
Abstract
Migraine is a common disabling primary headache disorder that is ranked as the most common neurological cause of disability worldwide. Women present with migraine much more frequently than men, but the reasons for this difference are unknown. Migraine heritability is estimated to up to 57%, yet much of the genetic risk remains unaccounted for, especially in non-European ancestry populations. To elucidate the etiology of this common disorder, we conduct a multiethnic genome-wide association meta-analysis of migraine, combining results from the GERA and UK Biobank cohorts, followed by a European-ancestry meta-analysis using public summary statistics. We report 79 loci associated with migraine, of which 45 were novel. Sex-stratified analyses identify three additional novel loci (CPS1, PBRM1, and SLC25A21) specific to women. This large multiethnic migraine study provides important information that may substantially improve our understanding of the etiology of migraine susceptibility.
Similar content being viewed by others
Introduction
Migraine is a common disabling disorder that can be accompanied by a wide range of symptoms of varying intensity, including headache pain that is often one-sided, and accompanied by nausea, sound and light sensitivity, and disturbed vision1,2,3. Epidemiological studies have repeatedly found that women have a substantially higher prevalence of migraine compared to men; however, the reasons for this difference are poorly understood1,4,5,6,7,8.
Migraine has a moderate genetic component, with heritability estimates ranging between 0.10 and 0.579,10,11,12,13,14,15,16, depending on the type of estimate (i.e., based on twins studies vs. single-nucleotide polymorphism (SNP)-based heritability). In the past decade, genome-wide association studies (GWASs) have reported 42 genetic loci associated with migraine at genome-wide significance17,18,19,20,21,22. The most recent large genetic study of migraine was conducted by the International Headache Genetics Consortium (IHGC), by combining 22 European ancestry GWASs in a meta-analysis, they identified 38 loci, including 28 novel loci awaiting independent replication21. To our knowledge, no studies have yet conducted a genetic analysis of migraine in large and ethnically diverse cohorts. Therefore, there is a clear need for research to illuminate the genetic underpinnings of migraine.
Here, we present a large and ethnically diverse human genetic study of migraine, including, for the first time to our knowledge, East Asian, African American, and Hispanic/Latino adult individuals. Our study utilizes data from 554,569 individuals (28,852 migraine cases) from the Genetic Epidemiology Research in Adult Health and Aging (GERA) cohort and the UK Biobank (UKB) cohort (Table 1). We also used GWAS summary statistics data from the study of Gormley et al.21, consisting of 375,752 participants (including 59,674 migraine cases) from the IHGC, which were publicly accessible. We conduct multiethnic GWAS meta-analyses, identifying several novel loci, including sex-specific ones. The associated loci provide potential causal variants, candidate genes, and relevant pathways underlying migraine susceptibility.
Results
Multiethnic meta-analysis of GERA and UKB
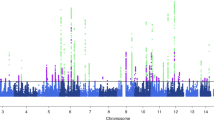
We first undertook GWAS analysis of migraine in the GERA cohort and UKB cohort, stratified by ethnic group, followed by a meta-analysis combining results from GERA and UKB. This combined meta-analysis identified 22 loci associated with migraine (P < 5 × 10−8), of which 10 were novel (Fig. 1, Table 2, Supplementary Fig. 1, and Supplementary Data 1). The effect estimates of the ten lead SNPs at novel loci were consistent across the two studies (Supplementary Fig. 2), and no significant heterogeneity was observed between them (Table 2).

The y-axis represents the –log10(P value); all P values derived from logistic regression model are two-sided. The red dotted line represents the threshold of P = 5 × 10−8, which is the commonly accepted threshold of adjustments for multiple comparisons in GWAS. Locus names in blue are for the novel loci and the ones in dark are for the previously reported ones.
Replication in the IHGC data
We then tested the ten lead SNPs representing each of the ten novel loci for replication in the most recent large genetic study of migraine conducted by the IHGC21. However, as the GWAS summary statistics data, publicly accessible, reported only the SNPs from the Gormley et al. study with a P value of less than 1.0 × 10−5, some of the strongest SNPs reported by Gormley et al. were different than ours. Six loci, including TMEM51, MIR4791-EFHB, LINC00472-RIMS1, FXN, GATA3-SFTA1P, and LINC00310-KCNE2, replicated at Bonferroni significance (P < 0.05/10 novel loci = 5.0 × 10−3) (Supplementary Data 2). Our lead SNPs within the remaining four novel loci (i.e., SLC45A1/RERE, MRGPRE-ZNF195, CALCB, and B3GNTL1-METRNL) were not reported in the publicly accessible GWAS summary statistic from the Gormley et al. study; however, those may replicate at a Bonferroni significance threshold in IHGC (1.0 × 10−5 ≤ P < 5.0 × 10−3) but were not publicly accessible.
Replication of previous migraine GWAS results
We also investigated the lead SNPs within 38 loci associated with migraine at a genome-wide significance level from the most recent and exhaustive GWAS of migraine conducted to date21 (Supplementary Data 3). Ten lead SNPs of the 36 available replicated at a genome-wide level of significance in our combined (GERA + UKB) multiethnic meta-analysis (including rs10218452 at PRDM16, rs2078371 near TSPAN2/NGF, rs1925950 at MEF2D, rs10166942 at TRPM8/HJURP, rs9349379 at PHACTR1, rs28455731 near GJA1, rs186166891 at C7orf10, rs6478241 at ASTN2, rs1024905 near FGF6, and rs11172113 at LRP1) (Supplementary Data 3). Furthermore, 14 additional SNPs replicated at Bonferroni significance (P < 0.05/36 = 1.39 × 10−3), and 4 showed nominal evidence of association (P < 0.05). In contrast, eight SNPs (including rs140002913 near NOTCH4, rs10155855 near DOCK4/IMMP2L, rs2506142 at NRP1, rs561561 at IGSF9B, rs75213074 near WSCD1/NLRP1, rs17857135 at RNF213, rs144017103 near CCM2L/HCK, and rs12845494 near MED14/USP9X) were not validated in the current combined (GERA + UKB) multiethnic meta-analysis (P > 0.05).
Ethnic-specific and conditional analyses
For ethnic groups represented in each cohort, we conducted ethnic-specific meta-analyses of each group. In the European ancestry (GERA non-Hispanic whites + UKB Europeans + IHGC Europeans only; 85,726 migraine cases and 803,292 controls) meta-analysis, we identified 73 loci, of which 35 were additional novel (Supplementary Data 4). To identify independent signals within the 73 genomic regions identified in the European-specific meta-analysis, we performed a multi-SNP-based conditional and joint association analysis (COJO)23, which revealed two additional independent SNPs within the known loci TSPAN2-NGF (rs2207237) and ADAMTSL4-ECM1 (rs7524797). Conducting a GWAS meta-analysis of East Asian-specific cohorts (GERA + UKB East Asian ancestry individuals only; 569 migraine cases and 6619 controls) and a GWAS meta-analysis of African-specific cohorts (GERA + UKB African ancestry individuals only; 504 migraine cases and 10,104 controls) did not result in the identification of genome-wide significant findings. We may have been underpowered to detect effects with statistical significance in those non-European ancestry meta-analyses.
Sex-specific analyses identified additional loci
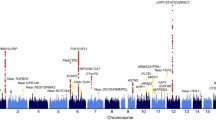
Next, we conducted meta-analyses (GERA + UKB) for migraine by sex. The women-specific (GERA + UKB) meta-analysis (22,500 migraine cases and 279,762 controls) identified 19 loci (P < 5.0 × 10−8), of which three, CPS1 on chromosome 2, PBRM1 on chromosome 3, and SLC25A21 on chromosome 14, were additional novel loci (Fig. 2 and Supplementary Data 5). CPS1 rs1047891, PBRM1 rs11718509, and SLC25A21 rs10150336 were significantly associated with migraine in women (P < 5.0 × 10−8) but not in men (P > 0.05) (Supplementary Fig. 3a–c). Furthermore, among the loci also identified in the multiethnic meta-analysis (GERA + UKB), we observed that ASTN2 locus was significantly differently associated in women and men (ASTN2 rs7858153: OR = 1.11 and P = 1.83 × 10−19 in women; OR = 1.02, P = 0.40 in men; Z = 3.59, P = 1.63 × 10−4) (Supplementary Fig. 3d). Regional association plots illustrate the sex-specific association signals (Supplementary Fig. 3). For each of the four women-specific lead SNPs, we compared the effect allele frequencies (EAF) between cases and controls in women and men separately, as well as between women and men cases and women and men controls (Supplementary Data 6). We found that the EAF of each SNP were near identical when comparing women cases to men cases, or when comparing women controls to men controls. This shows that the differences in association signals in the women and men meta-analyses were not due to the difference in case and/or control EAF in the women and men samples. The men-specific (GERA + UKB) meta-analysis (6352 migraine cases and 245,955 controls) did not result in the identification of additional novel genome-wide significant findings (Fig. 2 and Supplementary Data 7). To further evaluate the shared genetic basis of migraine between women and men, we compared the GWAS results from the two sex-specific meta-analyses by performing a linkage disequilibrium (LD) score regression (LDSC). We observed a high genetic correlation (rg) between women and men for migraine (rg = 0.76, P = 2.39 × 10−15).

Results from the meta-analysis combining women from GERA and UKB are presented on upper panel, while results from the meta-analysis combining men from GERA and UKB are presented on the lower panel. The y-axis represents the –log10(P value); all P values derived from logistic regression model are two-sided. The red dotted line represents the threshold of P = 5 × 10−8, which is the commonly accepted threshold of adjustments for multiple comparisons in GWAS. Locus names in black are for those previously reported. Locus names in bold (i.e., CPS1, PBRM1, and SLC25A21) are for the additional novel loci specific to women (compared to the multiethnic meta-analysis (GERA + UKB)). Novel loci significantly associated (P < 5 × 10−8) with migraine in women are highlighted in blue.
SNP prioritization and annotations
To prioritize variants within the 22 loci identified in the combined multiethnic (GERA + UKB) meta-analysis and within the 73 loci identified in the combined (GERA + UKB + IHGC) European-specific meta-analysis, we applied a Bayesian approach (CAVIARBF)24. For each of the associated signals, we computed each variant’s capacity to explain the identified signal within a 2 Mb window (±1.0 Mb with respect to the original top variant) and derived the smallest set of variants that included the causal variant with 95% probability (95% credible set). For the 22 loci identified in the combined multiethnic (GERA + UKB) meta-analysis, two sets included a unique variant (Supplementary Data 8). These include the previously reported intronic variant rs9349379 at PHACTR121, and the newly identified intergenic variant rs13087932 at MIR4791-EFHB with 100.0% and 97.2% posterior probability of being the causal variants, respectively, suggesting that these variants are more likely to be the true causal variants. For the 73 loci identified in the European-specific meta-analysis, four sets included a unique variant (Supplementary Data 9). In addition to rs9349379 at PHACTR1, we found that the intronic variants rs5763529 at ASCC2 and rs11172113 at LRP1, and the intergenic variant rs28451064 at LINC00310-KCNE2 were more likely to be the true causal variants with 100.0%, 99.9%, and 97.3% posterior probability, respectively.
Gene-based association analysis and gene prioritization
To identify additional genes associated with migraine at a gene level, we conducted a gene-based association analysis using the functional mapping and annotation of genetic associations (FUMA)25 integrative tool using the combined multiethnic GWAS meta-analysis (GERA + UKB) results. FUMA implements Multi-marker Analysis of GenoMic Annotation (MAGMA)26 gene-based analysis, which employs a multiple linear regression approach to properly incorporate LD between genetic variants and to detect multi-variant effects. As 19,933 genes were tested, the P value adjusted for Bonferroni correction was set as P < 2.51 × 10−6 (0.05/19,933). We found significant associations with migraine for 47 genes, with the strongest association for STAT6 (P = 1.24 × 10−23), followed by UFL1 (P = 7.26 × 10−19), and FHL5 (P = 1.25 × 10−18) (Supplementary Data 10). Out of the 47 genes, 9 were located outside the loci identified in the current study, including PRKCE, RCHY1, THAP6, MAPK9, RP11-508N12.4, LRCH1, PNKP, AKT1S1, and TBC1D17.
To prioritize genes within the 73 loci identified in the combined (GERA + UKB + IHGC) European-specific meta-analysis, we used the DEPICT27 integrative tool. DEPICT gene prioritization analysis detected 15 genes, of which 9 were within novel migraine-associated loci, to prioritize after false-discovery rate (FDR) correction (Supplementary Data 11). These included: LEPR on chromosome 1, TJP2 on chromosome 9, AMBRA1 on chromosome 11, HOXB2, HOXB3, HOXB6, and POLR2A on chromosome 17, TGFB1 on chromosome 19, and JAG1 on chromosome 20.
Biological pathway annotations and prioritization
While DEPICT gene-set enrichment analysis using independent genome-wide significant genetic variants from the combined (GERA + UKB + IHGC) European-specific meta-analysis did not detect pathways to prioritize after FDR correction (Supplementary Data 12), FUMA25 gene-set enrichment analysis based on the GWAS multiethnic meta-analysis (GERA + UKB) results highlighted many gene-sets involved in the flavonoid glucuronide biosynthetic process, the ascorbate and aldarate metabolic pathways, the response to endogenous stimulus or wounding, and the regulation of muscle contraction or system process (Supplementary Data 13).
FUMA tissue expression quantitative trait loci (eQTL) specificity analysis highlighted the sigmoid colon as the main tissue for which expression was affected by migraine-associated variants, followed by the esophagus muscularis and gastroesophageal junction (P < 9.43 × 10−4; Bonferroni significance after correcting for 53 GTEx tissues tested—Supplementary Fig. 4 and Supplementary Data 14). In contrast, DEPICT tissue-enrichment analysis using independent genome-wide significant genetic variants from the combined (GERA + UKB + IHGC) European-specific meta-analysis identified two tissues or cell type annotations to prioritize after FDR correction: the arteries (cardiovascular system), consistent with the previously reported Gormley et al. study21, and the serous membrane (Supplementary Data 15).
Genetic correlation between migraine and other phenotypes
Genome-wide genetic correlations of migraine were calculated with a total of 772 complex traits and diseases by comparing allelic effects using a LDSC with the European-specific migraine meta-analysis (GERA + UKB) summary statistics (Methods). A total of 75 significant genetic correlations were observed (P < 6.48 × 10−5 which corresponds to 0.05/772 phenotypes tested; Supplementary Data 16). Among those 75 genetic correlations, 38 reached genome-wide level of significance (Supplementary Fig. 5). The strongest positive correlations were observed with medications for pain relief, constipation, or heartburn; neck, shoulder or back pain; alcohol intake frequency; mood swings, anxiety, depression or neuroticism; and sleeplessness or insomnia. There was also a strong negative correlation with physical activity.
Discussion
In this study, a multiethnic meta-analysis combining the GERA and UKB cohorts and a meta-analysis of migraine across European ancestry individuals identified 79 loci, of which 45 were novel. Our multiethnic meta-analysis also validated 78% of the migraine-associated loci identified in the most recent and exhaustive GWAS of migraine conducted to date21. Furthermore, our sex-stratified analyses identified three additional novel loci (CPS1, PBRM1, and SLC25A21) specific to women.
The identified loci give new insight and additional evidence about the genes and pathways/systems underlying migraine susceptibility. For instance, we identified a new region associated with migraine at 17q21 and our DEPICT gene analysis prioritized three members of the Antp homeobox family genes (i.e., HOXB2, HOXB3, and HOXB6) at this region that encode proteins with a homeobox DNA-binding domain. Those three genes have been involved in the early development28,29,30,31,32 (i.e., hindbrain, nervous system, or epidermal development) and common variants in HOXB3 have been shown to be associated with motion sickness, which is a condition that shares underlying genetic factors with migraine33. Our DEPICT gene analysis also prioritized TGFB1 at the novel 19q13 migraine-associated locus. TGFB1 encodes the transforming growth factor beta 1 protein that is a multifunctional proinflammatory cytokine that regulates cell proliferation, differentiation, and growth. Early works suggested that TGFB1 could play a role in migraine susceptibility. Plasma level of TGFB1 has been shown to increase in patients with migraine during headache-free periods compared to healthy subjects without headache34. Another study investigated the TGFB1 genotype in pediatric migraine patients and reported significant differences between control and migraine patients35. Our DEPICT gene analysis also prioritized JAG1 at the novel 20p12 migraine-associated locus. JAG1 encodes that the jagged 1 protein is the ligand for the receptor notch 1, which is involved in signaling processes. JAG1 plays a role in the formation of blood cellular components36,37,38 and has been involved in the pathogenesis of patent foramen ovale, which is an atrial septal deformity associated with major causes of morbidity, including stroke and migraine39,40. Future investigations may provide insights into how these genes influence migraine susceptibility.
Our study also reported, for the first time to our knowledge, sex-specific loci associated with migraine susceptibility in women but not in men. Previous studies18,41 evaluated the concordance of genetic risk for migraine between women and men but did not reveal any heterogeneity in the effect sizes of the genome-wide significant loci. Among the four sex-specific loci identified in the current study, two, PBRM1 and ASTN2, have been involved in susceptibility to bipolar disorder and other psychiatric phenotypes. PBRM1 encodes polybromo 1, which is a subunit of ATP-dependent chromatin-remodeling complexes. Common genetic polymorphisms at PBRM1 (also named 3p21.1 locus) have been implicated in susceptibility to bipolar disorder, as well as major depression and schizophrenia42,43,44,45,46. ASTN2 encodes astrotactin 2 that is expressed in the brain and is involved in neuronal migration. Deletions of ASTN2 have been associated with schizophrenia and other psychiatric and neurodevelopmental disorders47,48,49. Similarly, our genetic correlation results indicate that migraine was significantly correlated with mood swings, anxiety, depression, or neuroticism, consistent with a previous genetic correlation analysis study50. Thus, these findings suggest that migraine shares common variation risk with psychiatric disorders, such as schizophrenia, for which genome-wide significant loci involved in brain function have been reported51,52.
Among the sex-specific loci associated with migraine susceptibility in women but not in men, less is known about the role of the identified CPS1 and SLC25A21 in regard to the biologic pathways underlying migraine. CPS1 encodes a mitochondrial enzyme named carbamoyl-phosphate synthase 1 that catalyzes synthesis of carbamoyl phosphate from ammonia and bicarbonate. Mutations in this gene have been associated with metabolic deficiencies such as urea cycle disorder and hyperammonemia53,54. Individuals with a CPS1 deficiency can present with a wide range of clinical manifestations, including headache, behavioral or psychiatric problems, learning disabilities, sleep disorder, periodic vomiting, seizures, coma, and even death55,56,57. SLC25A21 (also known as ODC1) encodes the solute carrier family 25 member 21, which is essential for ammonium fixation and lysine biosynthesis58. While rare mutations in this gene cause a syndromic neurometabolic disorder associated with macrocephaly, developmental delay, alopecia, and dysmorphic features59,60, common polymorphisms have been associated with smoking behaviors, and could contribute to Alzheimer’s disease61,62. Thus, our findings will help to better understand the biological mechanisms underlying migraine susceptibility, particularly in women, even if follow-up experimental studies are needed to validate the role of the identified genes in migraine susceptibility.
Our results also illustrate the association between disorders of the large bowel and migraine susceptibility, as our eQTL analysis highlighted the sigmoid colon tissue for which expression was affected by migraine-associated variants. Similarly, our multiethnic meta-analysis results identified variants in CALCB associated with migraine susceptibility. CALCB encodes the calcitonin-related polypeptide beta (CGRP), which has been shown to contribute to migraine63,64,65. Several monoclonal antibodies targeting CGRP or its receptor have been proven to be effective therapeutics for the preventive treatment of migraine66,67 and have been recently approved by the U.S. Food and Drug Administration68,69. In parallel, common genetic polymorphisms at CALCB have been reported to contribute to diverticular disease that is a common of intestinal neuromuscular function70. Of note, several epidemiologic studies reported that gastrointestinal disorders such as the “irritable bowel syndrome” are the most commonly reported comorbidities associated with migraine71,72. Our results suggest that the relationship between the intestine and migraine, also referred to “gut-brain axis,“ could have some genetic origin.
Our genetic correlation results identified the strongest positive correlations between migraine and neck, shoulder or back pain, suggesting shared genetic factors underlying those conditions. This result is consistent with early work demonstrating that migraine patients have higher pain responses in the splenius and temporalis muscles after a cognitive stress test compared to controls, and their muscle pain responses are regional (i.e., more pain in the neck and trapezius)73. Thus, our results support the concept that sensitization of pain pathways and the muscular system seem to be important in migraine susceptibility.
We recognize several potential limitations of our study. First, it is important to note phenotypic differences for migraine between the two study cohorts. Although migraine cases in GERA were identified in the Kaiser Permanente Northern California (KPNC) electronic health record system using our previously described and validated migraine probability algorithm (MPA)74, which is based on migraine-specific prescriptions and International Classification of Disease, Ninth (ICD9) or Tenth Revision (ICD10) diagnosis codes, most of the migraine cases in UKB were based on self-reported data. This may lead to phenotype misclassification that may have affected, for instance, the high positive genetic correlation between migraine and neck, shoulder, or back pain. However, our meta-analysis combining GERA and UKB results showed consistency of the SNPs effect estimates between cohorts. Furthermore, the previously reported associations21 were well validated in our multiethnic meta-analysis and our novel loci were well replicated in the most recent large genetic study of migraine conducted by the IHGC21. Second, because of the limited sample of men cases (compared to women), we may have been underpowered to detect effects with statistical significance in the men-specific analysis.
In conclusion, our results identified additional loci that contribute to migraine susceptibility and represent potential candidates for the development of new therapeutic targets for this common neurological cause of disability. Although these findings will help to better understand the etiology of migraine susceptibility, additional genetic studies are needed to validate these associations in more large cohorts.
Methods
GERA
The GERA cohort contains genome-wide genotype, clinical, and demographic data of over 110,000 adult members from mainly four ethnic groups (non-Hispanic white, Hispanic/Latino, East Asian, and African American) of the KPNC integrated healthcare system75,76. The Institutional Review Board of the Kaiser Foundation Research Institute approved all study procedures. Patients with migraine were identified in the KPNC electronic health record system using the previously validated MPA74 (Supplementary Data 17), which is based on migraine-specific prescriptions and ICD9 diagnosis code: 346.XX and ICD10: G43.XX. We defined migraine cases as patients with a score >10 on the MPA (any evidence of migraine). After excluding individuals with any evidence of headache without a migraine diagnosis, as well as individuals with a MPA score = 10, our control group included all the non-cases. In total, 11,320 migraine cases and 60,282 controls from GERA were included in this study.
Protocols for participant genotyping, data collection and quality control (QC) have been described in detail76. Briefly, GERA participants’ DNA samples were extracted from Oragene kits (DNA Genotek Inc., Ottawa, ON, Canada) at KPNC and genotyped at the Genomics Core Facility of the University of California, San Francisco. DNA samples were genotyped at over 665,000 genetic markers on four ethnic-specific Affymetrix Axiom arrays (Affymetrix, Santa Clara, CA, USA) optimized for European, Latino, East Asian, and African American individuals77,78. Genotype QC procedures and imputation were conducted on an array-wise basis76, after an updated genotyping algorithm with an advanced normalization step specifically for SNPs in batches not recommended or flagged by the outlier plate detector than has previously been done. Subsequently, variants were excluded if: >3 clusters were identified; the number of batches was <38/42 (EUR array), <3/5 (AFR), <3/6 (EAS), or <7/9 (LAT); and the ratio of expected allele frequency variance across packages was <100 (EUR), <50 (AFR), <100 (EAS), <200 (LAT). On the EUR array, variants were additionally excluded if heterozygosity >0.52 or <0.02, and if an association test between Reagent kit v1.0 and v2.0 had P < 10−4. Imputation was done by array, and we additionally removed variants with call rates <90%. Genotypes were then pre-phased with Eagle79 v2.3.2, and then imputed with Minimac380 v2.0.1, using two reference panels. Variants were preferred if present in the EGA release of the Haplotype Reference Consortium (HRC; n = 27,165) reference panel81, and from the 1000 Genomes Project Phase III release if not (n = 2504; e.g., indels)82.
UK Biobank
The UKB is a large prospective study following the health of approximately 500,000 participants from five ethnic groups (European, East Asian, South Asian, African British, and mixed ancestries) resident in the UK aged between 40 and 69 years at the baseline recruitment visit83,84. Demographic information and medical history were ascertained through touch-screen questionnaires. Participants also underwent a wide range of physical and cognitive assessments, including blood sampling. Migraine cases (N = 17,532) were defined as participants with a self-reported migraine (data field 20002 code 1265) and/or a diagnosis code for migraine (ICD10: G43). After excluding participants who self-reported headaches (data field 20002, code 1436) and/or who had a diagnosis code for headaches (ICD10: G44), the control group included 465,435 participants who were not cases. Phenotyping, genotyping, and imputation were carried out by members of the UKB team. Imputation to the Haplotype Reference Consortium reference panel has been described (www.ukbiobank.ac.uk). Following QC, over 10 million variants in 487,622 individuals were tested for migraine adjusting for age, sex, and genetic ancestry principal components (PCs). The analyses presented in this paper were carried out under UKB Resource project #14105.
The International Headache Genetics Consortium (IHGC)
GWAS summary statistics data (SNPs with P < 1.0 × 10−5) from the study of Gormley et al.21, consisting of 375,000 participants from the IHGC, were publicly accessible (http://eagle-i.itmat.upenn.edu/i/00000155-e1db-73aa-c956-e86e80000000).
GWAS and adjustment in GERA
We first analyzed each ethnic group (non-Hispanic white, Hispanic/Latino, East Asian, and African American) separately. We ran a logistic regression of migraine and each SNP using PLINK85 v1.9 (www.cog-genomics.org/plink/1.9/) adjusting for age, sex, and ancestry PCs, which were previously75 assessed within each ethnic group using Eigenstrat86 v4.2. We included as covariates the top ten ancestry PCs for the non-Hispanic whites, whereas we included the top six ancestry PCs for the three other ethnic groups. To adjust for genetic ancestry, we also included the percentage of Ashkenazi (ASHK) ancestry as a covariate for the non-Hispanic white sample analyses75. For comparison, the GWAS analyses were also conducted using a new approach accounting for relatedness that fits a whole genome regression model, implemented in REGENIEv2.0.287 (https://rgcgithub.github.io/regenie/).
GWAS meta-analyses
First, a meta-analysis of migraine was conducted in GERA to combine the results of the 4 ethnic groups using the R88 (https://www.R-project.org) package “meta.” Similarly, a meta-analysis was conducted in UKB to combine the results of the five ethnic groups. The meta-analysis GWAS results generated using REGENIE were similar compared to the results generated using PLINK (Supplementary Data 18 and Supplementary Fig. 6). Three ethnic-specific meta-analyses were also performed: (1) combining European-specific samples (i.e., GERA non-Hispanic whites + UKB Europeans + IHGC Europeans); (2) combining East Asian-specific samples (i.e., GERA and UKB East Asians); and (3) combining African-specific samples (i.e., GERA African Americans and UKB Africans). A meta-analysis was then conducted to combine the results from GERA and UKB. The overall (GERA + UKB) meta-analysis results generated using REGENIE were similar compared to the results generated using PLINK (Supplementary Fig. 6). Two sex-specific meta-analyses were also performed: (1) combining women from GERA and UKB; and (2) combining men from GERA and UKB. Fixed-effects summary estimates were calculated for an additive model. We also estimated heterogeneity index, I2 (0–100%) and P value for Cochrane’s Q statistic among different groups, and studies. For each locus, we defined the top SNP as the most significant variant within a 2 Mb window. Novel loci were defined as those that were located over 1 Mb apart from any previously reported locus17,18,20,21.
Conditional and joint (COJO) analysis
A multi-SNP-based COJO23 was performed on the combined European-specific (GERA non-Hispanic whites + UKB Europeans + IHGC Europeans) meta-analysis results to potentially identify independent signals within the 73 identified genomic regions. To calculate LD patterns, we used 10,000 randomly selected samples from GERA non-Hispanic white ethnic group as a reference panel. A P value less than 5.0 × 10−8 was considered as the significance threshold for this COJO analysis.
Variants prioritization
To prioritize variants within the identified genomic regions for follow-up functional evaluation, a Bayesian approach (CAVIARBF)24 was used, which is available publicly at https://bitbucket.org/Wenan/caviarbf. Each variant’s capacity to explain the identified signal within a 2 Mb window (±1.0 Mb with respect to the original top variant) was computed for each identified genomic region. Then the smallest set of variants that included the causal variant with 95% probability (95% credible set) was derived.
FUMA for gene-based association analysis and gene-set analysis
To prioritize genes and biological pathways, and highlight gene-set and tissue/cell enrichments within the 22 migraine-associated loci identified in the combined multiethnic (GERA + UKB) meta-analysis, we used the FUMA25 integrative tool. FUMA uses input GWAS summary statistics to compute gene-based P values (gene analysis) and gene-set P value (gene-set analysis) using the MAGMA26 tool, which employs multiple regression to obtain gene-based and gene-set P values. GWAS summary statistics from the combined multiethnic (GERA + UKB) meta-analysis served as input for the “SNP2GENE” function. We used 10,000 random samples from GERA non-Hispanic whites as a reference panel. This “SNP2GENE” function provides extensive functional annotation for all SNPs in genomic areas identified and prioritizes gene-set and genes (using the MAGMA gene-set association analysis and the MAGMA gene analysis, respectively) for the next FUMA function, which is “GENE2FUNC.” Then, this “GENE2FUNC” function annotates the prioritized genes in biological context. For gene analysis, the gene-based P value is computed for protein-coding genes by mapping SNPs to genes if SNPs are located within the genes. For gene-set analysis, the gene-set P value is computed using the gene-based P value for 4728 curated gene-sets (including canonical pathways) and 6166 GO terms obtained from MsigDB v5.2. For both analyses, the default MAGMA setting (SNP-wise model for gene analysis and competitive model for gene-set analysis) is used, and the Bonferroni correction (gene) or FDR (gene-set) was used to correct for multiple testing. For the MAGMA gene-based association analysis conducted on the combined (GERA + UKB) meta-analysis results, the P value adjusted for Bonferroni correction was set as P < 2.51 × 10−6 (0.05/19,933 genes tested).
FUMA tissue eQTL specificity
To highlight and visualize tissue eQTL enrichments within the 22 migraine-associated genomic regions identified in the combined multiethnic (GERA + UKB) meta-analysis, we used the FUMA25 integrative tool. FUMA is an integrative web-based platform that accommodates eQTL, and provides tissue enrichment results for each of 53 tissue types based on the genotype-tissue expression (GTEx) v6 RNA-seq data89.
DEPICT
To prioritize genes and biological pathways, and highlight gene-set and tissue/cell enrichments within the 73 migraine-associated loci identified in the combined (GERA + UKB + IHGC) European meta-analysis, we used the following integrative tool: DEPICT27. All independent genome-wide significant genetic variants (P < 5.0 × 10−8) served as input, and as the reference panel, we used 10,000 random samples from GERA non-Hispanic white ethnic group. Genes, gene-sets, and tissue/cell annotations that achieved a nominal significance level of 0.05 after FDR correction were subsequently prioritized.
Genetic correlations
To estimate the genetic correlation of migraine with more than 700 diseases/traits from different publicly available resources/consortia, we used the LD Hub web interface90, which performs automated LDSC. In the LDSCs, we included only HapMap3 SNPs with MAF > 0.01. Genetic correlations were considered significant after Bonferroni adjustment for multiple testing (P < 6.48 × 10−5 which corresponds to 0.05/772 phenotypes tested).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The GERA genotype data are available upon application to the KP Research Bank (https://researchbank.kaiserpermanente.org/). The combined multiethnic (GERA + UKB) meta-analysis GWAS summary statistics are available from the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics), study accession number GCST90000016. GWAS summary statistics data (SNPs with P < 1.0 × 10−5) from the study of Gormley et al.21 are publicly accessible (http://eagle-i.itmat.upenn.edu/i/00000155-e1db-73aa-c956-e86e80000000).
References
Steiner, T. J., Stovner, L. J. & Birbeck, G. L. Migraine: the seventh disabler. J. Headache Pain. 14, 1 (2013).
Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia 38, 1–211 (2018).
Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386, 743–800 (2015).
Wang, R. et al. Gender differences of cognitive function in migraine patients: evidence from event-related potentials using the oddball paradigm. J. Headache Pain. 15, 6 (2014).
Yu, S. et al. The prevalence and burden of primary headaches in China: a population-based door-to-door survey. Headache 52, 582–591 (2012).
Maleki, N. et al. Her versus his migraine: multiple sex differences in brain function and structure. Brain 135, 2546–2559 (2012).
Liu, J. et al. Gender-related differences in the dysfunctional resting networks of migraine suffers. PLoS One 6, e27049 (2011).
Burch, R. C., Loder, S., Loder, E. & Smitherman, T. A. The prevalence and burden of migraine and severe headache in the United States: updated statistics from government health surveillance studies. Headache 55, 21–34 (2015).
Polderman, T. J. et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 47, 702–709 (2015).
Ge, T., Chen, C. Y., Neale, B. M., Sabuncu, M. R. & Smoller, J. W. Phenome-wide heritability analysis of the UK Biobank. PLoS Genet. 13, e1006711 (2017).
Yang, Y. et al. Molecular genetic overlap between migraine and major depressive disorder. Eur. J. Hum. Genet. 26, 1202–1216 (2018).
Svensson, D. A., Larsson, B., Waldenlind, E. & Pedersen, N. L. Shared rearing environment in migraine: results from twins reared apart and twins reared together. Headache 43, 235–244 (2003).
Mulder, E. J. et al. Genetic and environmental influences on migraine: a twin study across six countries. Twin Res. 6, 422–431 (2003).
Schurks, M. Genetics of migraine in the age of genome-wide association studies. J. Headache Pain. 13, 1–9 (2012).
Cox, H. C. et al. Heritability and genome-wide linkage analysis of migraine in the genetic isolate of Norfolk Island. Gene 494, 119–123 (2012).
Honkasalo, M. L. et al. Migraine and concomitant symptoms among 8167 adult twin pairs. Headache 35, 70–78 (1995).
Anttila, V. et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat. Genet. 42, 869–873 (2010).
Anttila, V. et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet. 45, 912–917 (2013).
Chasman, D. I. et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat. Genet. 43, 695–698 (2011).
Freilinger, T. et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 44, 777–782 (2012).
Gormley, P. et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 48, 856–866 (2016).
Chang, X. et al. Common variants at 5q33.1 predispose to migraine in African-American children. J. Med Genet. 55, 831–836 (2018).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012). S1-3.
Chen, W. et al. Fine mapping causal variants with an approximate bayesian method using marginal test statistics. Genetics 200, 719–736 (2015).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 11, e1004219 (2015).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015).
Sauvageau, G. et al. Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc. Natl Acad. Sci. USA. 91, 12223–12227 (1994).
Manzanares, M. et al. Krox20 and kreisler co-operate in the transcriptional control of segmental expression of Hoxb3 in the developing hindbrain. EMBO J. 21, 365–376 (2002).
Chan, K. K. et al. Hoxb3 vagal neural crest-specific enhancer element for controlling enteric nervous system development. Dev. Dyn. 233, 473–483 (2005).
Sham, M. H. et al. The zinc finger gene Krox20 regulates HoxB2 (Hox2.8) during hindbrain segmentation. Cell 72, 183–196 (1993).
Komuves, L. G. et al. Changes in HOXB6 homeodomain protein structure and localization during human epidermal development and differentiation. Dev. Dyn. 218, 636–647 (2000).
Hromatka, B. S. et al. Genetic variants associated with motion sickness point to roles for inner ear development, neurological processes and glucose homeostasis. Hum. Mol. Genet. 24, 2700–2708 (2015).
Ishizaki, K. et al. Increased plasma transforming growth factor-beta1 in migraine. Headache 45, 1224–1228 (2005).
Saygi, S. et al. TGF-beta1 genotype in pediatric migraine patients. J. Child Neurol. 30, 27–31 (2015).
Varnum-Finney, B. et al. The Notch ligand, Jagged-1, influences the development of primitive hematopoietic precursor cells. Blood 91, 4084–4091 (1998).
Walker, L. et al. The Notch/Jagged pathway inhibits proliferation of human hematopoietic progenitors in vitro. Stem Cells 17, 162–171 (1999).
Karanu, F. N. et al. The notch ligand jagged-1 represents a novel growth factor of human hematopoietic stem cells. J. Exp. Med. 192, 1365–1372 (2000).
Elliott, G. C., Gurtu, R., McCollum, C., Newman, W. G. & Wang, T. Foramen ovale closure is a process of endothelial-to-mesenchymal transition leading to fibrosis. PLoS One 9, e107175 (2014).
Sztajzel, R., Genoud, D., Roth, S., Mermillod, B. & Le Floch-Rohr, J. Patent foramen ovale, a possible cause of symptomatic migraine: a study of 74 patients with acute ischemic stroke. Cerebrovasc. Dis. 13, 102–106 (2002).
Nyholt, D. R. et al. Concordance of genetic risk across migraine subgroups: impact on current and future genetic association studies. Cephalalgia 35, 489–499 (2015).
Vassos, E. et al. Replication study and meta-analysis in European samples supports association of the 3p21.1 locus with bipolar disorder. Biol. Psychiatry 72, 645–650 (2012).
Yang, Z. et al. The genome-wide risk alleles for psychiatric disorders at 3p21.1 show convergent effects on mRNA expression, cognitive function, and mushroom dendritic spine. Mol. Psychiatry 25, 48–66 (2020).
Scott, L. J. et al. Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc. Natl Acad. Sci. USA. 106, 7501–7506 (2009).
McMahon, F. J. et al. Meta-analysis of genome-wide association data identifies a risk locus for major mood disorders on 3p21.1. Nat. Genet. 42, 128–131 (2010).
Williams, H. J. et al. Most genome-wide significant susceptibility loci for schizophrenia and bipolar disorder reported to date cross-traditional diagnostic boundaries. Hum. Mol. Genet. 20, 387–391 (2011).
Wang, K. S., Liu, X. F. & Aragam, N. A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr. Res. 124, 192–199 (2010).
Arioka, Y., Kushima, I., Kubo, H., Mori, D. & Ozaki, N. Induced pluripotent stem cells derived from a schizophrenia patient with ASTN2 deletion. Stem Cell Res. 30, 81–84 (2018).
Gazzellone, M. J. et al. Uncovering obsessive-compulsive disorder risk genes in a pediatric cohort by high-resolution analysis of copy number variation. J. Neurodev. Disord. 8, 36 (2016).
Siewert, K. M. et al. Cross-trait analyses with migraine reveal widespread pleiotropy and suggest a vascular component to migraine headache. Int J. Epidemiol. 49, 1022–1031 (2020).
Elvsashagen, T. et al. The genetic architecture of human brainstem structures and their involvement in common brain disorders. Nat. Commun. 11, 4016 (2020).
Schmidt-Kastner, R., Guloksuz, S., Kietzmann, T., van Os, J. & Rutten, B. P. F. Analysis of GWAS-derived schizophrenia genes for links to ischemia-hypoxia response of the brain. Front Psychiatry 11, 393 (2020).
Xu, J., Zhang, A. & Huang, F. Biallelic mutations in carbamoyl phosphate synthetase 1 induced hyperammonemia in a neonate: a case report. Exp. Ther. Med. 20, 623–629 (2020).
Nitzahn, M. et al. Split AAV-mediated gene therapy restores ureagenesis in a murine model of carbamoyl phosphate synthetase 1 deficiency. Mol. Ther. 28, 1717–1730 (2020).
Fan, L. et al. Molecular, biochemical, and clinical analyses of five patients with carbamoyl phosphate synthetase 1 deficiency. J. Clin. Lab Anal. 34, e23124 (2020).
Haberle, J. et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J. Inherit. Metab. Dis. 42, 1192–1230 (2019).
Zhou, Q., Huang, H., Ma, L. & Zhu, T. The application of next-generation sequencing (NGS) in neonatal-onset urea cycle disorders (UCDs): clinical course, metabolomic profiling, and genetic findings in nine chinese hyperammonemia patients. Biomed. Res Int. 2020, 5690915 (2020).
Scarcia, P., Palmieri, L., Agrimi, G., Palmieri, F. & Rottensteiner, H. Three mitochondrial transporters of Saccharomyces cerevisiae are essential for ammonium fixation and lysine biosynthesis in synthetic minimal medium. Mol. Genet Metab. 122, 54–60 (2017).
Rodan, L. H. et al. Gain-of-function variants in the ODC1 gene cause a syndromic neurodevelopmental disorder associated with macrocephaly, alopecia, dysmorphic features, and neuroimaging abnormalities. Am. J. Med Genet A. 176, 2554–2560 (2018).
Bupp, C. P., Schultz, C. R., Uhl, K. L., Rajasekaran, S. & Bachmann, A. S. Novel de novo pathogenic variant in the ODC1 gene in a girl with developmental delay, alopecia, and dysmorphic features. Am. J. Med Genet A. 176, 2548–2553 (2018).
Minica, C. C. et al. Pathways to smoking behaviours: biological insights from the Tobacco and Genetics Consortium meta-analysis. Mol. Psychiatry 22, 82–88 (2017).
Sun, J. et al. Hidden risk genes with high-order intragenic epistasis in Alzheimer’s disease. J. Alzheimers Dis. 41, 1039–1056 (2014).
van Rossum, D., Hanisch, U. K. & Quirion, R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci. Biobehav Rev. 21, 649–678 (1997).
Russo, A. F. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev. Pharm. Toxicol. 55, 533–552 (2015).
Pellesi, L., Guerzoni, S. & Pini, L. A. Spotlight on anti-CGRP monoclonal antibodies in migraine: the clinical evidence to date. Clin. Pharm. Drug Dev. 6, 534–547 (2017).
Edvinsson, L. CGRP antibodies as prophylaxis in migraine. Cell 175, 1719 (2018).
Edvinsson, L., Haanes, K. A., Warfvinge, K. & Krause, D. N. CGRP as the target of new migraine therapies – successful translation from bench to clinic. Nat. Rev. Neurol. 14, 338–350 (2018).
King, C. T. et al. Discovery of the migraine prevention therapeutic aimovig (Erenumab), the first FDA-approved antibody against a G-protein-coupled receptor. ACS Pharm. Transl. Sci. 2, 485–490 (2019).
Tepper, S. J. History and review of anti-calcitonin gene-related peptide (CGRP) therapies: from translational research to treatment. Headache 58(Suppl 3), 238–275 (2018).
Schafmayer, C. et al. Genome-wide association analysis of diverticular disease points towards neuromuscular, connective tissue and epithelial pathomechanisms. Gut 68, 854–865 (2019).
El-Metwally, A. et al. The epidemiology of migraine headache in Arab countries: a systematic review. ScientificWorldJournal 2020, 4790254 (2020).
Arzani, M. et al. Gut-brain Axis and migraine headache: a comprehensive review. J. Headache Pain. 21, 15 (2020).
Leistad, R. B., Sand, T., Westgaard, R. H., Nilsen, K. B. & Stovner, L. J. Stress-induced pain and muscle activity in patients with migraine and tension-type headache. Cephalalgia 26, 64–73 (2006).
Pressman, A. et al. Prevalence of migraine in a diverse community–electronic methods for migraine ascertainment in a large integrated health plan. Cephalalgia 36, 325–334 (2016).
Banda, Y. et al. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics 200, 1285–1295 (2015).
Kvale, M. N. et al. Genotyping informatics and quality control for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics 200, 1051–1060 (2015).
Hoffmann, T. J. et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics 98, 79–89 (2011).
Hoffmann, T. J. et al. Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics 98, 422–430 (2011).
Loh, P. R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Birney, E. & Soranzo, N. Human genomics: the end of the start for population sequencing. Nature 526, 52–53 (2015).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Allen, N. E., Sudlow, C., Peakman, T., Collins, R. & Biobank, U. K. UK biobank data: come and get it. Sci. Transl. Med. 6, 224ed4 (2014).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Mbatchou J. et al. Computationally efficient whole genome regression for quantitative and binary traits. bioRxiv. 2020:2020.06.19.162354.
R: A Language and Environment for Statistical Computing. The R Foundation for Statistical Computing. 2014.
GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Zheng, J. et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017).
Acknowledgements
We are grateful to the Kaiser Permanente Northern California members who have generously agreed to participate in the Kaiser Permanente Research Program on Genes, Environment, and Health. Support for participant enrollment, survey completion, and biospecimen collection for the RPGEH was provided by the Robert Wood Johnson Foundation, the Wayne and Gladys Valley Foundation, the Ellison Medical Foundation, and Kaiser Permanente Community Benefit Programs. Genotyping of the GERA cohort was funded by a grant from the National Institute on Aging, National Institute of Mental Health, and National Institute of Health Common Fund (RC2 AG036607). H.C. and E.J. were supported by the National Eye Institute (NEI) grant R01 EY027004, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R01 DK116738, and by the National Cancer Institute (NCI) R01CA2416323. The Genetics and Comorbidity of Migraine study was funded by a grant from the National Institute for Neurological Disorders and Stroke (NINDS) R01NS080863.
Author information
Authors and Affiliations
Contributions
H.C., A.L.A., and A.R.P. contributed to study conception and design. T.J.H. and E.J. were involved in the genotyping and quality control of the GERA samples. T.J.H. performed the imputation analyses in the GERA cohort. A.S.J. and B.H.H. extracted phenotype and other data for the GERA subjects based on EHRs. J.Y. performed statistical analyses and in silico analyses. H.C., A.L.A., and A.R.P. interpreted the results of analyses. H.C. drafted the manuscript. H.C., T.J.H., E.J., A.L.A., and A.R.P. critically revised the manuscript for key intellectual content.
Corresponding authors
Ethics declarations
Competing interests
H.C. is an Editorial Board Member for Communications Biology, but was not involved in the editorial review of, or the decision to publish this article. The authors declare no other competing interests.
Additional information
Peer review information Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary handling editor: Brooke LaFlamme. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choquet, H., Yin, J., Jacobson, A.S. et al. New and sex-specific migraine susceptibility loci identified from a multiethnic genome-wide meta-analysis. Commun Biol 4, 864 (2021). https://doi.org/10.1038/s42003-021-02356-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02356-y
- Springer Nature Limited
This article is cited by
-
Identifying therapeutic target genes for migraine by systematic druggable genome-wide Mendelian randomization
The Journal of Headache and Pain (2024)
-
Genetics of migraine: where are we now?
The Journal of Headache and Pain (2023)
-
Lipids, lipid-modifying drug target genes and migraine: a Mendelian randomization study
The Journal of Headache and Pain (2023)
-
Identifying causal genes for migraine by integrating the proteome and transcriptome
The Journal of Headache and Pain (2023)
-
Migraine, chronic kidney disease and kidney function: observational and genetic analyses
Human Genetics (2023)