
Abstract
Background
Obsessive-compulsive disorder (OCD) is a heterogeneous neuropsychiatric condition, thought to have a significant genetic component. When onset occurs in childhood, affected individuals generally exhibit different characteristics from adult-onset OCD, including higher prevalence in males and increased heritability. Since neuropsychiatric conditions are associated with copy number variations (CNVs), we considered their potential role in the etiology of OCD.
Methods
We genotyped 307 unrelated pediatric probands with idiopathic OCD (including 174 that were part of complete parent-child trios) and compared their genotypes with those of 3861 population controls, to identify rare CNVs (<0.5 % frequency) of at least 15 kb in size that might contribute to OCD.
Results
We uncovered de novo CNVs in 4/174 probands (2.3 %). Our case cohort was enriched for CNVs in genes that encode targets of the fragile X mental retardation protein (nominal p = 1.85 × 10−03; FDR=0.09), similar to previous findings in autism and schizophrenia. These results also identified deletions or duplications of exons in genes involved in neuronal migration (ASTN2), synapse formation (NLGN1 and PTPRD), and postsynaptic scaffolding (DLGAP1 and DLGAP2), which may be relevant to the pathogenesis of OCD. Four cases had CNVs involving known genomic disorder loci (1q21.1-21.2, 15q11.2-q13.1, 16p13.11, and 17p12). Further, we identified BTBD9 as a candidate gene for OCD. We also sequenced exomes of ten “CNV positive” trios and identified in one an additional plausibly relevant mutation: a 13 bp exonic deletion in DRD4.
Conclusions
Our findings suggest that rare CNVs may contribute to the etiology of OCD.
Similar content being viewed by others


Background
Obsessive-compulsive disorder (OCD) is a heterogeneous neuropsychiatric condition characterized by persistent intrusive unwanted thoughts (obsessions) and repetitive behaviours (compulsions) [1]. It is among the most common neuropsychiatric disorders, with a lifetime prevalence of ~2 % [2]. Males and females are affected in similar numbers (1:1 sex ratio), although males typically experience symptoms earlier than do females [1, 2]. First-degree relatives of individuals with OCD are significantly more likely to have OCD than are relatives of controls [1]. Classification of individuals into more homogeneous subtypes has been used to increase the likelihood of identifying causal factors [3]. One such approach takes advantage of the bimodal distribution of age-of-onset, with one peak in childhood and another in mid-adulthood [4]. The 30–50 % of individuals who first experience symptoms before age 18 may represent a distinct subtype of OCD [5]. On average, these individuals have worse symptom severity scores, respond less to pharmacological interventions, and have higher rates of tic disorders and attention deficit hyperactivity disorder (ADHD) compared with individuals with adult onset OCD [6–8]. Most important, the duration of OCD symptoms in childhood is a significant predictor of how persistent these behaviours will be in adulthood, stressing the significance of early diagnosis and intervention [5, 6].
Higher heritability estimates in childhood-onset OCD relative to adult-onset OCD emphasize a more substantial role for genetic factors in these individuals, though the identification of specific genetic susceptibility variants requires additional investigation [1]. Studies of genetic linkage in large pedigrees and analyses of candidate genes have identified potentially contributory loci, but replication of these findings has been inconsistent [1]. The contribution of common variants to the disorder was explored via two genome-wide association studies (GWAS) [9, 10], and these studies only identified genes of suggestive relevance, possibly due to insufficient power. Altogether, findings from GWAS and candidate gene studies suggest that OCD is complex in origin, possibly involving perturbations in many genes, with suggestive etiological evidence for DLGAP1, BTBD3, SLC1A1, FAIM2, and PTPRD [1].
Gene dosage imbalances caused by rare copy number variations (CNVs) have been identified as plausible contributory factors in other neuropsychiatric conditions, particularly those that are de novo in nature [11–13]. Higher levels of de novo copy number variation are noted in individuals with a range of neuropsychiatric conditions compared to the general population and are typically a focal point of CNV studies of disease [14–16]. OCD traits often co-occur with such conditions, including in 30–40 % of individuals with autism spectrum disorder (ASD), [17] 7–17 % with schizophrenia, [18] and 11–21 % with bipolar disorder [19]. One study so far has examined the genome-wide contribution of rare CNVs to OCD [20]. It identified an association trend between deletions at 16p13.11 and OCD. The relatively large size cut-off used (500 kb) excluded the investigation of smaller CNVs.
To identify potentially contributory genetic factors in OCD, we conducted a CNV screen using high-resolution microarrays, which facilitated CNV calling down to 15 kb. We also performed exome sequencing of selected families, in search of additional contributory factors. Finally, we assessed the cohort with respect to its overall burden of mutation and enrichment of variants in functional gene sets.
Methods
Subjects and controls
Participants were recruited from four academic child psychiatry sites: The Hospital for Sick Children, McMaster University, University of Michigan, and Wayne State University. Subjects were enrolled via clinics (site clinics, other mental health providers and primary care physicians), the internet (e.g., www.umhealthresearch.org at University of Michigan), hospital and community bulletin boards, and paid and public service advertisements in local media.
All enrolled individuals (164 females and 143 males) had symptoms first identified before age 18 (mean, 7.9 ± 3.5 years). Respective institutional ethics review boards approved all procedures. Informed consent was provided by capable adolescents. For younger children, parents or other legal guardians provided written informed consent, and the children gave verbal assent prior to participating in the study. Criteria for diagnosis are in the Additional file 1.
Our unrelated population control data were from three cohorts: Cooperative Health Research in the Region Augsburg (KORA) [21], the collaborative genetic study of nicotine dependence (COGEND), [22] and the Ontario Population Genomics Platform (OPGP) [23]. The same quality control procedures and CNV calling algorithms applied to our subjects had been applied to these controls.
Detection of rare copy number variants
We genotyped all 174 trios and 58 additional unrelated probands using the CytoScan HD array, and 75 unrelated probands using the OMNI 2.5 M array. We employed multiple algorithms to call CNVs from the CytoScan HD and OMNI 2.5 M microarray data. We defined a stringent set of variants wherein each variant was called by at least two algorithms (Additional file 2: Table S1) [24]. We defined the ancestry and relatedness of the samples using PLINK [25] following methods previously described in other studies (see Additional file 1) [26, 27]. This information was used to exclude related probands or controls and detect any sample mismatches.
To define rare CNVs, we first computed frequency using a pooled set of stringent CNVs from cases and controls, matching for ancestry, platform, and sex (Additional file 1: Figure S1). We then removed those CNVs present at >0.5 % frequency, using the 50 % reciprocal overlap criteria [11, 28]. Next, we required that CNVs overlap a region that is at least 75 % copy number stable according to the CNV map of the human genome [28]. Finally, we used a cut-off of 15 kb and 10 probes for any remaining CNVs, yielding a comprehensive and high-quality list of rare CNVs from our case and control cohorts. We first ascertained CNVs from all samples, including individuals of non-European or mixed ancestry and utilized the entire control set. We then restricted our analysis to cases of European ancestry (for the subsequent gene set analysis). Those CNVs listed in Table 1 are not present in population controls unless stated otherwise in the table. We validated de novo events and possible risk variants in proband and parental samples (when available) using a secondary confirmation method (a SYBR-green-based real-time quantitative PCR (qPCR) or a TaqMan real-time PCR assay) [27, 29]. Primer sequences are available upon request from SWS. Twenty-five of 28 CNVs of interest were validated (including all in Table 1 and 4/5 that were tagged as being de novo). Suspected mosaicism in Case D was confirmed using digital droplet qPCR (Additional file 1: Figures S2 and S3). All coordinates refer to human genome assembly build GRCh37, hg19.
Burden of CNVs in cases and controls
We analysed the burden of all CNVs, and of deletions and duplications independently. We also looked at CNV burden in the following size ranges: >15 kb (all), 15–100, 100–500, >500 kb, and >1 Mb. A binomial logistic regression analysis determined whether the differences in gene rates (the average number of genes intersected by CNVs per subject) between cases and controls were the result of a biological mechanism or statistical chance. We corrected for the number of CNVs and the total length of CNVs in the subject in our model.
Gene set enrichment analysis
We tested gene set enrichment using the rare variants detected for individuals of European ancestry (Additional file 1: Figure S4). This analysis focused on events impacting protein-coding exons, and considered deletions and duplications separately. The analysis consists of a case-control burden test with correction for the different platforms and also for global CNV burden modelled as total CNV gene count by subject (i.e., penalization for large CNVs that may drive results in an unspecific way). The gene sets tested were comprised of genes associated with neurological functions (Additional file 3: Table S2) [11, 30]. Gene set burden enrichment was performed using a logistic regression analysis deviance test corrected for gene count and global burden (Additional file 1). The genotyping platform used for CNV calling was modelled as a covariate. We undertook the analysis twice; first without exclusion criteria, and second, by excluding neurodevelopmental risk loci previously described [20, 31, 32]. This provided an opportunity to see whether any enrichment signal was being driven by loci already recognized. For final results, we utilized 15 % as the Benjamini-Hochberg false discovery rate significance threshold.
Exome sequencing
For samples with notable CNV findings (Table 1), we selected those 10 families for which adequate sample material was available from the proband and both parents for additional exome sequencing. This was conducted using the Illumina HiSeq2500, following exome capture with Agilent SureSelect Human All Exon V5 target enrichment kit. We aligned sequences to the hg19 reference using Burrows-Wheeler Aligner (BWA) v0.5.9, and used the Genome Analysis Toolkit (GATK) v1.1–28 to detect single nucleotide and small insertion and deletion variants. Rare variants were defined as those with a frequency of <0.01 in 1000 genomes, National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server and Exome Aggregation Consortium (ExAc datasets; http://exac.broadinstitute.org/). A Residual Variation Intolerance Score (RVIS) [33] and Probability of Loss of Function (LoF) Intolerance score (pLI) [34] were retrieved for each variant discovered. We confirmed the presence of all variants in Table 2 in proband and parental samples using Sanger sequencing [35].
Results
Rare copy number variations
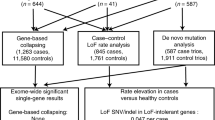
To explore the contribution of rare CNVs to OCD, we used a comprehensive CNV calling procedure with experimental validation of relevant variants. CNVs from 307 probands passed quality control, including 174 for whom DNA from both parents was also genotyped. Of these 307 probands, 259 were of European ancestry. Data from 1773 European controls were treated as those from the cases.
We uncovered 729 rare CNVs from these European cases (Additional file 2: Table S1), and 5182 from the European controls. This represented a mean of 2.81 and 2.92 rare CNVs per individual, respectively. We considered the number of genes intersected by CNVs in cases compared with controls (Additional file 1: Table S3), but found no statistically significant difference in this respect when looking at all CNVs, or at deletions and duplications independently. We also looked for differences in a particular CNV size range but found no greater global burden for rare CNVs in cases compared with controls (Additional file 1: Table S3).
We screened 174 parent-child trios of all ancestries for rare de novo CNVs. In four of these probands (2.3 %) (Table 1, cases A–D), we uncovered a single de novo CNV (sometimes in addition to inherited variants).
Various genomic syndromes have been observed in neurodevelopmental disorders including autism, schizophrenia, and epilepsy [13]. We sought to identify in our cohort rare CNVs that have been noted in some of these syndromes using the DECIPHER database containing expert-curated microdeletions and microduplications involved in developmental disorders [36]. We also compared our results with a list of OCD and ASD risk genes [1, 11, 32, 37]. We identified four individuals (Table 1, cases E–H), each with a CNV whose breakpoints were compatible with those in DECIPHER. We identified other CNVs in genes with possible relevance to OCD (Table 1, cases I–M), in gene targets of the enriched gene sets, particularly the fragile X mental retardation protein (FMRP) (cases N–P), and in a new plausible candidate, BTBD9 (cases Q–R).
Exome sequencing
In 10 of the trios where we discovered a potentially relevant CNV (i.e., de novo, or involving gene(s) associated with neurological function), we followed our CNV analysis with exome sequencing of the proband and parents. We focused our analysis on any de novo mutations or any predicted loss-of-function mutations in genes considered to play a role in neurological function. From these families, we identified six de novo variants and seven rare inherited loss-of-function mutations in genes with a neurological role (Table 2). We subsequently obtained RVIS and pLI scores for each of the genes to show how able they are to tolerate functional genetic variation and loss-of-function mutations, respectively. Only one of the genes identified from exome sequencing, AFF2, had an RVIS score in the tenth percentile (among the most intolerant genes) and a pLI score of above 0.9 (a score considered to be suggestive of pathogenic LoF mutation) [34].
Gene set enrichment
Our initial analysis sought to identify gene set enrichment, in cases compared with ethnically matched controls, from CNVs that impact a gene’s coding sequence (Table 3; Additional file 1: Table S4 and Additional file 4: Table S5). We found such enrichment for CNVs in FMRP target genes (nominal p = 1.85 × 10−03), both when we included or excluded known neurodevelopmental risk loci from the analysis. When such loci were included, cases were 1.75 times more likely to have one FMRP target impacted by a CNV than were controls, and 4.38 times more likely to have two FMRP targets impacted (Additional file 4: Table S5). Much of the signal seemed to be driven by duplications (nominal p = 0.014), though the limited sample size and false discovery rate (FDR) of 36 % cautions against over-interpretation. When considering all loci and a more relaxed FDR threshold (0.275) [38], cases were enriched with CNVs for genes involved in nervous system development (human neural function or pathway, nervous system development (GO)) (nominal p = 0.019) and the broader group encompassing genes involved in neurological function (human neural function or pathway, union, inclusive (GO, KEGG, NCI, Reactome)) (nominal p = 0.020). Similar results were generated when removing CNVs at known neuropsychiatric disease loci.
Notable case findings
De novo copy number variants (Cases A–D)
Case A has a de novo duplication intersecting VCX3B and KAL1 on chromosome X. KAL1 is involved in cell adhesion, neurite outgrowth, and axon guidance. VCX3B is expressed only in male germ cells. Her mother was diagnosed with an eating disorder.
We identified a de novo 165 kb deletion encompassing ADRA2C in Case B. ADRA2C regulates neurotransmitter release and modulates GABA release from the striatum. B also carries a large 1.7 Mb duplication impacting CNTN6 and part of CNTN4, inherited from his father who has learning difficulties. CNVs involving each have been noted in autism cases [11, 16].
Case C had onset of OCD at age 3 years, and also has Tourette syndrome and ADHD. Parental phenotypes were not provided. Multiple CNVs included a de novo deletion in a non-genic region, maternally-inherited deletions of NLGN1 and DPP6, and a PTPRN2 duplication also seen in his father. Exome sequencing also revealed three maternally-inherited variants: a 13 bp frameshift deletion of DRD4 (a dopamine receptor gene), a 4-amino-acid in-frame deletion of AFF2 (also known as FMR2) and a single-base insertion in MBD4. NLGN1 encodes a protein involved in forming excitatory synapses and maintaining synaptic plasticity [39]. Exonic CNVs of NLGN1 and DPP6 have been identified in individuals with autism [11, 40]. Duplications of PTPRN2 has been implicated in ADHD [12]. Rare mutations in AFF2 have been noted in ASD [41]. DRD4 has been previously associated with OCD [42].
Case D is mosaic for a large de novo duplication overlapping ZFAND4, FAM21C, and MARCH8. None of these genes has been previously implicated in neuropsychiatric disorders. She also carries a rare exonic duplication in SLIT1, inherited from her mother who was diagnosed with depression. SLIT1 is thought to play a role in axonal navigation and neuron projection.
CNVs known to be associated with curated DECIPHER syndromes
In female case E, we found a large 15q11-q13 duplication. About 1 in 500 children referred for genetic testing for developmental delay or autism has a CNV at this locus [43], and duplications of this region are among the most common genomic rearrangements seen in ASD probands, being present in 1–3 % of these individuals [44]. This locus includes the GABAA receptor subunit gene cluster, disturbances of which are proposed to increase the risk of developing anxiety disorders [45]. The duplication of this gene cluster may be an important contributor to the observed OCD phenotype of this individual.
From his unaffected mother, case F inherited a large duplication overlapping the 1q21.1 locus, and he shares the duplication with his male dizygotic twin who does not have an OCD diagnosis. Duplications of this locus are variably associated with both child- and adult-onset neuropsychiatric conditions and congenital abnormalities [46]. They have also been observed in some individuals with ASD [47].
Case G has OCD with generalized anxiety disorder, social anxiety disorder, and specific phobia. We discovered a large maternally-inherited deletion at 17p12—a locus associated with hereditary liability to pressure palsies (HNPP)—in this individual. Deletions of this locus usually manifest in patients as peripheral nerve problems [48], though this patient has no such symptoms. This deletion has been noted in asymptomatic individuals and may not be relevant to the OCD phenotype of this individual [48]. She also carries a paternally-inherited duplication impacting the first five exons of HCN1, a gene encoding ion channels that facilitate synaptic integration and plasticity. Phenotypic information was not available for either parent.
We identified a maternally-inherited duplication overlapping the 16p13.11 recurrent microduplication locus in case H. This locus is associated with neurocognitive disorders, and is implicated in individuals with autism [11] and some with OCD [20], though deletions (not duplications) were the significant finding in the latter.
CNVs in genes implicated in other neurodevelopmental disorders
In addition to OCD, case I suffers from major depressive disorder, social anxiety disorder, and a panic disorder. We found a duplication of the first non-coding exon of NLGN4X, inherited from his mother (who was not examined phenotypically). Functional studies of NLGN4X have shown that the gene plays important roles with respect to neuronal development and neurite formation [49]. Elsewhere, brothers with Tourette syndrome—one with autism as well, and the other with ADHD—had a three-exon deletion of NLGN4X [50]. Interestingly, their mother who also carried the deletion had anxiety and depression, suggesting that variants of NLGN4X might predispose to a range of neuropsychiatric phenotypes.
ASTN2, a gene that plays an important role in neuronal migration, was found deleted in two males. Case J was previously reported by our group in an individual with OCD and social anxiety [29]. Case K, the other individual with an ASTN2 deletion, has depression, ADHD, and Tourette syndrome. Both variants overlap the “critical region” at the 3’ end of the gene identified in a previous study [29].
In case L, we identified a duplication of TMEM132D, a gene implicated in panic disorder, and thought to contribute to anxiety phenotypes [51].
Case M has OCD with trichotillomania, and a deletion involving CDH7 and CDH19. Her parents were not interviewed. Both genes encode members of the cadherin family—proteins that play an important role in brain development.
CNVs involving FMRP target genes
In case N, we found a 1.5 Mb duplication involving PTPRD, a gene that is highly expressed at the presynaptic terminal and regulates glutamatergic synapse development, and is a target of FMRP. This variant completely duplicates five of the six main isoforms, suggesting that increased dosage of the gene product may contribute to the disorder phenotype. In a large GWAS for OCD, a single nucleotide polymorphism (SNP) near this gene was also the marker with the smallest p value [10].
Case O inherited from her unaffected father a single exon deletion from DLGAP2—another FMRP target. This is the first study to identify a CNV in DLGAP2 in an individual with OCD. However, previously, two SNPs in DLGAP2 were nominally associated with the orbitofrontal cortex white matter volume in children with OCD (although this association was not significant after correction for multiple comparison) [52].
The duplication impacting DLGAP1 in case P involves an exon that is considered to be “brain critical” [53]. This child also has Tourette syndrome, separation anxiety disorder, and oppositional defiant disorder. Though he inherited this variant from his unaffected father, his younger sister who carries the same variant was also diagnosed with OCD at age 4, and has social anxiety disorder, specific phobia, and transient tic disorder. Their mother possesses some features of OCD-like behaviour, but does not meet the criteria for diagnosis. This may suggest that other maternally inherited genetic factors also contribute to the phenotypes of the child.
CNVs impacting BTBD9
We identified two individuals with CNVs overlapping BTBD9. Case Q has a 107 kb exonic deletion inherited from her father (for whom phenotypic details were unavailable). Case R has OCD and Tourette syndrome, and inherited a larger duplication that overlaps the 5’ end of the gene. She inherited the duplication from her father who has seasonal affective disorder. This being the first time CNVs of BTBD9 have been identified in individuals with OCD, we expanded our analysis by also inspecting our unpublished microarray, exome, and whole-genome sequencing data in autism cases for CNVs or loss-of-function mutations in this gene. We found a previously unpublished male ASD proband with a paternally-inherited single-base-pair deletion, predicted to cause a frameshift and premature truncation of the BTBD9 protein. This individual also displayed obsessive compulsive tendencies. Though his father had no evidence of OCD (only writing difficulties), the child’s paternal grandfather had a host of psychiatric issues including depression, poor social skills, substance abuse, OCD, and Tourette syndrome. DNA was not available from the grandfather to test for segregation of the variant.
Discussion
We found de novo CNVs in 2.3 % of OCD cases—a rate lower than typically seen in other neurodevelopmental disorders such as schizophrenia (4.5 %) [14], bipolar disorder (4.3 %), [14] cerebral palsy (7.0 %) [26], or autism (4.7 %) [11], but slightly higher than for ADHD (1.7 %) [12]. The de novo rate in population controls from microarray is closer to 0.9–1.4 % [14, 54]. Although there is some variation between the different microarray platforms used, all are considered to be high-resolution arrays and all focus on CNVs in comparable size ranges to those we examined. As a result, a general comparison can be attempted with the caveat that this is a single study, and larger sample numbers for all disorders would even better highlight any differences that might exist. In addition, disparities in the selection of variants for validation and sample source might play a minor role in the differences observed. That said, there are also important biological reasons that could explain the differences inde novo rates in different disorders.
Individuals with autism, schizophrenia, and bipolar disorder have reduced reproductive rates [55] making it less likely that they pass on a newly acquired mutation; although reduced fecundity is also noted in OCD, it is only slightly lower than that of individuals without a neuropsychiatric condition [56]. Therefore, inherited genetic variation may play a more substantial role than de novo events in the underlying genetic architecture for OCD. We identified rare inherited CNVs at positions congruent with curated DECIPHER syndromic loci, and overlapping genes associated with other neurodevelopmental disorders, FMRP targets, or BTBD9. In 10/174 (5.7 %) of our trios, we identified an inherited genetic lesion that we propose might have contributed to the patient’s OCD phenotype. Four additional probands had possibly pathogenic copy number changes of unknown inheritance. In all, we identified candidate variants of potential significance to OCD in 18/307 cases (5.9 %).
Enrichment of CNVs in certain gene sets can highlight specific pathways or functional categories that contribute to OCD risk. In OCD cases compared with controls, we identified significant enrichment of rare CNVs intersecting genes involved in neurological function, particularly targets of FMRP (Table 3). Three CNVs overlapping FMRP targets—PTPRD, DLGAP1 and DLGAP2—were of particular interest in this cohort. Two SNPs near DLGAP1 showed the highest associations in a previous GWAS of OCD [9]. A marker near PTPRD exhibited the highest association with OCD in the second GWAS [10], and the locus has also been implicated in OCD by linkage studies [57, 58]. The enrichment in these targets is of particular relevance, given FMRP’s high expression in neurons and its role as a regulatory protein that alters the translation of proteins involved in synaptic function [59], making it essential for a range of cognitive processes. This concurs with studies of ASD and schizophrenia that suggest CNVs impacting FMRP targets increase risk for neuropsychiatric conditions [11, 60]. However, in contrast to the larger role for FMRP deletions in these disorders, duplications are potentially more relevant for OCD (statistically significant in our study, albeit with a high FDR). This may reflect an alternate mechanism underlying some of the relationship between genotype and phenotype.
Studies of OCD and of other neuropsychiatric conditions invoke a genetic model whereby the atypical development or maintenance of synaptic connections contributes to a neuropsychiatric phenotype [1, 61]. As a result, many of the same candidate genes are emerging across a range of neuropsychiatric disorders. Here, we have once again uncovered ASTN2 variants in individuals with a neuropsychiatric condition. A previous study [29] that has shown CNVs of this gene often are observed in ADHD, and one individual in our study has this diagnosis in addition to OCD. We also identified a potential new OCD risk gene, BTBD9, which encodes a protein with a BTB/POZ domain. Proteins containing this domain play a role in synaptic plasticity and neurotransmission and alter dendritic branching.
Conclusions
One particularly interesting area of further research would be to apply complementary microarray, exome sequencing, or whole-genome sequencing approaches [41, 62]. In autism, findings from exome sequencing and from CNVs are largely non-overlapping [63]. With this precedent, here, we undertook exome sequencing in 10 trios in our study, one of which revealed a plausibly relevant mutation (DRD4). This pilot study demonstrates the importance of using multiple approaches to investigate the genetic contribution to OCD.
We identified a number of CNVs that overlap risk loci for other neurodevelopmental conditions. As in McGrath et al. [20], we found no increased global CNV burden in cases compared with controls. Although we did not find 16p13.11 deletions in our study (the main finding in McGrath et al. [20]), we did identify a duplication at this locus. Most of our variants of interest were smaller than their 500 kb cut-off, stressing the importance of examining variation at all size ranges in OCD.
There are important caveats that must be accounted for when interpreting our study. The first is that our statistical power is constrained by our small sample size. Because of this, most of the rare variants that were identified in our cases only occurred once. As a result, we were required to rely to a greater extent on previously published work and functional studies to prioritize genes of interest. In addition, the small sample size resulted in higher false discovery rates than what we would wish, necessitating follow-up studies in larger cohorts of OCD patients. Unfortunately, in many of our families, we also lacked extensive phenotypic details in some patients and even more frequently, in parents, preventing more comprehensive genotype and phenotype correlation. That said, our study presents new avenues for further investigation and highlights some important candidate genes. Most importantly, we reaffirm the extensive genetic heterogeneity in neuropsychiatric conditions, while demonstrating overlap between genes uncovered here with those of other neurodevelopmental conditions.
Abbreviations
- ADHD:
-
Attention deficit hyperactivity disorder
- ASD:
-
Autism spectrum disorder
- CNV:
-
Copy number variation
- COGEND:
-
Collaborative genetic study of nicotine dependence
- DECIPHER:
-
Database of Genomic Variation and Phenotype in Humans Using Ensembl Resources
- FDR:
-
False discovery rate
- FMRP:
-
Fragile X mental retardation protein
- GATK:
-
Genome Analysis Toolkit
- GWAS:
-
Genome-wide association studies
- HNPP:
-
Hereditary liability to pressure palsies
- KORA:
-
Cooperative health research in the Region of Augsburg
- NHLBI:
-
National Heart, Lung, and Blood Institute
- OCD:
-
Obsessive-compulsive disorder
- OPGP:
-
Ontario population genomics platform
- qPCR:
-
Quantitative polymerase chain reaction
- SNP:
-
Single nucleotide polymorphism
References
Pauls DL, Abramovitch A, Rauch SL, Geller DA. Obsessive-compulsive disorder: an integrative genetic and neurobiological perspective. Nat Rev Neurosci. 2014;15(6):410–24. doi:10.1038/nrn3746.
Ruscio AM, Stein DJ, Chiu WT, Kessler RC. The epidemiology of obsessive-compulsive disorder in the National Comorbidity Survey Replication. Mol Psychiatry. 2010;15(1):53–63. doi:10.1038/mp.2008.94.
Leckman JF, Denys D, Simpson HB, Mataix-Cols D, Hollander E, Saxena S, et al. Obsessive-compulsive disorder: a review of the diagnostic criteria and possible subtypes and dimensional specifiers for DSM-V. Depress Anxiety. 2010;27(6):507–27. doi:10.1002/da.20669.
Anholt GE, Aderka IM, van Balkom AJ, Smit JH, Schruers K, van der Wee NJ, et al. Age of onset in obsessive-compulsive disorder: admixture analysis with a large sample. Psychol Med. 2014;44(1):185–94. doi:10.1017/S0033291713000470.
Stewart SE, Geller DA, Jenike M, Pauls D, Shaw D, Mullin B, et al. Long-term outcome of pediatric obsessive-compulsive disorder: a meta-analysis and qualitative review of the literature. Acta Psychiatr Scand. 2004;110(1):4–13. doi:10.1111/j.1600-0447.2004.00302.x.
Micali N, Heyman I, Perez M, Hilton K, Nakatani E, Turner C, et al. Long-term outcomes of obsessive-compulsive disorder: follow-up of 142 children and adolescents. Br J Psychiatry J Ment Sci. 2010;197(2):128–34. doi:10.1192/bjp.bp.109.075317.
Rosario-Campos MC, Leckman JF, Mercadante MT, Shavitt RG, Prado HS, Sada P, et al. Adults with early-onset obsessive-compulsive disorder. Am J Psychiatry. 2001;158(11):1899–903.
Bloch MH, Craiglow BG, Landeros-Weisenberger A, Dombrowski PA, Panza KE, Peterson BS, et al. Predictors of early adult outcomes in pediatric-onset obsessive-compulsive disorder. Pediatrics. 2009;124(4):1085–93. doi:10.1542/peds.2009-0015.
Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA, et al. Genome-wide association study of obsessive-compulsive disorder. Mol Psychiatry. 2013;18(7):788–98. doi:10.1038/mp.2012.85.
Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT, et al. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry. 2015;20(3):337–44. doi:10.1038/mp.2014.43.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–94. doi:10.1016/j.ajhg.2014.03.018.
Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Science translational medicine. 2011;3(95):95ra75. doi:10.1126/scitranslmed.3002464.
Cook Jr EH, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455(7215):919–23. doi:10.1038/nature07458.
Malhotra D, McCarthy S, Michaelson JJ, Vacic V, Burdick KE, Yoon S, et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72(6):951–63. doi:10.1016/j.neuron.2011.11.007.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–9. doi:10.1126/science.1138659.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–72. doi:10.1038/nature09146.
Leyfer OT, Folstein SE, Bacalman S, Davis NO, Dinh E, Morgan J, et al. Comorbid psychiatric disorders in children with autism: interview development and rates of disorders. J Autism Dev Disord. 2006;36(7):849–61. doi:10.1007/s10803-006-0123-0.
Achim AM, Maziade M, Raymond E, Olivier D, Merette C, Roy MA. How prevalent are anxiety disorders in schizophrenia? A meta-analysis and critical review on a significant association. Schizophr Bull. 2011;37(4):811–21. doi:10.1093/schbul/sbp148.
Amerio A, Odone A, Liapis CC, Ghaemi SN. Diagnostic validity of comorbid bipolar disorder and obsessive-compulsive disorder: a systematic review. Acta Psychiatr Scand. 2014;129(5):343–58. doi:10.1111/acps.12250.
Mcgrath LM, Yu D, Marshall C, Davis LK, Thiruvahindrapuram B, Li B, et al. Copy number variation in obsessive-compulsive disorder and tourette syndrome: a cross-disorder study. J Am Acad Child Adolesc Psychiatry. 2014;53(8):910–9. doi:10.1016/j.jaac.2014.04.022.
Verhoeven VJ, Hysi PG, Wojciechowski R, Fan Q, Guggenheim JA, Hohn R, et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat Genet. 2013;45(3):314–8. doi:10.1038/ng.2554.
Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet. 2007;16(1):24–35. doi:10.1093/hmg/ddl441.
Uddin M, Thiruvahindrapuram B, Walker S, Wang Z, Hu P, Lamoureux S et al. A high-resolution copy-number variation resource for clinical and population genetics. Genetics in medicine: official journal of the American College of Medical Genetics. 2014. doi:10.1038/gim.2014.178.
Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, Fitzgerald T, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol. 2011;29(6):512–20. doi:10.1038/nbt.1852.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi:10.1086/519795.
Oskoui M, Gazzellone MJ, Thiruvahindrapuram B, Zarrei M, Andersen J, Wei J, et al. Clinically relevant copy number variations detected in cerebral palsy. Nat Commun. 2015;6:7949. doi:10.1038/ncomms8949.
Gazzellone MJ, Zhou X, Lionel AC, Uddin M, Thiruvahindrapuram B, Liang S, et al. Copy number variation in Han Chinese individuals with autism spectrum disorder. J Neurodev Disord. 2014;6(1):34. doi:10.1186/1866-1955-6-34.
Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–83. doi:10.1038/nrg3871.
Lionel AC, Tammimies K, Vaags AK, Rosenfeld JA, Ahn JW, Merico D, et al. Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum Mol Genet. 2014;23(10):2752–68. doi:10.1093/hmg/ddt669.
Engchuan W, Dhindsa K, Lionel AC, Scherer SW, Chan JH, Merico D. Performance of case-control rare copy number variation annotation in classification of autism. BMC Med Genomics. 2015;8 Suppl 1:S7. doi:10.1186/1755-8794-8-S1-S7.
Rees E, Walters JT, Georgieva L, Isles AR, Chambert KD, Richards AL, et al. Analysis of copy number variations at 15 schizophrenia-associated loci. Br J Psychiatry J Ment Sci. 2014;204(2):108–14. doi:10.1192/bjp.bp.113.131052.
Marshall C, Howrigan D, Merico D, Thiruvahindrapuram B, Wu W, Greer D et al. A contribution of novel CNVs to schizophrenia from a genome-wide study of 41,321 subjects. bioRxiv. 2016. doi:10.1101/040493.
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9(8):e1003709. doi:10.1371/journal.pgen.1003709.
Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv. 2015. doi:10.1101/030338.
Lionel AC, Vaags AK, Sato D, Gazzellone MJ, Mitchell EB, Chen HY, et al. Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum Mol Genet. 2013;22(10):2055–66. doi:10.1093/hmg/ddt056.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84(4):524–33. doi:10.1016/j.ajhg.2009.03.010.
Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nature medicine. 2015. doi:10.1038/nm.3792.
Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012;8(8):e1002843. doi:10.1371/journal.pgen.1002843.
Hoy JL, Haeger PA, Constable JR, Arias RJ, McCallum R, Kyweriga M, et al. Neuroligin1 drives synaptic and behavioral maturation through intracellular interactions. J Neurosci Off J Soc Neurosci. 2013;33(22):9364–84. doi:10.1523/JNEUROSCI.4660-12.2013.
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–88. doi:10.1016/j.ajhg.2007.12.009.
Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. American journal of human genetics. 2013. doi:10.1016/j.ajhg.2013.06.012.
Millet B, Chabane N, Delorme R, Leboyer M, Leroy S, Poirier MF, et al. Association between the dopamine receptor D4 (DRD4) gene and obsessive-compulsive disorder. Am JMed Genet B Neuropsychiatr Genet. 2003;116B(1):55–9. doi:10.1002/ajmg.b.10034.
Moreno-De-Luca D, Sanders SJ, Willsey AJ, Mulle JG, Lowe JK, Geschwind DH, et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry. 2013;18(10):1090–5. doi:10.1038/mp.2012.138.
Cook Jr EH, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60(4):928–34.
Nuss P. Anxiety disorders and GABA neurotransmission: a disturbance of modulation. Neuropsychiatr Dis Treat. 2015;11:165–75. doi:10.2147/NDT.S58841.
Dolcetti A, Silversides CK, Marshall CR, Lionel AC, Stavropoulos DJ, Scherer SW, et al. 1q21.1 microduplication expression in adults. Genet Med Off J Am Coll Med Genet. 2013;15(4):282–9. doi:10.1038/gim.2012.129.
Bernier R, Steinman KJ, Reilly B, Wallace AS, Sherr EH, Pojman N, et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet Med Off J Am Coll Med Genet. 2016;18(4):341–9. doi:10.1038/gim.2015.78.
Mouton P, Tardieu S, Gouider R, Birouk N, Maisonobe T, Dubourg O, et al. Spectrum of clinical and electrophysiologic features in HNPP patients with the 17p11.2 deletion. Neurology. 1999;52(7):1440–6.
Shi L, Chang X, Zhang P, Coba MP, Lu W, Wang K. The functional genetic link of NLGN4X knockdown and neurodevelopment in neural stem cells. Human molecular genetics. 2013. doi:10.1093/hmg/ddt226.
Lawson-Yuen A, Saldivar JS, Sommer S, Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet EJHG. 2008;16(5):614–8. doi:10.1038/sj.ejhg.5202006.
Erhardt A, Akula N, Schumacher J, Czamara D, Karbalai N, Muller-Myhsok B, et al. Replication and meta-analysis of TMEM132D gene variants in panic disorder. Transl Psychiatry. 2012;2:e156. doi:10.1038/tp.2012.85.
Wu K, Hanna GL, Easter P, Kennedy JL, Rosenberg DR, Arnold PD. Glutamate system genes and brain volume alterations in pediatric obsessive-compulsive disorder: a preliminary study. Psychiatry Res. 2013;211(3):214–20. doi:10.1016/j.pscychresns.2012.07.003.
Uddin M, Tammimies K, Pellecchia G, Alipanahi B, Hu P, Wang Z, et al. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat Genet. 2014;46(7):742–7. doi:10.1038/ng.2980.
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70(5):863–85. doi:10.1016/j.neuron.2011.05.002.
Power RA, Kyaga S, Uher R, Maccabe JH, Langstrom N, Landen M, et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiat. 2013;70(1):22–30. doi:10.1001/jamapsychiatry.2013.268.
Baron-Cohen S. The maladapted mind: classic readings in evolutionary psychopathology. Hove, East Sussex, UK: Psychology Press; 1997.
Hanna GL, Veenstra-Vanderweele J, Cox NJ, Boehnke M, Himle JA, Curtis GC, et al. Genome-wide linkage analysis of families with obsessive-compulsive disorder ascertained through pediatric probands. Am J Med Genet. 2002;114(5):541–52. doi:10.1002/ajmg.10519.
Willour VL, Yao Shugart Y, Samuels J, Grados M, Cullen B, Bienvenu 3rd OJ, et al. Replication study supports evidence for linkage to 9p24 in obsessive-compulsive disorder. Am J Hum Genet. 2004;75(3):508–13. doi:10.1086/423899.
Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–61. doi:10.1016/j.cell.2011.06.013.
Szatkiewicz JP, O’Dushlaine C, Chen G, Chambert K, Moran JL, Neale BM, et al. Copy number variation in schizophrenia in Sweden. Mol Psychiatry. 2014;19(7):762–73. doi:10.1038/mp.2014.40.
Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 2012;22(3):229–37. doi:10.1016/j.gde.2012.03.002.
Stavropoulos DJ, Merico D, Jobling R, Bowdin S, Monfared N, Thiruvahindrapuram B, et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. Npj Genet Med. 2016;1:15012. doi:10.1038/npjgenmed.2015.12.
Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314(9):895–903. doi:10.1001/jama.2015.10078.
Acknowledgements
The authors thank Bhooma Thiruvahindrapuram, John Wei, Zhuozhi Wang, Jeffrey MacDonald, Chao Lu, Sergio Pereira, Ting Wang, Xiaolin Wang, Zhanqin Liu, Honglei Yu, Karen Ho, Zhizhou Hu, Sylvia Lamoureux, Alireza Mowjoodi, Anath Lionel, Ada Chan, Lia D’Abate, Thanuja Selvanayagam, and Arakel Minassian for technical assistance. We thank The Centre for Applied Genomics funded by Genome Canada/Ontario Genomics Institute for computational support.
Funding
This study was supported by grants from Genome Canada, Canada Foundation for Innovation, Canadian Institute for Advanced Research, Government of Ontario, Canadian Institutes of Health Research, National Institute of Mental Health, Ontario Brain Institute, The Hospital for Sick Children and University of Toronto McLaughlin Centre. PDA holds the Alberta Innovates Health Solutions (AIHS) Translational Health Chair in Child and Youth Mental Health. SWS holds the GlaxoSmithKline-CIHR Chair in Genome Sciences at The Hospital for Sick Children and University of Toronto.
Availability of data and materials
All relevant microarray data have been deposited in the Gene Expression Omnibus and can be accessed using accession number GSE76438.
Authors’ contributions
MJG, PDA, and SWS conceived and designed the project. MJG analysed and interpreted the microarray findings and oversaw experiments confirming these results. MZ and DM developed and executed the gene set enrichment protocol. SW and MU contributed to the informatics and exome sequencing analyses. CLB, RR, RJS, and MC provided and interpreted phenotypic details for the patients. SMS and JC prepared the samples and contributed to the experimental design. GLH, DRR, NS, KDF, and PDA assessed the patients. CRM advised on the study design and writing of the manuscript. MJG, MZ, JAB, PDA and SWS wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The study was approved by the Research Ethics Board of the Hospital for Sick Children, Toronto, Canada, REB File No. 1000011896 (to Dr. Arnold), the Research Ethics Board of McMaster University, Hamilton, Canada, Registration Number 12–050 (to Dr. Soreni), and by the Institutional Review Board of the University of Michigan, Ann Arbor, MI, USA OHRP IRB Registration Number IRB00000244 (to Dr. Hanna). Issues of consent to participate are included in these approvals.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1:
Supplementary Information. (DOCX 292 kb)
Additional file 2:
Table S1 (XLSX 63 kb)
Additional file 3:
Table S2 (XLSX 11 kb)
Additional file 4:
Table S5 (XLSX 1313 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gazzellone, M.J., Zarrei, M., Burton, C.L. et al. Uncovering obsessive-compulsive disorder risk genes in a pediatric cohort by high-resolution analysis of copy number variation. J Neurodevelop Disord 8, 36 (2016). https://doi.org/10.1186/s11689-016-9170-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11689-016-9170-9