
Abstract
Here we report an ABA triblock copolymer that can express microscopic autonomous formation and break-up of aggregates under constant condition to generate macroscopic viscoelastic self-oscillation of the solution. The ABA triblock copolymer is designed to have hydrophilic B segment and self-oscillating A segment at the both sides by RAFT copolymerization. In the A segment, a metal catalyst of chemical oscillatory reaction, i.e., the Belousov-Zhabotinsky (BZ) reaction, is introduced as a chemomechanical transducer to change the aggregation state of the polymer depending on the redox states. Time-resolved DLS measurements of the ABA triblock copolymer confirm the presence of a transitional network structure of micelle aggregations in the reduced state and a unimer structure in the oxidized state. This autonomous oscillation of a well-designed triblock copolymer enables dynamic biomimetic softmaterials with spatio-temporal structure.
Similar content being viewed by others
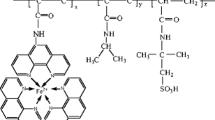

Introduction
Nanometer~sub-micron ordered spatial structures play a crucial role in living organisms; for example, the substrate-binding sites located inside enzyme proteins, selective ion transport channels within lipid bilayer membranes, compartmentalization regions formed by bilayer vesicles, etc. To mimic this scale of spatial structure, spontaneous self-assembly of amphiphilic block copolymers is frequently used1,2,3. A number of concepts to achieve valuable biomimetic materials from block copolymer, such as functional membrane4, vesicles5,6, nanoparticles7 and catalyst8, have been proposed.
Not only limited to the static spatial structure, microscopic temporal structures such as rhythm and oscillation are also known to influence macro dynamic processes in living organisms. These include energy transport and/or signal transduction via periodic formation of synaptic vesicles at nerve terminals9, periodic structural oscillation of fibroblast cells10, pulsation of cardiac muscle cells, amoeboid locomotion11,12,13,14,15, etc. In the straightforward movement of an amoeba, rhythmic sol-gel conversion occurs based on the hierarchical actin polymerization/depolymerization beneath the plasma membrane13,14,15. Protoplasma in the depolymerized sol state flow forward inside the cell and then reverse direction to fuse to gelled pseudopodium at the front of the cell. Concurrently, at the back of the cell, protoplasma in the form of polymerized gel immediately melts to a sol state and moves forward inside the cell again. Thus, the sol-gel transition is based on microscopic structure formation and break-up of the building blocks.
Block copolymers are often used as building blocks for forming self-assembled structures, as mentioned above. However, it is difficult to generate such an autonomous cyclic sol-gel transition from block copolymers because self-assembly forms under thermodynamically stable equilibrium state where the Gibbs free energy of the system is minimized. In spite to this difficulty, we recently succeeded in creating a unique spatio-temporal structure in non-equilibrium state of AB diblock copolymers under constant conditions by introducing a catalyst site of the Belousov-Zhabotinsky (BZ) reaction16, which is a well-known chemical oscillation reaction, into block copolymer architecture. The BZ reaction is often compared with the tricarboxylic acid (TCA) cycle, which is a key metabolic process that occurs in the living body and is recognized as a chemical model for understanding several non-equilibrium phenomena in nature. The overall process consists of oxidation of an organic substrate, such as malonic acid (MA), by an oxidizing agent in the presence of a strong acid with the aid of a metal catalyst. During the reaction, the redox state of a metal catalyst, such as ruthenium bipyridine (Ru(bpy)3), undergoes a spontaneous rhythmic change.
With the redox change, the block copolymers show autonomous disintegration and reconstruction under constant conditions, although the random copolymers undergo expansion-contraction and swelling-deswelling in the case of linear polymer and gel, respectively17,18,19,20. By using the AB diblock copolmyer, we realized artificial oscillation phenomena, such as self-oscillating formation and break-up of micelles21, vesicles24 and the unique periodic structural transition of chemically cross-linked bilayer vesicles25, at the nanometer~sub-micron scale. Further, by designing architecture of block copolymer, it is expected that the oscillations are converted to higher-ordered structural changes leading to changes in macroscopic solution property.
In this paper, we demonstrate autonomous viscosity oscillation coupled with periodic aggregation and dissociation of an ABA triblock copolymer under constant conditions. The ABA triblock copolymer comprises poly(ethylene oxide) (PEO) as a hydrophilic central segment with a random copolymer of N-isopropylacrylamide (NIPAAm), methacrylamide with a Ru(bpy)3 side-chain as the metal catalyst for the BZ reaction and N-(3-aminopropyl)methacrylamide (NAPMAm) as a precursor residue of the binding site of Ru(bpy)3 (Fig. 1(b)). The resultant triblock copolymers show low temperature unimolecular dissolution and high temperature micellization owing to the lower critical solution temperature (LCST)-type thermosensitivity of NIPAAm. The aggregation temperature strongly depends on the redox state of the Ru(bpy)3 incorporated into the self-oscillating segment because of hydrophilicity changes of the metal center (Fig. 1(a)). Therefore, the dilute triblock copolymer solution autonomously oscillates between unimolecular dissolution and aggregation with an appropriate amount of the BZ substrate under the bistable temperature at which the reduced block copolymer aggregates to form micelles while the oxidized polymer dissolves as individual polymer chains. By employing ABA triblock copolymer, micelle clusters are formed as transient precursors to the network structure. Then, during the oscillation, the microscopic structural changes are amplified to macroscopic viscosity oscillation. In this study, to analyze the spatio-temporal structure of the self-oscillating triblock copolymer, time-resolved dynamic light scattering (DLS) was used. From the comprehensive DLS analysis under various concentration conditions, the presence of micelle clusters was revealed as transient precursors to the network structure, which results in macroscopic viscosity oscillation with a large amplitude.

(a) Conceptual illustration of the periodic sol-gel transition based on autonomous oscillation of the ABA triblock copolymer. (b) Chemical structure of the self-oscillating AB diblock, ABA triblock and ABCBA pentablock copolymers.
Results and Discussion
Self-assembly of the block copolymers in equilibrium
A representative chemical structure of the self-oscillating ABA triblock copolymer is shown in Fig. 1(b). We first prepared a random copolymer of NIPAAm and NAPMAm and then attached the Ru(bpy)3 metal catalyst with an activated ester (i.e., Ru(bpy)3-NHS) to the amino side-chain of NAPMAm26,27. This strategy gives a significantly different chemical structure than that of the self-oscillating block copolymer prepared by one-step method using Ru(bpy)3 vinyl monomer in the earlier study21,22,23,24. The arrangement of NIPAAm and Ru(bpy)3 groups along the polymer chain is highly random because the electron densities of the propagation terminals of the polymers with NIPAAm and NAPMAm during polymerization process are comparable due to the similarity of the main-chain chemical structure; Ru(bpy)3-NHS is attached to the amino side-chain after random copolymerization.
Figure 2 compares the mean Rh of the ABA triblock copolymer (0.1 wt%) in reduced (Ru(bpy)32+) and oxidized (Ru(bpy)33+) states. During the heating process, the Rh significantly increases with increasing temperature because of the LCST nature of NIPAAm. This suggests that the triblock copolymer self-assembles into flower-like micelles with self-oscillating segment cores surrounded by well-solvated hydrophilic PEO shells looping-back to core at higher temperatures, being consistent with expectation28. The micellization temperature of the triblock copolymer is consistent with the previously reported phase transition temperature of PNIPAAm in block copolymers26,27,28,29,30,31,32,33,34,35,36,37,38. In oxidized state, the Rh at 20.3 °C below the lower critical micellization-type aggregation temperature (LCMT) is 10.4 nm, which is comparable with the sizes of the ABA single polymer chains with molecular weights of 225 kDa.

Relationship between hydrodynamic radius (Rh) and temperature of reduced and oxidized ABA triblock copolymers (0.1 wt%). The redox states were achieved by adding 3 mM Ce2(SO4)3 and 1.0 M HNO3 (reduced state, Ru(bpy)32+) and 3 mM Ce(SO4)2 and 1.0 M HNO3 (oxidized state, Ru(bpy)33+), respectively.
In both the reduced and oxidized states, increasing the temperature above LCMT results in an estimated Rh of >100 nm, which suggests network structures. In an amphiphilic diblock copolymer consisting of polybutadiene and PEO, vesicular morphology can occur when the weight fraction of the hydrophilic PEO is less than ~0.45. Similar effects on the self-assembled structure with changes in the weight fraction of the block copolymer were also reported for a block copolymer with NIPAAm segments at higher temperature than LCMT37,38. Another possible explanation for the large aggregate is micellar aggregation of the ABA triblock copolymer. Zhou and co-workers recently reported the thermo-reversible aggregation behavior of a well-defined ABC terpolymer consisting of poly(ethylene-alt-propylene), PEO and PNIPAAm as the hydrophobic, hydrophilic and thermo-sensitive segments, respectively39. They observed a significant increase in Rh to >100 nm as the Mn of the PNIPAAm chain increased with constant molecular weights of the other two blocks; the large Rh was attributed to micellar aggregation around the PEO shell. Increasing the temperature above the LCST minimizes the contact of PNIPAAm chains with water through the formation of larger multi-micellar aggregates. We also confirmed the reversibility of the size changes of the scatters indicating unimer-micelle transitions in both the reduced and oxidized states.
Autonomous oscillation of the block copolymers
Figure 3(a) shows photographs of the appearance of the ABA triblock copolymer solution (0.1 wt%) at room temperature with an appropriate amount of the substrate for the BZ reaction over time. The solution was irradiated with Ar laser (488 nm) from the left side of the sample. Periodic color changes between orange and light-green derived from the redox oscillation of Ru(bpy)3 are evident with periodic blinking of the laser light. When the sample is orange (reduced state), the Tyndall effect is clearly seen and indicates block copolymer aggregation, while no light scattering occurs in the light-green Ru(bpy)3 oxidized state. The transmittance of the ABA triblock copolymer solution at 570 nm under BZ reaction conditions also confirms that the turbidity of the solution oscillates autonomously (Fig. 3(b)), which suggests that block copolymer aggregation periodically changes as driven by the BZ reaction.

(a) Time course of the appearance of dilute ABA triblock copolymer (0.1 wt%) solution with an appropriate amount of BZ reaction substrate. The solution was magnetically stirred; the white object at the bottom of the solution is the stir bar. The sample was irradiated with green Ar laser (488 nm) from the left side of the sample to generate the Tyndall effect. (b) Self-oscillation of the transmittance of the dilute ABA triblock copolymer (0.1 wt%) solution at 20 °C. The concentration of the BZ substrates in the feed are as follows: [NaBrO3] = 0.2 M, [HNO3] = 0.3 M and [MA] = 0.025 M.
To quantitatively investigate dynamic aggregation behavior, we monitored the size oscillation of particles in the block copolymer solution under various BZ substrate and polymer concentrations using time-resolved DLS. Figure 4(a–c) show the representative oscillation behaviors of the scattering intensity, Rh and the relaxation time distribution function (G(Γ−1)) indicated by RGB color, respectively, for the ABA triblock copolymer. As a result of measuring the average scattering intensity every 2 s, the periodic scattering intensity oscillation was clearly observed (Fig. 4(a)-1, 2, 3,). From the oscillation waveform, it is clear that the decrease in scattering intensity that accompanies demicellization occurs much faster than the increase in intensity that accompanies micellization. This is explained by the characteristics of the BZ reaction where the process of autocatalytic oxidation or conversion of Ru(bpy)32+ to Ru(bpy)33+ occurs more rapidly than the opposite reduction process. Another possible explanation for the waveform may be a difference in the mechanism between the integration and reconstruction of aggregates, as discussed elsewhere21,30.

Oscillation behaviors of (a) normalized scattering intensity, (b) Rh and (c) the relaxation time distribution function, G(Γ−1), derived from CONTIN analysis during the BZ oscillation reaction for the ABA triblock copolymer.
MA acts as a reductant during the reaction. Increasing the concentration of MA in the feed from 0.025 to 0.1 M while keeping the concentrations of the other BZ substrates and polymer solutions constant increases the length of the reduced period and size of the aggregates (Fig. 4(a)-1, 2, (b)-1, 2, and Table S2). In the reduced period during oscillation, the decreased solubility of the self-oscillating segment is likely to result in aggregates. The higher MA concentration makes the block copolymer solution more reductive leading to the formation of aggregates being favored over unimolecular dissolution. Therefore, the reduction period is prolonged and the size of micelle is larger.
Furthermore, increasing the concentration of block copolymer results in the wave deformation to show a plateau in the reduced state and the aggregates form larger particles through the reduction process (Fig. 4(a)-2, 3, (b)-2, 3, and Table S2). This suggests that higher MA as well as triblock copolymer concentrations can promote the formation of a material-spanning network resulting in gelation or a highly viscous solution based on high molar mass aggregates when the solution is reduced. Incidentally, the waveform of normalized scattering intensity (Fig. 4(a)-1, 2, 3,) and Rh (Fig. 4(b)-1, 2, 3,) do not always correspond together. Although the exact reasons for the difference in waveforms between scattering intensity and Rh is still unclear, one possible explanation is described in terms of the consideration of form factor of aggregation structure in the Supplementary Information.
In an ABA triblock copolymer composed of B and A blocks with and without solvent selectivity, respectively, A chains basically need to overcome an energy penalty to dissociate and diffuse into the solvent matrix. The value of energy barrier depends on the incompatibility χN between A block and solvent from a physically cross-linked transient network theory40, where χ is the Flory-Huggins interaction parameter and N is the degree of polymerization. From this consideration, self-assembled A domains in the solution will be subjected to network dissociation more easily when the molecular weight of the A segment reduces, leading to rapid formation/break-up network structure. On the other hand, when the molecular weight of the A segment increases, robust viscoelastic fluid (high viscosity solution or high storage elastic modulus gel at reduced state, transiently) might be organized, as expected. In these ways, the molecular weight of the blocks will be trade-off and an important factor which will affect oscillation of micellization/demicellization.
Comprehensive observations of the period of the block copolymer oscillation as a function of the concentrations of the BZ substrates confirm that the oscillation period decreases at higher initial concentrations of BZ substrates (NaBrO3, HNO3 and MA) in the feed (Figure S4). The oscillation period for the ABA triblock copolymer can be described using the following relationship:

This tendency consistently agrees with other reports on BZ oscillation behavior, as judged by periodic color changes of the metal catalyst41,42. This result supports that the formation and break-up cycle of the ABA triblock copolymer is induced by the BZ oscillation reaction.
Another important aspect is the size distribution of the block copolymer aggregates during the oscillation. A series of Fig. 4(c) shows the result of time-resolved CONTIN analysis of the autocorrelation function obtained every 2 s. The time course of relaxation time distribution function, G(Γ−1), is indicated by RGB scale for relaxation time (Γ−1). Red indicates intense signals and blue indicates that no signal appears within the region of Γ−1. This analysis provides not only the average Γ−1 of the polymer in solution, but also the change in the distribution of these values over time. From the analysis, it can be seen that the distribution of Γ−1 is relatively broad and the signal intensity autonomously oscillates in all cases. The relaxation time distribution of the ABA triblock copolymer periodically changes more than one order of magnitude between the oxidized and reduced states as the BZ reaction occurs. In Fig. 4(c)-1, 2, 3, intense signals observed at around Γ−1 = 0.1 and 1 ms correspond to the unimer and micelle, respectively. More interestingly, in addition to the oscillation of the faster relaxation mode derived from unimers and micelles, slower relaxation is evident around 10 ms when the concentrations of both the polymer and MA in the feed are the highest (Fig. 4(c)-3). This suggests that the formation of large aggregates of micelle clusters during oscillation as a possible precursor to the network structure, which has not been recognized from the discussion on the average Γ−1, could be detected. This one order slower Γ−1 is not observed in self-oscillating AB diblock copolymer with corresponding design (Figure S5).
This result encouraged us to investigate viscosity oscillation driven by the BZ reaction under high concentrations of ABA triblock copolymer. We further studied the development of the microscopic oscillation mode in the aggregation state of the triblock copolymer to macroscopic viscoelastic oscillation. Viscosity measurements on the ABA triblock copolymer (polymer concentration = 0.5, 5 and 15 wt%) under BZ reaction conditions with higher MA concentrations were conducted (Fig. 5). Clearly, autonomous viscosity oscillation of the block copolymer solution is exhibited in all these cases. Increasing the concentration of ABA triblock copolymer results in monotonic increases in the oscillation amplitude as well as viscosity baseline. The oscillation amplitude of the 15 wt% ABA triblock copolymer solution is 1.5 mPa s with a viscosity baseline of 38 mPa s. This clearly indicates that the periodic changes in aggregate structure of the ABA triblock copolymer induce periodic changes of the molar mass of aggregation resulting in the macroscopic properties of the system. Here the amplitude of the viscosity is comparable to previous report43,44. One possible reason is that percolation of network structure was not achieved within 60 s, the period of the BZ reaction. In addition, under the BZ reaction oscillation condition, the redox state of incorporated Ru(bpy)3 into the random copolymer chain does not proceed to the complete oxidation or reduction states41. Aiming to increase the viscosity amplitude, we have synthesized ABCBA pentablock copolymer comprising self-oscillating A, hydrophilic B and hydrophobic (insoluble) poly(propylene oxide) C blocks. The C block is introduced at the middle of the block copolymer architecture as a hydrophobic core to induce micelle formation during the oxidized state. By forming micelle with multi-arms, it is expected that network structure is easily formed at reduced state and hence viscosity amplitude is enhanced. As shown in Fig. 5, the viscosity oscillation of the ABCBA pentablock copolymer exhibits abound 2.0 mPa s amplitude with a baseline of around 5 mPa s. Although the polymer concentration is as low as 1.0 wt%, the largest amplitude was observed. This result supports that more effective network formation and break-up occurs by introducing hydrophobic center into the ABA block copolymer. Due to poor solubility of the ABCBA pentablock copolymer into aqueous solution, the solubility was found to be as low as 1 wt%. Although further optimization for controlling solubility would be necessary, the ABCBA pentablock copolymer designed to insert hydrophobic C block achieves the largest viscosity amplitude and shows the potential to realize sol-gel oscillation with self-oscillating multiblock copolymer system.

Viscosity oscillation driven by the periodic association and dissociation of the ABA triblock copolymers (0.5, 5.0 and 15 wt%) and an ABCBA pentablock copolymer (1.0 wt%) during the BZ reaction.
We previously reported autonomous viscosity oscillation coupled with rhythmic coordination and discoordination cycles of the terminal terpyridine ligand of a branched PEO with a Ru metal center depending on the redox states43. Also, sub-micron ordered well-defined self-oscillating microgel particles have been reported to induce autonomous viscosity oscillation44 with the dispersion and flocculation cycles under BZ reaction conditions. In this study, we displayed another example of induced viscosity oscillation from periodic changes of molecular aggregates. We are currently elucidating the mechanism of viscosity oscillation in detail. Among several strategies we have reported so far, designing self-oscillating block polymers as reported here would be more effective to increase the viscosity amplitude and realize sol-gel transition, according to further studies on multi-blocking design we are progressing now, which will be reported in the future. Such viscoelastic oscillation of the polymer solution can potentially enable its use as a functional fluid. Moreover, oscillating soft materials from straightforward synthetic polymer that express viscoelastic movement similar to amoeba motion can potentially act as model systems for simulation studies to achieve mechanical motion in living soft materials45.
In conclusion, we demonstrated autonomous viscosity oscillation induced by aggregation of ABA triblock copolymers without on–off switching. The target self-oscillating ABA triblock copolymer was successfully prepared by RAFT random copolymerization of NIPAAm and NAPMAm from PEO-base macro CTA followed by post-modification of Ru(bpy)3 activated ester to the amino residue of the NAPMAm side-chain. The ABA triblock copolymer exhibited a much wider range of bistable temperature between the reduced and oxidized states than previously reported self-oscillating AB diblock copolymers. Time-resolved DLS measurements for the oscillation at dilute block copolymer concentrations were carried out under various BZ substrate conditions. The comprehensive investigations confirmed the presence of the slower relaxation mode caused by micelle aggregation, which is a possible precursor of the polymer network, in addition to the faster relaxation modes of the unimer and micelle. The slower component appeared only under higher concentrations of MA (i.e., the reductant of the BZ reaction) and with higher concentrations of ABA triblock copolymer. In these conditions, remarkable viscosity oscillations were achieved.
Methods
Materials
S-1-Dodecyl-S’-(α,α’-dimethyl-α”-acetic acid)trithiocarbonate (CTA, purity >99%) was purchased from Trylead Corporation. Telechelic (–OH terminated) poly(ethylene oxide) (PEO), semi-telechelic PEO monomethyl ether, poly(ethylene oxide)99-b-poly(propylene oxide)69-b-poly(ethylene oxide)99 (F127) and 2,2′-azobisisobutyronitrile (AIBN) were purchased from Sigma-Aldrich. Anhydrous dichloromethane, 1,4-dioxane and oxalyl chloride were purchased from Wako Chemicals. N-(3-Aminopropyl)methacrylamide (NAPMAm) was purchased from Polyscience. N-isopropylacrylamide (NIPAAm) was generously donated by the Kojin Corporation and purified via two times recrystallizations from a toluene/hexane (1:10 by weight) mixed solvent. AIBN was recrystallized from methanol prior to use. PEO and F127 was dried by azeotropic distillation using toluene prior to use. Bis(2,2′-bipyridine)(1-(4′-methyl-2,2′-bipyridine-4-carbonyloxy)-2,5-pyrrolidinedione)ruthenium(II) bis(hexafluorophosphate) (Ru(bpy)3-NHS) was synthesized according to the previously reported procedure29. All other chemical reagents were from Wako Chemicals and used as received unless otherwise noted.
Synthesis of the Multiblock copolymer
Here, we describe a representative procedure for synthesis of the ABA triblock copolymer23,30. The triblock copolymer was synthesized following the procedure shown in Figure S2 in Supporting Information. First, the PEO-CTA macro-chain transfer agent was prepared as follows: Oxalyl chloride (0.74 mL, 8.6 × 10−3 mol) and CTA (0.63 g, 1.7 × 10−3 mol) were mixed in anhydrous dichloromethane (30 mL) under an Ar atmosphere and stirred at room temperature until gas evolution stopped (~2 h). Excess reagents were then removed under vacuum and the residue was redissolved in anhydrous dichloromethane (100 mL) followed by the addition of telechelic PEO (6.0 g, 1.7 × 10−4 mol). The reaction was allowed to proceed for 24 h at room temperature after which the contents were reprecipitated two times using toluene as a good solvent and hexane as a poor solvent, respectively and dried under vacuum at room temperature. The conversion for esterification of both end-groups was monitored by 1H-NMR spectroscopy. The reaction was carried out quantitatively. Yield: 5.5 g, 91%.
Next, a round-bottom flask was charged with PEO-CTA (4.67 g, 1.34 × 10−4 mol), NIPAAm (40.6 g, 3.6 × 10−1 mol), NAPMAm (2.0 g, 1.1 × 10−2 mmol) ([NIPAAm]/[NAPMAm] = 97/3 mol%) and methanol (solvent, 280 mL) and purged with Ar for 30 min. AIBN (25 mg, 1.5 × 10−4 mol) was separately dissolved in 10 mL of methanol (solvent) in another round-bottom flask and purged with Ar for 30 min at room temperature. All procedure was conducted under Ar atmosphere. An aliquot of the AIBN solution (3.5 mL, 5.3 × 10−5 mmol) was added to the monomer solution. RAFT random copolymerization was carried out at 60 °C for 48 h. The products were concentrated under reduced pressure and purified by three reprecipitations from toluene as a good solvent and hexane as a poor solvent.
Following the RAFT random copolymerization, the dodecyl trithiocarbonate residues derived from the CTA attached to the ABA polymer terminus were removed according to the following procedure: All obtained ABA triblock copolymer and appropriate amount of AIBN were dissolved in 290 mL of ethanol and the solution was purged with Ar. The cleavage reaction was performed at 80 °C for 10 h under an Ar atmosphere. The product was concentrated under reduced pressure and the resulting residue was purified by reprecipitation three times from toluene as a good solvent and hexane as a poor solvent and drying under vacuum at room temperature overnight. Subsequently, The ABA triblock copolymer (2.00 g, 1.7 × 10−5 mol) was dissolved in DMSO (30 mL) and added to a DMSO solution containing Ru(bpy)3-NHS (1.70 g, 1.7 × 10−3 mol). Triethylamine (7.8 × 10−5 mL, 5.6 × 10−4 mol) was added to the solution and the solution was stirred for 48 h at room temperature. The obtained polymer was dialyzed against water and finally lyophilized for recovery. The structure and average molecular weight of the polymer were determined by a combination of 1H NMR spectroscopy and gel permeation chromatography (GPC) using dimethyl formamide (DMF) containing 1 mol L−1 LiBr as the carrier solvent (Figure S2). The total number average molecular weight (Mn) of the triblock copolymer was calculated from the ratio of the integrated signal intensities of the PEO main chain and second block (δ = 3.7 ppm from NIPAAm, δ = 2.9–3.4 ppm from NAPMAm and δ = 3.6 ppm from PEO). The NAPMAm composition was calculated using the ratio of the integrated signal intensities of NIPAAm and NAPMAm (Figure S3). The columns (Tosoh) for GPC analyses were calibrated using PEO standards. The characterization results for the polymers are summarized in Table S1. The Mn of the ABA polymer was 225 kDa. The AB diblock copolymer and the ABCBA pentablock copolymer were prepared similarly to the procedure mentioned above except that semi-telechelic PEO or F127 was used as a precursor instead of telechelic PEO.
Measurements
All methods for the measurements of optical transmittance oscillation, dynamic light scattering measurements and rheological measurements are noted in supplementary information.
Additional Information
How to cite this article: Onoda, M. et al. Multiblock copolymers exhibiting spatio-temporal structure with autonomous viscosity oscillation. Sci. Rep. 5, 15792; doi: 10.1038/srep15792 (2015).
References
Riess, G. Micellization of block copolymers. Prog. Polym. Sci. 28, 1107 (2003).
Hamley, I. W. Block copolymer in solution: Fundamentals and applications. John Wiley & Sons, Ltd., Chichester (2005).
Alexandridis, P. & Lindman, B. Amphiphilic block copolymers: Self-assembly and applications. Elsevier, Amsterdam (2000).
Kowal, J., Zhang, X., Dinu, I. A., Palivan, C. G. & Meier, W. Planar Biomimetic Membranes Based on Amphiphilic Block Copolymers. ACS Macro Lett. 3, 59–63 (2014).
Jain, S. & Bate, F. S. On the Origins of Morphological Complexity in Block Copolymer Surfactants. Science 300, 460–464 (2003).
Kim, Y. R. et al. Synthetic Biomimetic Membranes and Their Sensor Applications. Sensor 12, 9530–9550 (2012).
Yu. H., Qin, X., Nunes, S. P. & Peinemann, K. V. Biomimetic block copolymer particles with gated nanopores and ultrahigh protein sorption capacity. Nat. Commun. 5, 4110–4119 (2014).
Cha, J. N., Stucky, G. D., Morse, D. E. & Deming, T. J. Biomimetic synthesis of ordered silica structures mediated by block copolypeptides. Nature 403, 289–292 (2000).
Südof, T. C. The synaptic vesicle cycle: a cascade of protein–protein interactions. Nature 375, 645–653 (1995).
Shalbreux, G., Joanny, J. F., Prost, J. & Pullarkat, P. Shape oscillations of non-adhering fibroblast cells. Phys. Biol. 4, 268–284 (2007).
Mast, S. O. Mechanics of Locomotion in Amoeba. Proc. Natl. Acad. Sci. 9, 258–261 (1923).
Mast, S. O. Structure, movement, locomotion and stimulation in amoeba. J. Morphol. Physiol. 41, 347–425 (1926).
Charras, G. T. & Paluch, E. Blebs lead the way: how to migrate without lamellipodia. Nat. Rev. Mol. Cell Biol. 9, 730–736 (2008).
Pollard, T. D. & Borisy, G. G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 (2003).
Nishigami, Y. et al. Reconstruction of Active Regular Motion in Amoeba Extract: Dynamic Cooperation between Sol and Gel States. PLoS One 8, e70317 (2013).
Epstein, I. R. & Pojman, J. A. An introduction to nonlinear chemical dynamics. Oxford University express, New York (1998).
Yoshida, R., Sakai, T., Ito, S. & Yamaguchi, T. Self-oscillation of polymer chains with rhythmical soluble-insoluble changes. J. Am. Chem. Soc. 124, 8095–8098 (2002).
Yoshida, R., Takahashi, T., Yamaguchi, T. & Ichijo, H. Self-oscillating gel. J. Am. Chem. Soc. 118, 5134–5135 (1996).
Yoshida, R., Self-Oscillating Gels Driven by the Belousov–Zhabotinsky Reaction as Novel Smart Materials. Adv. Mater. 22, 3463–3483 (2010).
Yoshida, R. & Ueki, T. Evolution of self-oscillating polymer gels as autonomous polymer systems. NPG Asia Mater. 6, e107 (2014).
Ueki, T., Shibayama, M. & Yoshida, R. Self-oscillating micelles. Chem. Commun. 49, 6947–6949 (2013).
Ueki, T. & Yoshida, R. Recent aspects of self-oscillating polymeric materials: designing self-oscillating polymers coupled with supramolecular chemistry and ionic liquid science. Phys. Chem. Chem. Phys. 16, 10388–10397 (2014).
Ueki, T., Onoda, M., Tamate, R., Shibayama, M. & Yoshida, R. Self-oscillating AB diblock copolymer developed by post modification strategy. Chaos 25, 064605 (2015).
Tamate, R., Ueki, T., Shibayama, M. & Yoshida, R. Self-Oscillating Vesicles: Spontaneous Cyclic Structural Changes of Synthetic Diblock Copolymers. Angew. Chem. Int. Ed. 53, 11248–11252 (2014).
Tamate, R., Ueki, T. & Yoshida, R. Self-Beating Artificial Cells: Design of Cross-Linked Polymersomes Showing Self-Oscillating Motion. Adv. Mater. 27, 837–842 (2015).
Masuda, T. et al. Self-Oscillating Polymer Brushes. Angew. Chem. Int. Ed. 52, 7468–7471 (2013).
Masuda, T. et al. Control of swelling–deswelling behavior of a self-oscillating gel by designing the chemical structure. RSC Adv. 5, 5781–5787 (2015).
Balsara, M. P., Tirrell, M. & Lodge, T. P. Micelle formation of BAB triblock copolymers in solvents that preferentially dissolve the A block. Macromolecules 24, 1975–1986 (1991).
Peek, B. M. et al. Synthesis of redox derivatives of lysine and related peptides containing phenothiazine or tris(2,2′-bipyridine)ruthenium(II). Int. J. Protein Res. 38, 114–123 (1991).
Ueki, T., Nakamura, Y., Lodge, T. P. & Watanabe, M. Doubly Thermosensitive Self-Assembly of Diblock Copolymers in Ionic Liquids. Macromolecules 45, 7566–7573 (2012).
Brown, W. Dynamic Light Scattering: The Method and Some Applications. Clarendon Press, Oxford, England (1993).
Koppel, D. E. Analysis of Macromolecular Polydispersity in Intensity Correlation Spectroscopy: The Method of Cumulants. J. Chem. Phys. 57, 4814–4820 (1972).
Provencher, S. W. A Constrained Regularization Method for Inverting Data Represented by Linear Algebraic or Integral Equations. Comput. Phys. Commun. 27, 213–227 (1982).
Provencher, S. W. CONTIN: A General Purpose Constrained Regularization Program for Inverting Noisy Linear Algebraic and Ingegral Equations. Comput. Phys. Commun. 27, 229–242 (1982).
Schild, H. G. Poly(N-isopropylacrylamide): experiment, theory and application. Prog. Polym. Sci. 17, 163–249 (1992).
Sundararaman, A., Stephan, T. & Grubbs, R. B. Reversible Restructuring of Aqueous Block Copolymer Assemblies through Stimulus-Induced Changes in Amphiphilicity. J. Am. Chem. Soc. 130, 12264–12265 (2008).
Zhao, J., Zhang, G. & Pispas, S. Morphological transitions in aggregates of thermosensitive poly(ethylene oxide)-b-poly(N-isopropylacrylamide) block copolymers prepared via RAFT polymerization. J. Polym. Sci. Part A. 47, 4099–4110 (2009).
Qin, S., Geng, Y., Discher, D. E. & Yang S. Reversible Restructuring of Aqueous Block Copolymer Assemblies through Stimulus-Induced Changes in Amphiphilicity. Adv. Mater. 18, 2905–2909 (2006).
Zhou, C., Hillmyer, M. A. & Lodge, T. P. Micellization and Micellar Aggregation of Poly(ethylene-altpropylene)-b-poly(ethyleneoxide)-b-poly(N-isopropylacrylamide) Triblock Terpolymers in Water. Macromolecules 44, 1635–1641 (2011).
Tanaka, F. & Edwards, S. F., Viscoelastic properties of physically crosslinked networks. 1. Transient Network Theory, Macromolecules 25, 1516 (1992).
Yoshida, R., Onodera, S., Yamaguchi, T. & Kokufuta, E. Aspects of the Belousov−Zhabotinsky Reaction in Polymer Gels. J. Phys. Chem. A 103, 8573–8578 (1999).
Ueki, T., Watanabe, M. & Yoshida, R. Belousov–Zhabotinsky Reaction in Protic Ionic Liquids. Angew. Chem. Int. Ed. 51, 11991–11994 (2012).
Ueki, T. et al. Autonomous viscosity oscillation via metallo-supramolecular terpyridine chemistry of branched poly(ethylene glycol) driven by the Belousov–Zhabotinsky reaction. Soft Matter 10, 1349–1355 (2014).
Suzuki, D., Taniguchi, H. & Yoshida, R. Autonomously Oscillating Viscosity in Microgel Dispersions. J. Am. Chem. Soc. 131, 12058–12059 (2009).
Kuksenok, O., Deb, D., Dayal, P. & Balazs, A. C. Modeling chemoresponsive polymer gels. Annu. Rev. Chem. Biol. Eng. 5, 35–54 (2014).
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research (No. 237791 and No. 26620164 to T. U. and No. 22245037 to R. Y.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Contributions
M.O. performed all experiments and authored the manuscript. T.U., M.S. and R.Y. conceived and directed the study, as well as authored the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Onoda, M., Ueki, T., Shibayama, M. et al. Multiblock copolymers exhibiting spatio-temporal structure with autonomous viscosity oscillation. Sci Rep 5, 15792 (2015). https://doi.org/10.1038/srep15792
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15792
- Springer Nature Limited
This article is cited by
-
Creation of softmaterials based on self-oscillating polymer gels
Polymer Journal (2022)
-
Amoeba-like self-oscillating polymeric fluids with autonomous sol-gel transition
Nature Communications (2017)