also had more S cycling related genes (7) than Control Group (1), including genes for adenylylsulfate reductase, sulfide oxidation, sulfite reductase and sulphur oxidation. Control Group had increased dsrB for sulfite reductase.
Bloom Group had more stress genes related to N limitation (1 to 0) while Control Group had more related to P limitation (1 to 5). Bacteriophage genes involved in lysis and replication were high in Control Group (5 genes) than Bloom Group (2 genes), indicating the bloom might decrease viral infection of bacteria.
Relationship between significantly changed functional genes and environmental parameters
The 288 significantly changed genes represented the main microbial functional responses to this bloom. To test how environmental variables contributed to the functional gene differences, CCA was performed using all 288 genes and measured seawater and phytoplankton properties (Fig. 5a), including chemical oxygen demand (COD), nitrite-nitrogen (N2), nitrate-nitrogen (N3), dissolved inorganic phosphorus (DIP), chlorophyll a (CHLa) and total algal density (TALG).

CCA of significantly changed gene data with selected environmental variables (a) and partial CCA-based variation partitioning analysis (VPA) (b). The selected environmental variables include chemical oxygen demand (COD), nitrite nitrogen (N2), nitrate nitrogen (N3), dissolved inorganic phosphorus (DIP), chlorophyll a (CHLa) and total algal density (TALG).
In the CCA ordination plot (Fig. 5a), the first two canonical axes explained 41.0% and 13.7% of the constrained variation, respectively, in the significantly changed functional genes. The bloom and control samples were well separated along the first canonical axis (CCA1). The projection of environmental variables indicated that bloom samples (especially for the day1, 3, and 5 when the algal density was higher than day 4) showed a positive correlation with DIP, N3, TALG, CHLa and COD, while control samples were negatively correlated with these variables. The length of the arrows indicated that the impacts of TALG, CHLa and COD were greater than those of other variables.
Variation partitioning analysis (VPA) was conducted to test the seawater properties (COD, N2, N3 and DIP) and phytoplankton (CHLa and TALG) in influencing the significantly changed genes in the CCA analysis (Fig. 5b). The seawater and phytoplankton variables explained 44.6% and 34.0% of the variation in the genes, respectively, and their interaction explained 10.6%, leaving 10.8% unexplained.
Genes linked to the Akashiwo sanguinea community
Among the significantly changed genes, 67 showed significant (p < 0.05) correlations with A. sanguinea density; genes within C, N, S, P cycling and stress categories are shown in Fig. 6. The bacterial C degradation genes (AceA-isocitrate lyase, pectinase and phenol oxidase) showed significantly positive correlations with A. sanguinea while a gene for methane monooxygenase showed an inverse correlation. Four genes (nasA, nirA, nirS and nosZ) for assimilatory N reduction and denitrification were significantly correlated with A. sanguinea. The positive correlation with nirS and nosZ suggest that this bloom enhanced the denitrification potential of the free-living microbes. CysJ and dsrA for sulfite reductase showed higher relative abundances in bloom samples and had significantly positive correlations with A. sanguinea. This suggests that the sulfite reduction potential was enhanced during the bloom. The P cycling gene ppk had a lower relative abundance in the bloom and showed an inverse correlation with A. sanguinea.

A subset of significantly changed genes in the bloom compared to the control site.
*0.01 ≤ P < 0.05; **p < 0.01.
The stress response genes, pstA and phoA, for phosphate limitation, showed significantly negative correlations with A. sanguinea abundance. The heat shock genes-sigma 24, 32 and 70 showed significantly positive correlations with A. sanguinea while ctsR showed an inverse correlation, and the marine ecological function of these genes are still unclear. Bacteriophage related genes for replication and lysis showed a significantly inverse correlation with A. sanguinea, indicating the bloom might weaken potential of viruses to infect bacteria.
Discussion
Previously, we revealed how the free-living bacterial community responded to an Akashiwo sanguine bloom based on 16S rDNA sequencing25. This is a parallel study, examining functional gene changes in the communities examined in the earlier paper.
In this study, the ~10,000 detected probes covered a wide range of functional gene categories, indicating the community contained a high degree of microbial functional potential. This laid the foundation for the community’s resilience to natural disturbances.
Bloom doesn’t impact the functional potential of free-living microbial communities as greatly as expected
A. sanguinea blooms in the Xiamen Sea Area appeared for the first time in 20084 and have subsequently frequently occurred. Marine bacterial communities have been reported to be greatly influenced by A. sanguinea blooms4,25, including the bacterial abundance and community structure. But these studies were limited in their ability to reveal the influence of the blooms on bacterial and any other microbial functions. We hypothesized that the bloom would have a significant impact on free-living microbial function as compared to those in the control site. The overall community structures showed significant differences (Table 1) which were more pronounced between bloom and control areas than over time. But few individual genes/gene groups showed significant differences. Less than 10% of the detected probes were significantly different between the two sites. Most of the bloom-enriched genes were involved in organic matter degradation, which is unsurprising since high concentrations of phytoplankton can produce abundant organic matter from sunlight and inorganic matter42.
The microbial community functional structure appeared to be more stable within the bloom compared to the control, consistent with our previous results which based on bacterial community25. This could result from both environmental conditions and the bloom itself43, providing relatively stable environmental conditions necessary for bloom formation and sustainability44,45,46. In addition, the abundant algae within a bloom release organic matter42,47, which serves as a C source for the microbes within the bloom and influences environmental conditions. COD, N2, N3, DIP and TALG appeared to be strong drivers in selecting the microbial communities.
N and P important for bloom communities
Microbial communities within the bloom were enriched in N and P cycling genes, suggesting these elements are particularly important in this environment. Changes in many genes suggested the N cycling potential was accelerated in the bloom, especially for utilization of NO2− (denitrification) and NO3− (dissimilatory reduction of nitrate to ammonium), nitrogen fixation, and ammonium production. N limitation stress genes were among the bloom enriched genes, suggesting that N consumption in this field was quite large and that extraneous N input and biological N fixation did not meet demand. N limitation could be one reason for the subsequent bloom termination. In addition, the bloom may have increased the transfer potential of nitrate to N2 by enhancing the intensity of denitrification related genes (nosZ, nirS, nirK narG). The total nitrogen dynamic in mixed cultures of algae and other microorganisms hasn’t been reported. One report indicated that denitrification increased and total nitrogen concentration decreased 25.97–28.16% in Microcystis aeruginosa cultures48. Additional research into the influence of blooms on N cycling is needed.
Little is known about nutrient utilization by A. sanguinea. Raphael et al.49 reported that the uptake rates for ammonium are much higher than for NO3− in single substrate experiments. Similar results were obtained for N-specific uptake rates of natural assemblages dominated by A. sanguinea in California49, suggesting ammonium is a preferred N source of A. sanguinea. The production potential of ammonium from N2 and nitrate might be enhanced in the free-living microbial community by enhancing the intensity of nifH, nasA, nir, nirA, napA and nrfA genes, which might have contributed to the duration of this bloom. This potential beneficial effect for the algae from the microbial community is in contrast to other studies that have observed an inhibition or lethality of bacteria and phage towards algae during blooms42,50,51,52,53,54.
The control site contained more P limitation stress genes, suggesting P limitation may have prevented bloom formation in the control site. In addition, there was more DIP in the bloom40. P is required for the fast growth and bloom formation of A. sanguinea25. Polyphosphate kinase (ppk) catalyzes the formation of polyphosphate (polyP), a process by which microbes could remove P from water55. The decrease in ppk as A. sanguinea increased may be conducive to the bloom duration.
Novel impacts associated with the bloom
Changes in some of the functional genes detected in this study suggest that the bloom had surprising influences on the microbial communities.
Bacteria might be protected from phage
There were fewer lysis and replication related genes of bacterial phage in the bloom suggesting that phytoplankton blooms might reduce viral infections in bacteria. Studies on the infection dynamics of bacteriophage influenced by algal blooms are limited, but Maranger et al.56 found a disequilibrium between phage and bacterial growth and abundance during a spring ice algal bloom. During the bloom, the virus-to-bacteria ratios decreased although virus abundance increased. The authors speculated that proliferation of phage-resistant bacteria was one reason. Another study of a mesotrophic lake in Germany57 found that phage-induced mortality was lower during phytoplankton blooms (<11%) than after a bloom (18–21%). These results suggest that the bloom environment protects bacteria from bacterial phage. Maintaining a high abundance of bacteria may help to avoid accumulation of the massive amounts of organic matter produced by blooms, and may be an ecological strategy to maintain balance.
Bacteria activated glyoxylate cycle for isocitrate metabolism.
The AceA gene which encodes isocitrate lyase showed a significant positive correlation with A. sanguinea, indicating the glyoxylate cycle was more active in bloom-bacteria than in non-bloom bacteria. Rinta-Kanto et al. found bloom bacterioplankton transcribed more copies of genes for organic acid utilization, but transcripts for isocitrate dehydrogenase were less abundant in the bloom transcriptome19. It’s possible that the energy storage for bacteria within the bloom was enough, so more-than-usual isocitric acid entered into glyoxylate cycle by isocitrate lyase catalyzing, instead of entering tricarboxylic acid cycle by isocitrate dehydrogenase catalyzing (the main energy production process). And this resulted in higher net carbon assimilation.
Conclusion
We applied the GeoChip to the study of a phytoplankton bloom and found that the A. sanguinea bloom did not change the free-living microbial function potential as greatly as expected. Community differences were more pronounced between bloom and control areas than over time. Most significantly changed genes showed an increase in intensity within the bloom, especially for the genes related to carbon degradation. N or P availability appeared to be important for both start and termination of the bloom. The microbes in the bloom appeared to contribute to bloom duration by promoting ammonium accumulation and decreasing the competitive adsorption of biological Pi removal from water. In addition, the bloom may reduce infection activity of bacteriophage by decreasing the intensity of replication and lysis related genes.
Although this study was based on DNA, which provides indirect ecological evidence of functional potential, results were consistent with transcription studies in this field. Changes in gene abundance suggest that the bloom enhanced microbial ammonia accumulation and the transfer of nitrate to N2, reduced infection activity of bacteriophage, and that isocitrate was actively shuttled through the glyoxylate cycle in bacteria. These findings provide new research directions for the study of bloom dynamics and provides a deeper insight into the relationship between bloom algae and other microbes in marine systems. Continuing research is focused on RNA studies.
Additional Information
How to cite this article: Yang, C. et al. A comprehensive insights into functional profiles of free-living microbial community responses to a toxic Akashiwo sanguinea bloom. Sci. Rep. 6, 34645; doi: 10.1038/srep34645 (2016).
References
Mague, T. H. Extracellular release of carbon by marine phytoplankton; a physiological approach. Limnol Oceanogr 25, 262–279 (1980).
Tada, Y., Taniguchi, A., Sato-Takabe, Y. & Hamasaki, K. Growth and succession patterns of major phylogenetic groups of marine bacteria during a mesocosm diatom bloom. J. Oceanogr. 68, 509–519 (2012).
Zubkov, M. V. et al. Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ Microbiol. 3, 304–311(308) (2001).
Yang, C. et al. Bacterial community dynamics during a bloom caused by Akashiwo sanguinea in the Xiamen sea area, China. Harmful Algae 20, 132–141 (2012).
Sheik, A. R. et al. Responses of the coastal bacterial community to viral infection of the algae Phaeocystis globosa. ISME Journal 8, 212–225 (2014).
Oren, A. & Gurevich, P. Characterization of the dominant halophilic archaea in a bacterial bloom in the dead sea. FEMS Microbiol. Ecol. 12, 249–256 (1993).
Higgins, J. L., Kudo, I., Nishioka, J., Tsuda, A. & Wilhelm, S. W. The response of the virus community to the SEEDS II mesoscale iron fertilization. Deep-Sea Research Part II 56, 2788–2795 (2009).
Yager, P. L. et al. Dynamic bacterial and viral response to an algal bloom at subzero temperatures. Limnol Oceanogr. 46, 790–801 (2001).
Hyun, J. H. & Kim, K. H. Bacterial abundance and production during the unique spring phytoplankton bloom in the central Yellow Sea. Mar Ecol Prog. 252, 77–88 (2003).
Mioni, C. E., Poorvin, L. & Wilhelm, S. W. Virus and siderophore-mediated transfer of available Fe between heterotrophic bacteria: characterization using an Fe-specific bioreporter. Aquat. Microb. Ecol. 233–245 (2005).
Gobler, C. J. et al. Release and bioavailability of C, N, e Se, and Fe following viral lysis of a marine chrysophyte. Limnol Oceanogr 42, 1492–1504 (1997).
Wilhelm, S. W. & Suttle, C. A. Viruses and nutrient cycles in the sea - Viruses play critical roles in the structure and function of aquatic food webs. Bioscience 49, 781–788, doi: 10.2307/1313569 (1999).
Gonzalez, J. M. et al. Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl Environ Microbiol 66, 4237–4246, doi: 10.1128/Aem.66.10.4237-4246.2000 (2000).
Riemann, L. Dynamics of bacterial community composition and activity during a mesocosm diatom bloom. Appl Environ Microbiol. 66, 578–587 (2000).
Rooney-Varga, J. N. Links between phytoplankton and bacterial community dynamics in a coastal marine environment. Microbial Ecol. 49, 163–175 (2005).
Larsen, A. et al. Spring phytoplankton bloom dynamics in Norwegian coastal waters: Microbial community succession and diversity. Limnol Oceanogr. 49, 180–190 (2004).
Morris, R. M., Longnecker, K. & Giovannoni, S. J. Pirellula and OM43 are among the dominant lineages identified in an Oregon coast diatom bloom. Environ Microbiol. 8, 1361–1370(1310) (2006).
Teeling, H. et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608- (2012).
Rinta‐Kanto, J. M., Sun, S., Sharma, S., Kiene, R. P. & Moran, M. A. Bacterial community transcription patterns during a marine phytoplankton bloom. Environ Microbiol. 14, 228–239(212) (2012).
Howard, E. C. et al. Changes in dimethylsulfoniopropionate demethylase gene assemblages in response to an induced phytoplankton bloom. Appl Environ Microbiol. 77, 524–531 (2011).
Li, N. et al. Metagenome of microorganisms associated with the toxic Cyanobacteria Microcystis aeruginosa analyzed using the 454 sequencing platform. Chin. J. Oceanol. Limnol. 29, 505–513, doi: 10.1007/s00343-011-0056-0 (2011).
Cooper, E. D., Bentlage, B., Gibbons, T. R., Bachvaroff, T. R. & Delwiche, C. F. Metatranscriptome profiling of a harmful algal bloom. Harmful Algae 37, 75–83, doi: 10.1016/j.hal.2014.04.016 (2014).
Yoch, D. C. Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide. Appl Environ Microbiol. 68, 5804–5815 (2002).
Zubkov, M. V. et al. Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ Microbiol. 3, 304–311, doi: 10.1046/j.1462-2920.2001.00196.x (2001).
Yang, C. et al. Illumina sequencing-based analysis of free-living bacterial community dynamics during an Akashiwo sanguine bloom in Xiamen sea, China. Scientific reports 5, 8476, doi: 10.1038/srep08476 (2015).
He, Z. Development of functional gene microarrays for microbial community analysis. Curr. Microbiol. 23, 49–55 (2012).
Wu, L. Microarray-based characterization of microbial community functional structure and heterogeneity in marine sediments from the Gulf of Mexico. Appl Environ Microbiol. 74, 4516–4529 (2008).
Mason, O. U. et al. Prokaryotic diversity, distribution, and insights into their role in biogeochemical cycling in marine basalts. ISME Journal 3, 231–242, doi: 10.1038/ismej.2008.92 (2009).
Wang, F. P. et al. GeoChip-based analysis of metabolic diversity of microbial communities at the Juan de Fuca Ridge hydrothermal vent. P Natl Acad Sci USA 106, 4840–4845, doi: 10.1073/pnas.0810418106 (2009).
Lu, Z. M. et al. Microbial gene functions enriched in the Deepwater Horizon deep-sea oil plume. ISME Journal 6, 451–460, doi: 10.1038/ismej.2011.91 (2012).
Bai, S. J. et al. GeoChip-based analysis of the functional gene diversity and metabolic potential of soil microbial communities of mangroves. Appl Microbiol Biot. 97, 7035–7048, doi: 10.1007/s00253-012-4496-z (2013).
Kimes, N. E., Van Nostrand, J. D., Weil, E., Zhou, J. Z. & Morris, P. J. Microbial functional structure of Montastraea faveolata, an important Caribbean reef-building coral, differs between healthy and yellow-band diseased colonies. Environ Microbiol. 12, 541–556, doi: 10.1111/j.1462-2920.2009.02113.x (2010).
Gilbert, J. A. et al. Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. Plos One 3, e3042 (2008).
Ahn, S. J., Costa, J. & Emanuel, J. R. PicoGreen quantitation of DNA: effective evaluation of samples pre-or psost-PCR. Nucleic Acids Res. 24, 2623–2625 (1996).
Lu, Z. et al. GeoChip-based analysis of microbial functional gene diversity in a landfill leachate-contaminated aquifer. Environ Sci Technol. 46, 2142–2142 (2012).
Hazen, T. C. et al. Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science 330, 204–208, doi: 10.1126/science.1195979 (2010).
Rodríguez-Martínez, E. M., Pérez, E. X., Schadt, C. W., Zhou, J. & Massol-Deyá, A. A. Microbial diversity and bioremediation of a hydrocarbon-contaminated aquifer (Vega Baja, Puerto Rico). Inter J Env Res Pub Heal 3, 292–300 (2006).
Liang, Y. et al. Microbial functional gene diversity with a shift of subsurface redox conditions during in situ uranium reduction. Appl Environ Microbiol. 78, 2966–2972 (2012).
Tu, Q. et al. Fungal communities respond to long-term elevated CO2 by community reassembly. Appl Environ Microbiol. 81 (2015).
R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria URLhttp://www.R-project.org (2013).
Dixon, P. VEGAN, a package of R functions for community ecology. J Veg Sci. 14, 927–930 (2003).
Buchan, A., LeCleir, G. R., Gulvik, C. A. & Gonzalez, J. M. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nature Rev Microbiol. 12, 686–698, doi: 10.1038/Nrmicro3326 (2014).
Wong, K. T. M., Lee, J. H. W. & Hodgkiss, I. J. A simple model for forecast of coastal algal blooms. Estuar Coast Shelf 74, 175–196 (2007).
Cullen, J. J. & Macintyre, J. G. Behavior, physiology and the niche of depth-regulating phytoplankton. Physiological Ecology of Harmful Algal Blooms Springer-Verlag, Berlin, Heidelberg, Anderson, D. M., Cembella, A. D., Hallegraeff, G. M. (1998).
Watanabe, M. et al. Generation of a Chattonella antiqua bloom by imposing a shallow nutricline in a mesocosm. Limnol Oceanogr. 40, 1447–1460 (1995).
Paerl, H. Nutrient and other environmental controls of harmful cyanobacterial blooms along the freshwater-marine continuum. Adv. Exp. Med. Biol. 619, 217–237 (2008).
Gobler, C. J. & Sanudo-Wilhelmy, S. A. Cycling of colloidal organic carbon and nitrogen during an estuarine phytoplankton bloom. Limnol Oceanogr. 48, 2314–2320 (2003).
Jian, W. U. Effect of the growth of Microcystis aeruginosa on the pH value and the nitrogen transformation in the medium. J. Lake Sci. 21, 123–127 (2009).
Kudela, R. M., Lane, J. Q. & Cochlan, W. P. The potential role of anthropogenically derived nitrogen in the growth of harmful algae in California, USA. Harmful Algae 8, 103–110 (2008).
Li, Y. et al. Toxicity of algicidal extracts from Mangrovimonas yunxiaonensis strain LY01 on a HAB causing Alexandrium tamarense. J. Hazard. Mater. 278, 372–381, doi: 10.1016/j.jhazmat.2014.06.032 (2014).
Zhang, H. J. et al. Cell death in a harmful algal bloom causing species Alexandrium tamarense upon an algicidal bacterium induction. Appl Microbiol Biot. 98, 7949–7958, doi: 10.1007/s00253-014-5886-1 (2014).
Vardi, A. et al. Viral glycosphingolipids induce lytic infection and cell death in marine phytoplankton. Science 326, 861–865, doi: 10.1126/science.1177322 (2009).
Rosenwasser, S. et al. Rewiring host lipid metabolism by large viruses determines the fate of Emiliania huxleyi, a Bloom-Forming Alga in the Ocean. Plant Cell 26, 2689–2707, doi: 10.1105/tpc.114.125641 (2014).
Vardi, A. et al. Host-virus dynamics and subcellular controls of cell fate in a natural coccolithophore population. Proceedings of the National Academy of Sciences of the United States of America 109, 19327–19332, doi: 10.1073/pnas.1208895109 (2012).
Docampo, R. Acidocalcisomes and polyphosphate granules. Microbiology Monographs 53–70 (2006).
Maranger, R., Bird, D. F., Juniper, S. K. & Maranger, R. Viral and bacterial dynamics in Arctic sea ice during the spring algal bloom near Resolute, NWT, Canada. Mar Ecol Prog 111, 121–127 (1994).
Hennes, K. P. & Simon, M. Significance of bacteriophages for controlling bacterioplankton growth in a mesotrophic lake. Appl Environ Microbiol. 61, 333–340 (1995).
Acknowledgements
This work was financed by the National Nature Science Foundation of China (Nos: 41576109, 41376119, 31500095, 40930847), are gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Decision to publish: T.Z. and J.Z. Conceived and designed the experiments: C.Y., Y.L., Y.Z., X.L., W.Z., Y.T., J.Z. and T.Z. Performed the experiments: C.Y., Y.L., Y.Z. and X.Q.L. Analyzed the data: C.Y., Y.L., J.D.V.N., Z.H. and L.W. (C.Y. and Y.L. prepared Figures 1–4. J.D.V.N., Z.H. and L.W. prepared Figures 5 and 6). Contributed reagents/materials/analysis tools: X.L., W.Z., Y.T., J.Z. and T.Z. Wrote the paper: C.Y. and Y.L. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yang, C., Li, Y., Zhou, Y. et al. A comprehensive insight into functional profiles of free-living microbial community responses to a toxic Akashiwo sanguinea bloom. Sci Rep 6, 34645 (2016). https://doi.org/10.1038/srep34645
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34645
- Springer Nature Limited
This article is cited by
-
Temporal variability of free-living microbial culturability and community composition after an Akashiwo sanguinea bloom in Shenzhen, China
Ecotoxicology (2021)
-
New insights into the impacts of suspended particulate matter on phytoplankton density in a tributary of the Three Gorges Reservoir, China
Scientific Reports (2017)
-
Planktonic algal bloom significantly alters sediment bacterial community structure
Journal of Soils and Sediments (2017)