
Abstract
HIV-1 reverse transcriptase offers a key target for antiviral therapy. However, the rapid emergence of drug-resistant mutations in reverse transcriptase as well as the poor pharmacokinetic properties of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) limits their clinical use. Starting from a previous piperidine-substituted thiophene[3,2-d]pyrimidine compound (K-5a2), here we explore the chemical space around the thiophene ring located in the solvent-exposed regions of the NNRTI binding pocket in detail. Bioisosterism-based structural modification leads to the discovery of a number of compounds as potent in vitro reverse transcriptase inhibitors, providing improved drug resistance profiles compared to the listed drug Etravirine. Furthermore, 14a and 19a are identified as lead compounds with good solubility, appropriate ligand efficiency, and lower cytochrome P450 liability. Compound 19a exhibits useful in vivo pharmacokinetic properties in rat and safety in mice, suggesting that it may have the potential to be an effective drug candidate for treating AIDS.
Similar content being viewed by others
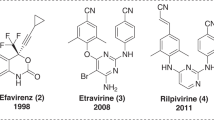


Introduction
HIV-1 reverse transcriptase (RT) represents one of the most successful molecular targets for novel development of precise medicine to treat AIDS patients1. Two generations of non-nucleoside RT inhibitors (NNRTIs) have been awarded FDA approval2. The second-generation NNRTIs, Etravirine (ETV) and Rilpivirine (RPV) (Fig. 1), were designed to suppress most of the RT-resistant mutations caused by treatment of the first-generation NNRTIs, especially the K103N and Y181C mutation3,4. Although ETV and RPV exhibited higher genetic barriers to emergence of drug resistance, they generally failed to interact with the most refractory mutations E138K and RES056 (K103N + Y181C)5,6. In addition, hypersensitivity reactions or other adverse effects have been reported with second-generation NNRTIs2,7.

Chemical structures of the leads and the design strategy in this study. a The chemical structures of the second-generation NNRTIs Etravirine (ETV), Rilpivirine (RPV) and our previously reported lead compounds K-5a2 3 and 25a 4. b Further optimization of the lead K-5a2 via bioisosteric replacement strategy. The yellow/green parts highlight the structural modifications of the central scaffold
Our previous efforts have led to the development of two novel NNRTIs 3 (K-5a2) and 4 (25a) with a piperidine-substituted thiophene[3,2-d]pyrimidine scaffold8,9. Both compounds exhibit highly potency toward wild-type (WT) strain and numerous clinically observed variants compared to ETV. However, the activity of 3 against the most challenging double mutation RES056 is inferior to that of ETV. Although 4 proved to be capable of inhibiting all of the resistant RT-mutants with single-nanomole activities, it also showed greater cytotoxicity (CC50 = 2.30 μM). This may be caused by the cyanovinyl group, as it can act as a Michael acceptor and potentially result in covalent modification of proteins and nucleic acids10. In addition, both 3 and 4 exhibit extremely low aqueous solubility, far below that of the typical range (4−4000 μg/mL) for oral drugs. The low solubility results in poor bioavailability of 3 (F = 22.9%) and 4 (F = 16.2%). This has prompted the search for novel HIV-1 inhibitors with improved drug resistance profiles and pharmacokinetic properties.
With the aim to provide valuable insights into the favorable structural features for further structure-based design, we have determined the crystal structures of HIV-1 WT and some mutant RT variants in complex with 3. The co-crystal structures demonstrated that the hydrophobic interactions of the left wing, the network of the main chain of hydrogen bonds formed between the NNRTIs and NNRTIs binding pocket (NNIBP), and its structural flexibility all play important roles in maintaining its high potency against mutant strains. These high-resolution co-crystal structures illustrate the molecular details of the binding mode, and provide valuable insights into favorable structural features that can be employed for designing novel HIV-1 NNRTIs that are broadly active against drug-resistant HIV-1 RT variants11.
The application of structural mimics of extensive scaffolds or peripheral substituents, commonly designated as bioisosteric replacement strategy, is a common approach in contemporary drug design and discovery. Guided by previous structural biology studies, we kept the hydrophobic interactions and hydrogen bonds unchanged and modified the thiophene[3,2-d]pyrimidine core ring to the furo[3,2-d]pyrimidine and thiazolo[4,5-d]pyrimidine ring in the first round of the present study (Fig. 1). The rational is that the introduction of oxygen atom may develop novel hydrogen bonds (hydrogen bond receptor) with Glu138 in NNIBP. Moreover, the replacement of a CH moiety with a nitrogen atom in heteroaromatic ring systems can significantly alter the physicochemical properties and intra- and intermolecular interactions, which may yield improved pharmacological and pharmacokinetics properties12. Following the disclosure of thiazolo[4,5-d]pyrimidine derivatives as potent HIV-1 inhibitors with potent activity against mutant HIV-1 strains RES056 in the first round, we also investigated the effects of substitution on the thiazolo[4,5-d]pyrimidine core to thiazolo[5,4-d]pyrimidine in the second round.
Here we report that the center ring can have dramatic effects on potency and pharmacokinetic properties. Especially, the compound 19a is shown to exhibit favorable in vivo pharmacokinetic properties in rat and safety in mice, suggesting that it may lead to an effective drug candidate for treating AIDS.
Results
Synthesis and characterization of the target compounds
The designed library and synthesis were based on our previously reported methods8 and are described in Fig. 2 and Supplementary Figs. 1, 2. 2,4-Dichlorofuro[3,2-d]pyrimidine (5) was selected as the starting material and was reacted with 4-hydroxy-3,5-dimethylbenzonitrile to yield intermediate 6. Then nucleophilic substitution of 6 with 4-(tert-butoxycarbonyl)-aminopiperidine was conducted in the presence of potassium carbonate, and the product was subsequently treated with trifluoroacetic acid to provide the key intermediate 8. Then target compounds 9a–f were obtained by another nucleophilic substitution between 8 and 4-picolyl chloride hydrochloride or substituted benzyl chloride (or bromine). The synthetic procedures of 14a–f and 19a–f were similar to that of 9a–f, only with the difference that 5,7-dichlorothiazolo[4,5-d]pyrimidine and 5,7-dichlorothiazolo[5,4-d]pyrimidine were used as starting materials (Supplementary Figs. 1, 2).

The synthetic route of 9a–f. Full details can be found in the Supplementary Methods
In vitro assay of anti-HIV activities in MT-4 cells
The newly designed derivatives 9a–f and 14a–f were evaluated for their activity against WT HIV-1 (IIIB), the most challenging double mutant HIV-1 strain RES056, and a HIV-2 strain (ROD) in the MT4 cell line. ETV was selected as control drug. The values of EC50 (anti-HIV potency), CC50 (cytotoxicity), SI (selectivity index, CC50/EC50 ratio), and RF (fold-resistance factor, EC50 (mutant strains)/EC50 (WT strain)) of the target compounds were summarized.
As depicted in Table 1, ten of the newly synthesized twelve compounds showed high potency against the WT HIV-1 strain with low nanomolar EC50 values ranging from 1.1 to 3.4 nM, which are superior to that of the reference drug ETV (EC50 = 5.1 nM). In the case of double mutant HIV-1 strain RES056, 9a–f with furo[3,2-d]pyrimidine scaffold showed a marked decrease of potency and 9d turned out to be the most potent inhibitor with an EC50 value of 100.2 nM, but still being inferior to that of ETV (EC50 = 45.4 nM). Among the derivatives in sub-series 14 with the thiazolo[4,5-d]pyrimidine scaffold, 14a, 14c, and 14d could potently inhibit RES056 with EC50 values of 26.1, 27.4 and 24.7 nM respectively, being about twofold more potent than ETV. Moreover, 14a and 14d also demonstrated with lower cytotoxicity (CC50 = 25.1 and 30.3 μM, respectively) and higher SI values toward WT (SI = 11071 and 16818) and mutant HIV-1 strains (SI = 961 and 1227).
Considering that the compounds of sub-series 14 with thiazolo[4,5-d]pyrimidine scaffold exhibited more active potency than compounds of sub-series 9 with furo[3,2-d]pyrimidine scaffold, we further replaced the thiazolo[4,5-d]pyrimidine with thiazolo[5,4-d]pyrimidine utilizing the bioisosterism strategy, with the aim to achieve more active potency against the double mutant strain RES056. As shown in Table 1, all the novel compounds were demonstrated with single-nanomole activity against WT HIV-1 strain. Moreover, 19a–d exhibited more potent activity against RES056 than ETV, with EC50 values from 18.1 to 28.1 nM. Among them, 19a (EC50 = 18.3 nM) and 19c (EC50 = 18.1 nM) were the most potent inhibitors against RES056, being about 2-fold potent than that of ETV. All the results confirmed that the thiazolo[5,4-d]pyrimidine scaffold was favorable for improving the activity against RES056.
The structure-activity relationship (SARs) could be concluded clearly from their activities. Detailed comparison of the activities of 9a–d vs 9e, 14a–d vs 14e, and 19a–d vs 19e indicated that hydrophilic substituents harboring hydrogen bond donors or acceptors (Ar = 4-SO2NH2-Ph, 4-SO2CH3-Ph, 4-CO2NH2-Ph, and pyridine-4-yl) at the 4-position in the phenyl ring of the Ar could strengthen the interaction with the NNRTI binding site compared to hydrophobic substituent (Ar = 4-NO2-Ph). Meanwhile, nitro-substitution-bearing compounds showed increased cytotoxicity compared to other hydrophilic substituted compounds, such as the CC50 values of 9e, 14e and 19e can up to 5.24, 5.17, and 5.68 μM, respectively. In addition, pairwise comparison of the activities of compounds 9b vs 9 f and 19b vs 19 f confirmed the critical role of the para-substitution in improving the activity against RES056 over meta-substitution. Moreover, comparison of the activities of sub-series 9, sub-series 14 and sub-series 19 leads to the conclusion that the activity of compounds toward mutant strain RES056 were also greatly influenced by their central scaffolds, thiazolo[5,4-d]pyrimidine and thiazolo[4,5-d]pyrimidine display significantly improved drug resistance profiles, which were superior to that of furo[3,2-d]pyrimidine. It is well to be reminded that compound 14b (EC50 > 15560 nM) exhibited sharply reduced activity, compared to the other derivatives, though they are structurally similar molecules. This seems to be another good example of an activity cliff13,14, which may be related to the flexibility of the binding pocket and the induced-fit mechanism of the NNRTIs8.
Furthermore, compounds 14a, 14c–d and 19a–d, which exhibited high potency against WT and RES056 HIV-1 strains, were tested for their potency against a panel of NNRTI-resistant strains, including single-mutant strains L100I, K103N, Y181C, Y188L, E138K, and double-mutant strain F227L + V106A. The results are depicted in Table 2 and Supplementary Table 1. As for mutant HIV-1 strain L100I, 14c turned out to be the most potent inhibitor with EC50 value of 2.70 nM, being about 2.2-fold than that of ETV (EC50 = 6.0 nM). In the case of K103N, Y181C, 188 L and E138K, 19b yielded the most active potency (EC50 = 1.6, 5.1, 7.2 and 5.2 nM, respectively), which is more active than that of ETV (EC50 = 3.3 nM, 14.5 nM, 20.8 nM and 9.7 nM, respectively). Moreover, the other compounds also exhibited comparable activity as ETV toward these single mutant HIV-1 strains. Against F227L + V106A, 14a, 14c, and 19a–c showed comparable potency (EC50 = 15.2–20.3 nM) with ETV (EC50 = 19.7 nM), while compounds 14d and 19d with pyridine-4-yl group at the Ar region exhibiting a decreased activity with EC50 values of 32.9 and 30.9 nM, respectively. This indicates that a pyridine-4-yl group at the Ar position is unable to maintain compounds activity toward resistance variants F227L + V106A effectively.
Recombinant HIV-1 reverse transcriptase inhibitory assays
To validate the binding target, some potent compounds were evaluated for their ability to inhibit recombinant WT HIV-1 RT enzyme. As shown in Table 3, all the selected compounds displayed highly inhibitory activity with IC50 values of 0.037–0.050 μM, which were superior to that of EFV (IC50 = 0.181 μM) and keep the same magnitude with that of ETV (IC50 = 0.011 μM). The results confirmed the viewpoint that the newly synthesized derivatives showed high affinity to HIV-1 RT and acted as classical NNRTIs. However, the results demonstrated that the antiviral activities of these compounds were inconsistent with their RT inhibitory potencies. These differences are considered to be due to template-specific variation in the relationship between HIV-RT-RNA binding affinity and polymerase processivity and have been observed in most NNRTI series.
Molecular modeling studies
In a previous study, we crystalized HIV-1 RT complexes with the K-5a2 ligand15 which is very similar to the 14a, 19a, and 19c compounds. Here, we consider prediction of the binding modes of these compounds through molecular modeling studies in different mutants. Molecular docking followed by MM-GBSA simulations were performed for 14a, 19a, and 19c in eight mutants and WT. From several studies, the NNIBP is well characterized as being more hydrophobic and comprises of amino acids L100, V106, T107, V108, V179, Y181, Y188, G190, F227, W229, L234, and Y31815,16. Initially, the binding mode of K-5a2 and ETV was compared in the WT. As expected, both compounds showed very similar binding pose (Supplementary Figs. 3, 4) and possess π-π interactions and hydrogen bonding with Trp229 and Lys101/Lys102 residues, respectively. In addition, compound K-5a2 makes additional hydrogen bonding between sulfonamide and residues such as Lys104 and Val106. Subsequently, the binding mode of the compounds used in the modeling study were also investigated in various mutants, however, for clarity, only the highly potent compound 19a is discussed. Figure 3 shows the best binding poses of 19a.

Molecular modeling studies. Comparison of binding mode of ETV (pink stick), K-5a2 (cyan stick) and 19a (green stick) is shown in the NNIBP and the clinically relevant mutants are shown in spheres. Binding mode of 19a in WT and various mutants is shown and its key intercations are highlighted (residues which form hydrogen bonding and hydrophobic interactions are shown in black and green boxes)
Overall, 19a showed the same binding mode as K-5a2 and ETV. The sulfonamide group is involved in hydrogen bonding with Val106 (or Ala106) and Lys104 in all mutants, including when Val106 was mutated to Ala106. Interestingly, thiazolopyrimidine showed better interaction with the NNIBP hydrophobic residues as compared to ETV, and particularly when it is substituted with thiazolopyrimidine, it makes π-H interactions with Val179 in all the mutant structures. In most cases, the binding affinity of 19a was not affected by mutations, except for double mutants. For instance, the binding affinity remains almost the same in the K103N mutant, due to complementary hydrogen bonding between Asn103 with one of the nitrogen atoms of the thioazolopyrimidine ring of 19a as observed in Lys103.
Solubility and lipophilic efficiency
In order to verify the drug-like properties of the promising compounds, the physicochemical properties of 14a, 14c, 14d, and 19a–d were determined. All the selected compounds were tested for their solubility at three different pH values (7.4, 7.0, and 2.0) with HPLC methods. The results are shown in Table 4. All compounds exhibited good solubility at pH 2.0 (S > 178.3 μg/mL). Especially, their solubility have been greatly improved compared to K-5a2 and ETV at pH 7.0 and pH 7.4. Among them, 14d has the best solubility (S = 33.8 μg/mL, pH = 7.0; S = 30.7 μg/mL, pH = 7.4). The results are in accord with the purpose of our design.
To further screen compounds with good physicochemical properties, seven compounds 14a, 14c,d and 19a–d were calculated their activity efficiency, including lipophilic efficiency (LE), ligand lipophilic efficiency (LLE), and ligand efficiency dependent lipophilicity (LELP), which are regarded as important metrics to measure the balance between in vitro activity and in vivo drug-like properties17. As depicted in Table 4, all the selected compounds satisfied the acceptable levels for LE and LLE (LE > 0.3 and LLE > 5)18, which means these compounds could combine their lipophilicity and in vitro potency well. Moreover, 14a, 14d, 19a, and 19c were also demonstrated with appropriate LELP value (LELP < 10), supplying a possibility that these compounds have more favorable ADME profiles than the lead K-5a2 and the approved drug ETV.
In vitro effects on CYP enzymatic inhibitory activity
The drug metabolism in vivo was mainly accomplished by the metabolizing enzymes in the liver, especially the cytochrome P450 (CYP450), which including CYP1A, CYP3A, CYP2C, and CYP2D619. On the other hand, the drugs could also inhibit the activity of CYP450 and down-regulate the expression of their genes, which may lead to metabolism-mediated drug-drug interactions (DDI) when co-administration different drugs and cause side effects20,21. Among these CYP450 isozymes, CYP3A4M is a major xenobiotic metabolizing enzyme, and it contributes to the metabolism of approximately 50% of prescribed drugs22. Considering the activity of 14d against double mutant strain F227L + V106A was inferior to that of ETV, so only the lead 14a, 19a, and 19c were evaluated for their CYP drug metabolizing enzymatic inhibitory activity. As depicted in Table 5, 14a and 19a yield lower inhibitory activity to all tested CYP isozymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4M) with higher IC50 values (IC50 > 3 μM), suggesting that 14a and 19a are anticipated to have a favorable drug interaction profile with respect to key CYP isozymes. In addition, 19c exhibited a moderate inhibitory activity to CYP3A4M with an IC50 value of 2.78 μM, which demonstrated that 19c has a strong CYP3A4 inhibition activity and may cause strong DDI.
Pharmacokinetic study
Considering the promising activities, favorable physiochemical properties and lower CYP enzymatic inhibitory activity of 14a and 19a, they were further evaluated for their pharmacokinetics (PK) profiles by Wistar rats PK models following intravenous (iv) and oral administration (po) (Table 6 and Supplementary Fig. 5). The results demonstrated that 14a and 19a showed a suitable clearance (CL = 10.2 and 16.3 L/h/kg, respectively) and half-life (T1/2 = 2.01 and 0.93 h, respectively) for a single 2 mg/kg iv dose. Furthermore, after a single 20 mg/kg po dose of 14a and 19a, they reached the maximum concentration (Tmax) at 0.90 and 2.8 h with a Cmax of 272 and 2982 ng/mL, respectively. Moreover, both of them displayed an acceptable oral bioavailability (F) for a drug candidate, and the oral bioavailability of 19a up to 83.8%.
Safety assessment
We evaluated the acute toxicity of 14a and 19a in Kunming mice. After oral gavage administrations with a dose of 2000 mg/kg, there was no death and no abnormity of body weight change compared to control group in the following one week (Supplementary Fig. 6). The results support the great potential of 14a and 19a as novel NNRTI drug candidates with low acute toxicity.
The subacute toxicity experiments of 14a and 19a were carried out to further evaluate their in vivo safety. No apparent signs associated with animal toxicity and no behavioral abnormalities were observed during the treatment period of po administered mice treated with 50 mg/kg of 14a and 19a every day for 7 days. Furthermore, hematoxylin and eosin (H&E) staining was utilized for histological analysis of the major organs. As displayed in Fig. 4, no apparent physiological abnormalities or lesions were observed in the heart, liver, spleen, lungs, and kidneys, suggesting the negligible side toxicity of 14a and 19a.

The result of subacute toxicity. H&E stained images of major organs (heart, liver, spleen, lung, and kidney) collected from various groups (blank, 14a and 19a). The scale bar was 50 µm
Compounds with a high affinity for the hERG potassium channel could induce QT interval prolongation, which is frequently related to potentially risk for cardiotoxicity23. So we next evaluated their hERG inhibition activity, and Terfenadine was selected as reference drug (Supplementary Tables 3, 4). As shown in Supplementary Figs. 7, 14a and 19a showed an IC50 of 0.18 and 0.12 μM against the potassium channel respectively. Although the IC50 values indicated that 14a and 19a has a risk of cardiotoxicity, their hERG inhibition activity lower than that of terfenadine (IC50 = 0.022 μM)9, which gave us much confidence for the further development.
Discussion
Although second-generation NNRTIs achieved success in suppressing HIV-1 replication and reducing viral loads, poor aqueous solubility, dose-limiting toxicity, and the rapid generation of drug-resistant mutations in HIV-1 RT remains a major impediment to effective anti-HIV treatment. The application of structural mimics of a range of scaffolds or peripheral substituents, commonly designated as bioisosteres, is a common approach in contemporary drug design and discovery. With ETV as a lead, two piperidine-substituted thiophene[3,2-d]pyrimidine derivatives, K-5a2 and 25a, with potent activity against WT and mutant HIV-1 strains were recently discovered as drug candidates in our lab. However, both of them encountered some deficiency, including weak activity against RES056 (K-5a2), higher cytotoxicity (25a) and low bioavailability (K-5a2 and 25a), so there still is an urgent need for searching novel NNRTIs with improved antiviral potency against WT HIV and mutant strains, reduced adverse effects and more favorable pharmacokinetic profiles.
In current study, with K-5a2 as lead, series of anti-HIV compounds were designed and synthesized guided by structural biology studies and bioisosteric replacement strategy. Especially, we detailed the results of our efforts to optimize the in vivo and in vitro properties of derivatives prepared from the furo[3,2-d]pyrimidine, thiazolo[4,5-d]pyrimidine and thiazolo[5,4-d]pyrimidine core. The results of in vitro assay of anti-HIV activities demonstrated that most of the synthesized compounds showed more active potency than ETV. In the case of RT variants bearing prevalent drug-resistant mutations, compounds with thiazolo[4,5-d]pyrimidine and thiazolo[5,4-d]pyrimidine core also displayed greater potency than ETV, such as 14a, 14c, and 19a–c. The SARs could be concluded that hydrophilic substituents harboring hydrogen bond donors or acceptors (–SO2NH2, –CONH2, and –SO2CH3) can significantly improve drug resistance profiles.
Moreover, the selected potent compounds exhibited an IC50 values of 0.037–0.050 μM in the recombinant HIV-1 RT enzyme assays and confirmed the binding target of these compounds is RT. Molecular modeling studies illustrate the molecular details of the binding poses of 19a, extensive hydrophobic interactions and network of main chain hydrogen bonds formed between the NNRTIs and NNIBP, and provide reasonable explanation for the improved drug resistance profiles, which give an effective evidence that the NNRTIs could retain effective activities against mutant HIV-1 strains by taking advantage of the structural flexibility of the inhibitors, plasticity of the NNIBP and the hydrogen bonding between the NNRTIs and the main chains of NNIBP residues.
Furthermore, 14a and 19a were identified as promising hit compounds after screening of compounds solubility, ligand efficiency calculations, and CYP enzymatic inhibition activity. Especially, 19a with favorable pharmacokinetic properties and its bioavailability reaches up to 83.8%. Moreover, both compounds showed good safety profiles, including acute toxicity, subacute toxicity and cardiotoxicity. In conclusion, the current study confirmed a potential anti-HIV-1 drug candidate with improved drug resistance profiles and favorable druggability, and the pre-clinical studies are under way.
Methods
Synthetic procedures
See Supplementary Methods and Supplementary Figs. 1, 2. For NMR spectra see Supplementary Figs. 8–16.
Anti-HIV activity test studies
See Supplementary Methods and Supplementary Table 1.
HIV-1 RT inhibitory assays and cytochrome P450 inhibition assay
Molecular modeling methods
See Supplementary Methods, Supplementary Table 2, and Supplementary Tables 3, 4.
Pharmacokinetics assays and acute toxicity experiment
See Supplementary Methods and Supplementary Figs. 5, 6
hERG activity assay procedures
See Supplementary Methods, Supplementary Tables 3, 4, and Supplementary Fig. 7
Laboratory animals
The authors declare that all experimental work complied with the institutional guidelines on animal studies (care and use of laboratory animals).
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information.
References
Bec, G. et al. Thermodynamics of HIV-1 reverse transcriptase in action elucidates the mechanism of action of non-nucleoside inhibitors. J. Am. Chem. Soc. 135, 9743–9752 (2013).
Zhan, P., Pannecouque, C., De Clercq, E. & Liu, X. Anti-HIV drug discovery and development: current innovations and future trends. J. Med. Chem. 59, 2849–2878 (2016).
Wainberg, M. A., Zaharatos, G. J. & Brenner, B. G. Development of antiretroviral drug resistance. New Engl. J. Med. 365, 637–646 (2011).
Lehman, D. A. et al. Low-frequency nevirapine resistance at multiple sites may predict treatment failure in infants on nevirapine-based treatment. J. Acquir. immune Defic. Syndr. 60, 225–233 (2012).
Beyrer, C. & Pozniak, A. HIV drug resistance - an emerging threat to epidemic control. New Engl. J. Med. 377, 1605–1607 (2017).
Namasivayam, V. et al. The journey of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) from lab to clinic. J. Med. Chem. 62, 4851–4883 (2018).
Wu, P. Y. et al. Multicenter study of skin rashes and hepatotoxicity in antiretroviral-naive HIV-positive patients receiving non-nucleoside reverse-transcriptase inhibitor plus nucleoside reverse-transcriptase inhibitors in Taiwan. PLoS ONE 12, e0171596 (2017).
Kang, D. et al. Design, synthesis, and evaluation of thiophene[3,2-d]pyrimidine derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors with significantly improved drug resistance profiles. J. Med. Chem. 59, 7991–8007 (2016).
Kang, D. et al. Structure-based optimization of thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 non-nucleoside reverse transcriptase inhibitors with improved potency against resistance-associated variants. J. Med. Chem. 60, 4424–4443 (2017).
Lee, W. G. et al. Picomolar inhibitors of HIV reverse transcriptase featuring bicyclic replacement of a cyanovinylphenyl group. J. Am. Chem. Soc. 135, 16705–16713 (2013).
Zhang, S. et al. Efficient drug discovery by rational lead hybridization based on crystallographic overlay. Drug Disco. Today 24, 805–813 (2018).
Pennington, L. D. & Moustakas, D. T. The necessary nitrogen atom: a versatile high-impact design element for multiparameter optimization. J. Med. Chem. 60, 3552–3579 (2017).
Dagmar, S., Ye, H., Dilyana, D. & Jürgen, B. Recent progress in understanding activity cliffs and their utility in medicinal chemistry. J. Med. Chem. 57, 18–28 (2014).
Stumpfe, D. & Bajorath, J. Exploring activity cliffs in medicinal chemistry. J. Med. Chem. 55, 2932–2942 (2012).
Yang, Y. et al. Structural basis for potent and broad inhibition of HIV-1 RT by thiophene[3,2-d]pyrimidine non-nucleoside inhibitors. eLife 7, e36340 (2018).
Schafer, W. et al. Non-nucleoside inhibitors of HIV-1 reverse transcriptase: molecular modeling and X-ray structure investigations. J. Med. Chem. 36, 726–732 (1993).
Freeman-Cook, K. D., Hoffman, R. L. & Johnson, T. W. Lipophilic efficiency: the most important efficiency metric in medicinal chemistry. Future Med. Chem. 5, 113–115 (2013).
Tarcsay, A., Nyiri, K. & Keseru, G. M. Impact of lipophilic efficiency on compound quality. J. Med. Chem. 55, 1252–1260 (2012).
Lynch, T. & Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. physician 76, 391–396 (2007).
Isoherranen, N., Kunze, K. L., Allen, K. E., Nelson, W. L. & Thummel, K. E. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab. Dispos.: Biol. fate Chem. 32, 1121–1131 (2004).
Kanayama, N., Kanari, C., Masuda, Y., Ohmori, S. & Ooie, T. Drug-drug interactions in the metabolism of imidafenacin: role of the human cytochrome P450 enzymes and UDP-glucuronic acid transferases, and potential of imidafenacin to inhibit human cytochrome P450 enzymes. Xenobiotica; Fate Foreign Compd. Biol. Syst. 37, 139–154 (2007).
Guengerich, F. P. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 39, 1–17 (1999).
Vandenberg, J. I. et al. hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478 (2012).
Acknowledgements
We gratefully acknowledge financial support from the Key Project of NSFC for International Cooperation (No.81420108027), the National Natural Science Foundation of China (NSFC Nos. 81273354, 81573347), China Postdoctoral Science Foundation (2018M640641), Young Scholars Program of Shandong University (YSPSDU No. 2016WLJH32), Key research and development project of Shandong Province (No. 2017CXGC1401), Natural science foundation of Shandong Province (ZR2019BH011) and KU Leuven (GOA 10/014).
Author information
Authors and Affiliations
Contributions
D.K., C.P., P.Z., and X.L. conceived the project. D.K., Z.W., D.F., H.Z., G.W., F.W., Z.Z., and L.J. finished the compounds synthesis and structure confirmation. D.K., T.Z., and X.Z. designed the pharmacokinetics assays and acute toxicity experiment. D.K. and Z.W. performed the CYP450 inhibition assay. D.K., B.H., and Y.T. performed the data analysis. V.P. and J.K. performed the molecular modeling study. E.D.C. and C.P. performed the evaluation of compounds activity. C.P., P.Z., and X.L. provided the resources, supervision and funding assistance. All authors critically evaluated the manuscript prior to submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, D., Zhao, T., Wang, Z. et al. Discovery of piperidine-substituted thiazolo[5,4-d]pyrimidine derivatives as potent and orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitors. Commun Chem 2, 74 (2019). https://doi.org/10.1038/s42004-019-0174-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0174-8
- Springer Nature Limited