
Abstract
Graphene nanoribbons (GNRs) of precise size and shape, critical for controlling electronic properties and future device applications, can be realized via precision synthesis on surfaces using rationally designed molecular precursors. Fluorine-bearing precursors have the potential to form GNRs on nonmetallic substrates suitable for device fabrication. Here, we investigate the deposition temperature-mediated growth of a new fluorine-bearing precursor, 6,11-diiodo-1,4-bis(2-fluorophenyl)-2,3-diphenyltriphenylene (C42H24F2I2), into helically shaped polymer intermediates and chevron-type GNRs on Au(111) by combining scanning tunneling microscopy, X-ray photoelectron spectroscopy, and density functional theory simulations. The fluorinated precursors do not adsorb on the Au(111) surface at lower temperatures, necessitating an optimum substrate temperature to achieve maximum polymer and GNR lengths. We compare the adsorption behavior with that of pristine chevron precursors and discuss the effects of C-H and C-F bonds. The results elucidate the growth mechanism of GNRs with fluorine-bearing precursors and establish a foundation for future synthesis of GNRs on nonmetallic substrates.

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Graphene nanoribbons (GNRs) feature moderate and tunable band gaps due to quantum confinement and edge effects1, distinguishing them from graphene and rendering them well suited for a range of nanoscale device applications2,3,4,5. Precision syntheses of GNRs with chemically defined width, periodicity, topology, doping, and functionalization are crucial for realizing their intended properties6,7,8,9,10. Within the realm of topological variations, the studies of chevron-type GNRs have garnered significant attention ever since their initial report by Cai et al.11 A gamut of chevron-type GNRs has been synthesized in a bottom-up manner on several metal surfaces under ultra-high-vacuum conditions12,13,14,15,16,17, as well as via large-scale chemical vapor deposition18 and solution-phase processes19,20,21. As band-gap engineering of GNRs has evolved from width and geometry modulation1,22,23 and heteroatomic substitution and functionalization24,25,26 to topological phases27,28,29,30 and spin control31, it is becoming ever more desirable to be able to fine-tune the electronic characteristics of the GNRs in question.
Unlike nitrogen and sulfur dopings15,16,18,32, which are used primarily to tune the electronic properties of GNRs, fluorine (F)-substitution is utilized not only to adjust the electronic properties14,33 but also to facilitate inter- and intra-molecular aryl–aryl coupling through HF elimination on both metal and metal oxide surfaces34,35. Careful precursor design allows F atoms to be located out of the region of cyclodehydrogenation such that F is retained in the fully cyclized GNRs14. However, fluorine atoms close to the cyclodehydrogenation region can undergo C-F bond scission during the reaction process, resulting in non-functionalized GNRs33. C-F bond cleavage can be particularly harnessed to grow carbon-based nanostructures directly on metal oxides36,37. For instance, it was recently demonstrated that F-bearing precursors can be used in a new approach based on HF elimination to grow atomically precise GNRs on rutile TiO2, a nonmetallic substrate suitable for device fabrications38,39. The F-substitution in a chevron precursor thus provides a promising avenue to synthesize GNRs with enhanced diversity and tunability of electronic properties.
A first step in this regard is optimizing the self-assembly and growth of the polymers and GNRs derived from the F-bearing chevron precursors. Owing to the many functional groups or heteroatoms with which precursors can be equipped, appropriate conditions are required for enhancing the coverage and length of GNRs on a substrate. We present here growth conditions that facilitate and inhibit the growth of chevron-type GNRs starting from a new precursor bearing internal fluorine atoms. Additionally, we compare the adsorption behavior to that of pristine chevron precursors, analyzing the effects of C-H and C-F bonds. Our results contribute to a deeper understanding of GNR synthesis and pave the way for future advancements in fabricating GNRs on nonmetallic substrates.
Results and discussion
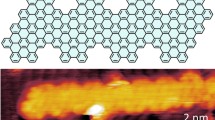
We synthesized a new molecular precursor, 6,11-diiodo-1,4-bis(2-fluorophenyl)-2,3-diphenyltriphenylene (C42H24F2I2, 1), with fluorine substitution placed at internal ortho-positions. The synthetic details are provided in the Supplementary Methods, with additional nuclear magnetic resonance (NMR) spectra presented in Supplementary Figs. 1-7. This precursor has the potential to form chevron-type GNRs on a nonmetallic substrate suited for device fabrication but here we explore the effects of fluorination on GNR growth on Au(111) surface, as illustrated in Fig. 1, by following a two-step thermally triggered on-surface reaction process.

Fluorine-bearing iodinated precursor 1 forms helically shaped polymers (poly-1) on Au(111) with increasing coverages from room temperature (RT) to 100 °C and 180 °C. Heating the polymers to 400 °C induces the formation of archetypical chevron GNR 2.
We find that adsorption of the precursor 1 to the substrate is largely dependent upon the temperature of the substrate during deposition. After deposition with the substrate at room temperature, molecules are only observed along the step edges (Fig. 2a, b). Organic molecules like precursor 1 have been reported to selectively decorate step edges40,41. The precursors 1 that do adsorb form very short oligomers, with individual and bright lobe-like features decorating the step edge which we attribute to the lost iodine atoms (Fig. 2b, green circle). Keeping the substrate at 100 °C during deposition is sufficient to consistently deiodinate the precursors and allows assembly on the Au(111) terraces as observed in Fig. 2c, d. However, the effectiveness of the deiodination, polymerization, and assembly processes seems to be enhanced at higher temperatures such as 180 °C (Fig. 2e, f), where much greater coverage and average polymer length are achieved. This behavior is in contrast to previously reported behavior for pristine brominated or iodinated chevron precursors on Au(111)42 (see Supplementary Fig. 8 as well).

All samples were created with identical deposition durations and crucible temperatures, with the only variation being in substrate temperature during deposition. a, c, e Large-scale and b, d, f small-scale scans of polymer assemblies from depositions performed at 22, 100, and 180 °C substrate temperatures, respectively. The green circle in b indicates a bright spot due to iodine that remains on the surface. Scan parameters: a -1.95 V, 30 pA; b 1.9 V, 10 pA; c -0.75 V, 30 pA; d -0.25 V, 10 pA; e, f -1.5 V, 15 pA.
Polymers of 1 are observed to be similar in appearance to previously reported STM images of non-functionalized chevron-type polymers11,12 (see Supplementary Fig. 9)—both poly-1 and previously reported chevron polymers appear to adopt a \(\pi\)-\(\pi\) interaction-stabilized assembly mode which favors the growth of small 2D islands43. However, a new helical feature is identified for the polymer intermediates from both experiments and density functional theory (DFT) calculations (Supplementary Figs. 10 and 11). To also discern the relative orientations of the fluorine atoms in the polymers, i.e., towards or away from the surface, we have calculated the energies and simulated STM images of these possible configurations, further discussed below and shown in Supplementary Fig. 12, respectively. The GNRs arising from the cyclization step demonstrate no structural features to be attributed to lingering fluorine atoms, unlike edge-fluorinated GNRs14, suggesting that the expected HF elimination has occurred by the time cyclodehydrogenation and cyclodehydrofluorination are complete.
In order to better understand the behavior of the precursor 1 upon deposition—particularly if polymers and GNRs retained the monomers’ fluorine functionalization—we employed X-ray photoelectron spectroscopy (XPS) to characterize a polymer sample grown at 180 °C on an Au(111)/mica thin film. Figure 3 shows the results of these experiments. This sample was transferred from the growth chamber of the STM system, in air, to a separate chamber for the XPS measurements. Measurements were acquired immediately upon introduction to the XPS chamber, 72 h afterwards, and then upon annealing to 400 °C. This was performed to acquire information regarding the short-term air and vacuum stability of the polymer sample as well as whether fluorine atoms survived through the polymerization and GNR formation steps.

The peaks corresponding to fluorine and the C[C2F] moiety are present after the initial polymer growth but disappear following cyclodehydrogenation. The entire 400 °C spectrum was offset vertically from the as-grown sample data for enhanced visual clarity.
As can be seen in Fig. 3, the sample presents clear signatures of fluorine in the form of the F 1s peak and the shoulder adjacent to the C 1s peak, attributed to the carbon in C2F, utilized by Panighel et al. as a model for a fluorinated benzene moiety14,44. The I 3d5/2 peak is also observed in the polymer sample at room temperature, consistent with our STM results as well as previously reported XPS results45, where iodine was found to persist in the form of Au-I up to approximately 350 °C. Annealing to 400 °C induces the expected cyclodehydrogenation and cyclodehydrofluorination reactions in the fluorinated polymers, resulting in chevron-type GNRs and desorption of iodine-attributed features from the surface. In the XPS spectra, this is marked by the disappearance of both the I 3d5/2 and F 1s peaks. The C2F shoulder in the C 1s peak is likewise lost in this transition, which supports our conclusion that the chevron GNRs observed in STM possessed no lingering fluorine atoms. Additional individual elemental spectra can be found in Supplementary Figs. 13-16.
Here we note that the polymer intermediates are observed to manifest a helical conformation. This new feature of helicity, a special case of axial chirality, has not been reported in previous bottom-up synthesis of chevron-type GNRs11,15,16,18. Consistent with earlier STM images of non-functionalized chevron-type polymers11,12,43, each polymer chain is visualized as a “stripe” shape, featuring low-lying triphenylene cores flanked by periodic bright regions attributed to the peripheral phenyl rings. Upon closer examination, these polymers in fact exhibit distinct helical signatures on the phenyl side groups. As shown in Fig. 4, the line profiles along the top and bottom fringes of a representative polymer chain, where the peripheral phenyl rings are situated, display asymmetric low-high and high-low features, respectively, unequivocally indicating a helical conformation. The precursor 1 can polymerize into chains with four possible helicities, including P- and M-, meso- and nonhelical conformations (Supplementary Fig. 11). By comparing the experimental line profiles with those generated from our DFT calculations, we find that the observed polymer intermediates belong to a P-conformation (Supplementary Fig. 11a, b) instead of an M-conformation (Supplementary Fig. 11c, d). In contrast, meso-chains would show asymmetric high-low feature for both traces (Supplementary Fig. 11e, f), while nonhelical chains would show two peaks of equal heights in the profiles (Supplementary Fig. 11g, h). Further investigations are needed to either identify the M-conformation or justify its absence. Additionally, the helical signatures are found more pronounced under lower bias condition and become diminished when a higher bias magnitude is applied (Supplementary Fig. 10). Evidence from previous studies showed that asymmetric bright spots in STM images appeared at lower bias conditions11,16.

a STM image of fluorinated chevron polymers acquired at −0.3 V, 55 pA. b Height profile under the two white lines in a, demonstrating the characteristic asymmetric appearance of the phenyl fringes on two sides along a representative polymer chain. The top trace exhibits a left peak lower in intensity than the right, while the bottom trace exhibits a left peak higher than the right. This is in good agreement with polymers with P-type helicity, shown in optimized geometry in c, where the polymer is given in a perspective view without Au substrate for clarity. d is a simulated p-type polymer along which traces e have been drawn as in a, revealing the same intrinsic peak shape observed in the experimental height profiles in b. Yellow lines have been added as a guide to the eye for the location within each subunit that exhibits the greatest apparent height. The scale bar in the bottom-right of d is 4 nm to coincide with a.
To better understand the effect of the growth temperatures, we now compare the growth of GNRs derived from the samples deposited at different temperatures. Figure 5 shows example STM images of the 180 °C (Fig. 5a) and 100 °C (Fig. 5c) samples before heating to 400 °C for cyclodehydrogenation plus cyclodehydrofluorination. Figure 5b, d show that the greatest average GNR length, greatest individual GNR length, and greatest total GNR coverage are obtained at the 180 °C polymerization temperature, with both average and greatest individual lengths reaching nearly double that observed at the 100 °C polymerization temperature. To shed light on this difference, we carried out DFT energy calculations for fluorine-bearing polymers adsorbed on Au(111) surface (optimized structures available in Supplementary Data 1). The fluorine-bearing polymer with C-F bonds pointing up (Fup) is found to have a negative adsorption energy of −2.96 eV, whereas the other configuration with C-F bonds pointing down (Fdn) has a positive adsorption energy of +0.16 eV, per unit cell of polymer. Due to the internal ortho-positions of F substitution, the preferred adsorption of Fup over Fdn is opposite to the previously reported trend for F atoms located on the polymer/GNR edges14,33. This large difference in adsorption strengths may contribute to the reduced fraction of surface coverage experimentally observed at lower temperature (Fig. 2). On the other hand, the adsorption energy of pristine polymers is also found to be negative at −0.10 eV, which is however weaker than the fluorine-bearing polymers in the Fup configuration, due to the same interfacial contact between C-H bonds and Au substrate, and the smaller molecular weight and accordingly weaker van der Waals interactions. Consequently, the stronger adsorption and larger molecular weight of the F-bearing precursors in the Fup configuration dictate a higher temperature needed for the precursors to overcome the barrier and to diffuse on the Au(111) surface. Previous experimental studies have shown that the polymerization of chevron-type monomers is controlled by diffusion42. Obviously, the temperature is not too high to dissemble the 2D polymer islands stabilized by inter-digited \(\pi\)-\(\pi\) interactions. We conclude from these results that the higher temperature of the surface is more conducive to the diffusion of molecules on contact with the terraces, affording longer and more plentiful polymers and thus GNRs.

a Large-scale scan of GNRs grown from precursors deposited at 180 °C substrate temperature. Scan parameters: −1.75 V, 25 pA. Scale bar 30 nm. b Pareto plot of GNR lengths from the sample shown in a, with the most populated bin ranging from 12 to 17 nm, mean length 17.9 nm, and longest individual GNR at 55.6 nm. c Representative scan of GNRs grown from precursors deposited at 100 °C substrate temperature. Scan parameters: −1.95 V, 20 pA. Scale bar 15 nm. d Pareto plot of GNR lengths from the sample shown in c, with the most populated bin ranging from 7 to 9 nm, mean length 9.57 nm, and longest individual GNR at 28.23 nm.
Conclusions
The fluorine-bearing precursors C42H24F2I2 are shown capable of growing high-quality chevron GNRs on Au(111), but the adsorption and self-assembly of these precursors are heavily dictated by the substrate temperature during deposition. Whereas the pristine chevron precursor can be deposited in appreciable amounts at room temperature via sublimation42 or even direct contact transfer43, the fluorinated 2I-2F-precursor is not observed to adsorb upon the Au(111) surface at lower temperatures. The fraction of sublimated molecules that adsorb on the Au(111) surface (and consequently the maximum possible polymer/GNR length) increases with increasing substrate temperature until approximately 180 °C, after which no appreciable marginal improvement is observed. We attribute the difference in adsorption behavior to the included fluorine atoms, as the pristine chevron precursor (with either bromine or iodine as a halogen group) is able to adsorb on room-temperature Au(111) without issue. Polymer intermediates are shown to be helically shaped, however the helical structure is not retained in the final chevron-type GNR products. This transient feature may still have implications for the synthesis of other helically shaped materials on surfaces.
Materials/methods
Graphene nanoribbon growth
The Au(111) single crystal was cleaned by repeated cycles of Ar+ sputtering and annealing at 740 K. Molecular precursor 1 was degassed at 170 °C overnight in a Knudsen cell. Detailed synthetic procedures of the precursor are described in the Supplementary Information. Deposition was performed with a crucible temperature of 200 °C. Polymerization appeared to occur sporadically at room temperature, but isolated precursors were never observed, nor were polymers on terraces. Reliable polymerization was achieved by annealing at 100 °C for 10 min (holding that temperature during precursor deposition), and full cyclization could be achieved by subsequently annealing at 400 °C for 15 min to yield chevron GNR 2. Increasing the substrate temperature to around 180 °C during and after deposition significantly increased the proportion of adsorbed molecules as well as the average polymer (and thus GNR) length.
Scanning tunneling microscopy
STM characterization was performed with a homemade variable-temperature system at 110 K under UHV conditions with a clean, commercially available PtIr tip. All STM images were acquired in constant-current mode. The bias voltage was applied to the sample bias with respect to the tip.
X-ray photoelectron spectroscopy
XPS measurements were made in ultrahigh vacuum chamber with an operating pressure of 2\(\times\)10-10 torr. X-rays were generated with a SPECS XR50M source using a Mg anode operated at 15 kV and 400 mA electron current. X-rays were then passed through a SPECS Focus 500 monochromator to narrow the photon energy distribution and remove satellites. Photoemitted electrons from the sample were analyzed in a SPECS Phoibos 150 hemispherical analyzer operated at 40 eV pass energy with lenses set for Medium Area. The sample normal was oriented toward the electron detector.
Density functional theory calculations
Atomic structures were optimized with the massively parallel real-space multigrid (RMG) code [https://github.com/RMGDFT/rmgdft]. The RMG parallelizes over nodes, using all CPU cores and all GPUs in each node. The calculations used the PBE exchange-correlation functional46 and van der Waals nonlocal correlation correction47. The real space grids were chosen so that the equivalent plane wave cutoff energies were 100 Ry for the wave functions and 400 Ry for the charge density. Adsorption energies were calculated by taking the difference between the total energy of the polymers adsorbed on the Au(111) surface and the sum of the total energies of the free polymers and Au(111) surface. The STM images were calculated with Tersoff-Hamann scheme48 at constant current mode.
Data availability
Supplementary Data 1 contains atomic coordinates of the optimized pristine and fluorinated polymer intermediates adsorbed on Au(111) surface. Correspondence and requests for materials should be addressed to J.H. or A.-P.L.
References
Son, Y.-W., Cohen, M. L. & Louie, S. G. Energy gaps in graphene nanoribbons. Phys. Rev. Lett. 97, 216803 (2006).
Han, W., Kawakami, R. K., Gmitra, M. & Fabian, J. Graphene spintronics. Nat. Nanotechnol. 9, 794–807 (2014).
Chen, Z., Lin, Y. M., Rooks, M. J. & Avouris, P. Graphene nano-ribbon electronics. Phys. E: Low.-Dimensional Syst. Nanostruct. 40, 228–232 (2007).
Celis, A. et al. Graphene nanoribbons: Fabrication, properties and devices. J. Phys. D: Appl. Phys. 49, 143001 (2016).
Tao, C. et al. Spatially resolving edge states of chiral graphene nanoribbons. Nat. Phys. 7, 616–620 (2011).
Gu, Y., Qiu, Z. & Mullen, K. Nanographenes and graphene nanoribbons as multitalents of present and Future Materials Science. J. Am. Chem. Soc. 144, 11499–11524 (2022).
Chen, Z., Narita, A. & Mullen, K. Graphene nanoribbons: on-surface synthesis and integration into electronic devices. Adv. Mater. 32, 2001893 (2020).
Wang, H. et al. Graphene nanoribbons for quantum electronics. Nat. Rev. Phys. 3, 791–802 (2021).
Ma, C. et al. Seamless staircase electrical contact to semiconducting graphene nanoribbons. Nano Lett. 17, 6241–6247 (2017).
Ma, C. et al. Controllable conversion of quasi-freestanding polymer chains to graphene nanoribbons. Nat. Commun. 8, 14815 (2017).
Cai, J. et al. Atomically precise bottom-up fabrication of graphene nanoribbons. Nature 466, 470–473 (2010).
Linden, S. et al. Electronic structure of spatially aligned graphene nanoribbons on Au(788). Phys. Rev. Lett. 108, 216801 (2012).
Teeter, J. D. et al. Epitaxial growth of aligned atomically precise chevron graphene nanoribbons on Cu(111). Chem. Commun. (Camb.) 53, 8463–8466 (2017).
Panighel, M. et al. Stabilizing edge fluorination in graphene nanoribbons. ACS Nano 14, 11120–11129 (2020).
Bronner, C. et al. Aligning the band gap of graphene nanoribbons by monomer doping. Angew. Chem. Int. Ed. 125, 4422–4425 (2013).
Cai, J. et al. Graphene nanoribbon heterojunctions. Nat. Nanotechnol. 9, 896–900 (2014).
Teeter, J. D. et al. On‐surface synthesis and spectroscopic characterization of laterally extended chevron graphene nanoribbons. ChemPhysChem 20, 2281–2285 (2019).
Chen, Z. et al. Synthesis of graphene nanoribbons by ambient-pressure chemical vapor deposition and device integration. J. Am. Chem. Soc. 138, 15488–15496 (2016).
Vo, T. H., Shekhirev, M., Lipatov, A., Korlacki, R. A. & Sinitskii, A. Bulk properties of solution-synthesized chevron-like graphene nanoribbons. Faraday Discuss 173, 105–113 (2014).
Liu, X. et al. Chevron-type graphene nanoribbons with a reduced energy band gap: Solution synthesis, scanning tunneling microscopy and electrical characterization. Nano Res. 13, 1713–1722 (2020).
Pour, M. M. et al. Laterally extended atomically precise graphene nanoribbons with improved electrical conductivity for efficient gas sensing. Nat. Commun. 8, 820 (2017).
Han, M. Y., Özyilmaz, B., Zhang, Y. & Kim, P. Energy band-gap engineering of graphene nanoribbons. Phys. Rev. Lett. 98, 206805 (2007).
Palma, C.-A. et al. Sub-nanometer width armchair graphene nanoribbon energy Gap atlas. J. Phys. Chem. Lett. 6, 3228–3235 (2015).
Nguyen, G. D. et al. Bottom-Up Synthesis of N = 13 Sulfur-Doped Graphene Nanoribbons. J. Phys. Chem. C. 120, 2684–2687 (2016).
Pawlak, R. et al. Bottom-up Synthesis of Nitrogen-Doped Porous Graphene Nanoribbons. J. Am. Chem. Soc. 142, 12568–12573 (2020).
Kawai, S. et al. Multiple heteroatom substitution to graphene nanoribbon. Sci. Adv. 4, eaar7181 (2018).
Cao, T., Zhao, F. & Louie, S. G. Topological phases in graphene nanoribbons: junction states, spin centers, and quantum spin chains. Phys. Rev. Lett. 119, 076401 (2017).
Lee, Y. L., Zhao, F., Cao, T., Ihm, J. & Louie, S. G. Topological phases in cove-edged and chevron graphene nanoribbons: geometric structures, Z2 invariants, and junction states. Nano Lett. 18, 7247–7253 (2018).
Rizzo, D. J. et al. Topological band engineering of graphene nanoribbons. Nature 560, 204–208 (2018).
Joost, J.-P., Jauho, A.-P. & Bonitz, M. Correlated topological states in graphene nanoribbon heterostructures. Nano Lett. 19, 9045–9050 (2019).
Sun, Q. et al. Coupled spin states in armchair graphene nanoribbons with asymmetric zigzag edge extensions. Nano Lett. 20, 6429–6436 (2020).
Vo, T. H. et al. Nitrogen-doping induced self-assembly of graphene nanoribbon-based two-dimensional and three-dimensional metamaterials. Nano Lett. 15, 5770–5777 (2015).
Hayashi, H. et al. Experimental and theoretical investigations of surface-assisted graphene nanoribbon synthesis featuring carbon–fluorine bond cleavage. ACS Nano 11, 6204–6210 (2017).
Fan, Q. et al. Biphenylene network: A nonbenzenoid carbon allotrope. Science 372, 852–856 (2021).
Amsharov, K. Y., Kabdulov, M. A. & Jansen, M. Facile Bucky-bowl synthesis by regiospecific cove-region closure by HF elimination. Angew. Chem. Int. Ed. 51, 4594–4597 (2012).
Sharapa, D., Steiner, A.-K. & Amsharov, K. The Mechanism of Cyclodehydrofluorination on γ-Alumina. Phys. Status Solidi B 255, 1800189 (2018).
Kolmer, M. et al. Fluorine-programmed nanozipping to tailored nanographenes on rutile TiO2 surfaces. Science 363, 57–60 (2019).
Kolmer, M. et al. Rational synthesis of atomically precise graphene nanoribbons directly on metal oxide surfaces. Science 369, 571–575 (2020).
Zuzak, R. et al. On-surface synthesis of nanographenes and graphene nanoribbons on titanium dioxide. ACS Nano 17, 2580–2587 (2023).
Vladimirova, M. et al. Supramolecular self-assembly and selective step decoration on the Au(111) surface. Europhys. Lett. 56, 254–260 (2001).
Ma, C. et al. Step edge-mediated assembly of periodic arrays of long graphene nanoribbons on Au(111). Chem. Commun. 55, 11848–11851 (2019).
Bronner, C. et al. Iodine versus bromine functionalization for bottom-up graphene nanoribbon growth: role of diffusion. J. Phys. Chem. C 121, 18490–18495 (2017).
Teeter, J. D. et al. Dense monolayer films of atomically precise graphene nanoribbons on metallic substrates enabled by direct contact transfer of molecular precursors. Nanoscale 9, 18835–18844 (2017).
Clark, D. T., Kilcast, D., Adams, D. B. & Musgrave, W. K. R. An ESCA study of the molecular core binding energies of the fluorobenzenes. J. Electron Spectrosc. Relat. Phenom. 1, 227–250 (1972–1973).
Di Giovannantonio, M. et al. On-surface growth dynamics of graphene nanoribbons: the role of halogen functionalization. ACS Nano 12, 74–81 (2018).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Dion, M., Rydberg, H., Schröder, E., Langreth, D. C. & Lundqvist, B. I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 92, 246401 (2004).
Tersoff, J. & Hamann, D. R. Theory of the scanning tunneling microscope. Phys. Rev. B 31, 805–813 (1985).
Acknowledgements
This research was conducted at the Center for Nanophase Materials Sciences (CNMS), which is a DOE Office of Science User Facility. The electronic characterization was funded by ONR grants N00014-10-1-2302. The synthesis of the GNR precursors was supported by the ONR via N00014-19-1-2596. The supercomputer time was provided by DOE at the Oak Ridge Leadership Computing Facility and at the National Energy Research Scientific Computing Center (Contract No. DE-AC02-05CH11231 using NERSC award BES-ERCAP0027465).
Author information
Authors and Affiliations
Contributions
J.D.T. and M.S. contributed equally to this work. A.-P.L. conceived the project and designed the experiments; J.H. designed the theory tasks. J.D.T., C.T., A.P.B., and K.H. performed on-surface synthesis and characterizations; M.S. and A.S. conducted molecule synthesis; W.L., J.H., and J.B. performed the theoretical calculations. J.D.T., W.L., J.H. and A.-P.L. wrote the paper with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Teeter, J.D., Sarker, M., Lu, W. et al. Deposition temperature-mediated growth of helically shaped polymers and chevron-type graphene nanoribbons from a fluorinated precursor. Commun Chem 7, 193 (2024). https://doi.org/10.1038/s42004-024-01253-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-024-01253-9
- Springer Nature Limited