
Abstract
Liquid biopsies are emerging as an alternative source for pediatric cancer biomarkers with potential applications during all stages of patient care, from diagnosis to long-term follow-up. While developments within this field are reported, these mainly focus on dedicated items such as a specific liquid biopsy matrix, analyte, and/or single tumor type. To the best of our knowledge, a comprehensive overview is lacking. Here, we review the current state of liquid biopsy research for the most common non-central nervous system pediatric solid tumors. These include neuroblastoma, renal tumors, germ cell tumors, osteosarcoma, Ewing sarcoma, rhabdomyosarcoma and other soft tissue sarcomas, and liver tumors. Within this selection, we discuss the most important or recent studies involving liquid biopsy-based biomarkers, anticipated clinical applications, and the current challenges for success. Furthermore, we provide an overview of liquid biopsy-based biomarker publication output for each tumor type based on a comprehensive literature search between 1989 and 2023. Per study identified, we list the relevant liquid biopsy-based biomarkers, matrices (e.g., peripheral blood, bone marrow, or cerebrospinal fluid), analytes (e.g., circulating cell-free and tumor DNA, microRNAs, and circulating tumor cells), methods (e.g., digital droplet PCR and next-generation sequencing), the involved pediatric patient cohort, and proposed applications. As such, we identified 344 unique publications. Taken together, while the liquid biopsy field in pediatric oncology is still behind adult oncology, potentially relevant publications have increased over the last decade. Importantly, steps towards clinical implementation are rapidly gaining ground, notably through validation of liquid biopsy-based biomarkers in pediatric clinical trials.
Similar content being viewed by others
Introduction
Pediatric cancer
Cancer is characterized by uncontrolled malignant cell proliferation following accumulated (epi)genetic aberrations1. In children between 0 and 19 years, globally 289,500 new cases were diagnosed in 20202. Fortunately, increased insight in cancer biology and tumorigenesis together with international clinical trials enabled advances in treatment and supportive care and led to improved survival rates3,4. Indeed, in the US, the 5-year overall survival (OS) rate for children improved from 58% during the 1970s to 85% during 2012 through 20185. Nonetheless, and despite being a rare disease, cancer remains the leading cause of disease-related death in children over one year of age in high-income countries worldwide6. Moreover, despite of the rising cure rates, significant long-term side effects exist, for which reduction of therapy burden is also an important target7.
Pediatric and adult cancers are distinctly different regarding cause, occurrence, (epi)genetic properties, and outcome8. The most common pediatric cancer types include leukemias, central nervous system tumors, lymphomas, neuroblastoma, sarcomas (bone and soft tissue tumors), renal tumors, and germ cell tumors5. In contrast, breast, prostate, lung, and colorectal cancers are the most common types among adults5. Pediatric cancers generally have a low mutational burden with few recurrent hotspots and are characterized by epigenetic dysregulation and chromosomal structural variations (SVs), including copy number alterations (CNAs), and translocations8,9,10,11. Adult cancers are characterized by a high number of somatic mutations and insertion/deletion errors8,10. It is suggested that the mutational landscape of adult cancers is a result of the accumulated effects of aging and long-term exposure to physical, chemical, and biological environmental factors6,12,13,14. In contrast, pediatric cancer origins are thought to primarily involve accumulations of (epi)genetic aberrations during embryonal development, and inheritance of pathogenic germline variants10,11,15,16.
Common cancer treatments include surgery, systemic treatment (chemotherapy, targeted therapy, hormonal therapy, and immunotherapy), and radiation therapy, depending on cancer type and stage17. Treatment-related side-effects such as immunosuppression and infection, nephrotoxicity, cardiotoxicity, ototoxicity, decreased fertility, pain, endocrine insufficiency, nausea and vomiting, and psychosocial effects have a significant impact on children and their quality of life18,19. Therefore, treatment should be optimally tailored for every child to maximize the likelihood of cure while minimizing short and long-term side effects. Currently, tumor biopsies are still the golden standard for disease diagnosis, tumor biology analysis, and therapeutic decision-making. However, these biopsies are invasive, can be difficult to obtain, and do not comprise full tumor heterogeneity20. This limits feasibility of tumor biopsies for serial sampling, monitoring of disease and treatment efficacy, and identification of treatment resistant clones3. Furthermore, patients often receive advanced imaging during diagnosis and disease monitoring, exposing them to ionizing radiation and/ or general anesthesia21. These imaging techniques are not sensitive enough to detect minimal residual disease and do not allow analysis of molecular changes during treatment22. Indeed, many children still relapse despite showing complete remission based on clinical imaging3. Relapse arises from drug-resistant minimal residual disease (MRD) cells throughout treatment at levels below morphological detection limits, and is the primary cause of pediatric cancer-related death4.
Taken together, pediatric cancers are clearly distinct cancers with unique diagnostic and therapeutic challenges, and there is an urgent need for alternative less-invasive biomarkers for diagnosis, disease monitoring, therapeutic decision-making, and early relapse detection3,6.
Liquid biopsies
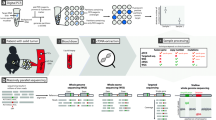
Malignant solid tumors are dynamic and continuously changing their microenvironment to enable growth and spread23,24. This includes shedding intact tumor cells and tumor-derived genetic material into the body fluids24. Sampling of these fluids, also known as a ‘liquid biopsy’, can be used for the detection and analysis of such tumor-derived material. Peripheral blood (plasma and serum) and cerebrospinal fluid (CSF) are the most established liquid biopsy fluids, with CSF being the most common for brain tumors25,26. Other body fluids such as bone marrow, urine, feces, saliva, sputum, pleural effusions, and ascites are also potentially useful liquid biopsies, depending on the tumor type26,27,28. Liquid biopsies provide access to different molecular analytes of which cell-free DNA (cfDNA)-derived circulating tumor DNA (ctDNA) is the most widely used3,4,8,26,28,29,30,31,32,33. In addition, RNA (cell-free RNA (cfRNA), messenger RNA (mRNA), microRNA (miRNA), long noncoding RNA (lncRNA)), circulating tumor cells (CTCs) including derived material, extracellular vesicle (EV) content, defined immune cell profiles, proteins, tumor-educated platelets, fibroblasts, and endothelial cells have also been described as analytes within liquid biopsies33,34,35 (Supplementary Tables 2–9). These analytes provide a source of potential biomarkers for cancer detection and monitoring3. A graphical overview of a liquid biopsy workflow is presented in Fig. 1.

Frequent anatomical locations where pediatric solid tumors commonly present are indicated. Liquid biopsy matrices (e.g., peripheral blood, bone marrow, and CSF) are sampled and processed for isolation of analytes (e.g., ctDNA, CTCs, RNA (mRNA, miRNA, lncRNA), immune cells, and EVs) that are measured and characterized by specific methods (mostly PCR-based or sequencing-based) for biomarker analysis. The AI vector generator function from Adobe Illustrator was used for detailed visualization of this figure.
Gradually, liquid biopsies are emerging as a more sensitive and potentially cheaper alternative to conventional tissue biopsies32. Clinical applications include (early) cancer detection, detection of therapeutic targets, real-time monitoring of therapeutic efficacy, monitoring tumor evolution and resistance mechanisms, early detection and prediction of relapse, and guidance of treatment decisions9,33,36. This offers the potential to monitor disease burden complementary or independently of radiologic or surgical findings. Also, while a tissue biopsy only provides information about the sampled region, a liquid biopsy captures the diverse tumor landscape, mitigating intra-tumor heterogeneity and temporal heterogeneity (differences among primary tumor and relapses)3,8. Finally, liquid biopsies are less-invasive than tissue biopsies and could avoid sedation, making them especially interesting for pediatric oncology8,32. Taken together, liquid biopsies can overcome several limitations associated with tissue biopsies.
Liquid biopsies have already been implemented in adult oncology as Food and Drug Administration (FDA) approved screening and diagnostic assays, mostly based on the detection of specific recurring mutations using quantitative PCR (qPCR) and next-generation sequencing (NGS) approaches37,38,39,40. Examples of approved methods include CellSearch CTC detection, Cobas EGFR mutation test v2, Guardant 360 CDx, FoundationOne Liquid CDx, and Archer LIQUIDPlex40,41. However, these specific panels cannot be translated to pediatric cancers due to the earlier discussed differences with adult cancer8. Indeed, at the time of publication, no liquid biopsy-based tests had been FDA-approved in pediatric oncology. Unique challenges such as sampling limitations and hence scarce input material (especially in young children), and readouts beyond mutational burden are provided8. Also, analytical method validation through large randomized controlled trials is required before clinical implementation3,8. Nonetheless, the field of liquid biopsies in pediatric oncology is rapidly expanding, and key findings for different tumor types are occasionally reviewed in the literature (Fig. 2a, Table 1, and Supplementary Table 10).

In (a), the number of unique articles (y-axis) is plotted over time (x-axis), for studies and review articles. All articles used for this plot are also included in Table 1. All articles in the ‘studies’ plot are also included in (b) and Supplementary Tables 2–9. All articles in the ‘reviews’ plot are also included in Supplementary Table 10. In (b), The number of published studies (y-axis) is plotted over time (x-axis), for each tumor type. Studies that included pediatric patients for more than one tumor type are indicated in each respective plot. All studies used for this plot are also included in (a), Table 1, and Supplementary Tables 2–9.
In this review, we provide a comprehensive overview of liquid biopsy-based biomarkers within the literature for the most frequent pediatric solid tumors, including neuroblastoma, renal tumors, germ cell tumors, osteosarcoma, Ewing sarcoma, rhabdomyosarcoma, non-rhabdomyosarcoma soft tissue sarcomas, and liver tumors. As such, hematological malignancies (leukemias and lymphomas) and central nervous system tumors were not included here.
Comprehensive literature analysis
An overview of research articles on liquid biopsies in pediatric solid tumors was generated through an advanced search in PubMed (see Supplementary Table 1 for search parameters, selection criteria, and exclusion criteria). Generally, studies were included if they investigated liquid biopsy-based biomarkers in pediatric patients suffering from one or more solid tumor types within our selection. Review articles focusing on liquid biopsies in one or more of these pediatric tumor types were included as well. Articles (studies and reviews) focusing on liquid biopsies within adult tumors or pediatric tumors outside our selection were not included.
Following our literature search, we identified 344 unique publications (283 studies and 61 reviews). We provide a historical overview of these articles, as well as a historical overview of studies investigating liquid biopsy-based biomarkers in pediatric patients, per tumor type (Fig. 2a, b, respectively). Studies that included pediatric patients with more than one tumor type were included in each respective tumor type plot in Fig. 2b. Research on liquid biopsy-based biomarkers within pediatric solid tumors clearly started in the early 90 s, with many articles having been published over the last 10 years.
In the following chapters, per tumor type, we briefly describe the respective malignancy, followed by involved genetic aberrations and a review of the literature on liquid biopsy implementation. For each tumor type, we provide a comprehensive overview of studies that included pediatric patients of the respective tumor type (Supplementary Tables 2–9). Per individual study, we include the investigated liquid biopsy-based biomarkers, analytes, matrices, detection methods, number of pediatric patients included, and suggested application of the biomarker(s). The neuroblastoma and osteosarcoma chapters are the only exceptions due to the excessively high number of publications on these tumor types, and we provided more generic tables instead (focusing on ctDNA-derived biomarkers for neuroblastoma, and different liquid biopsy analytes for osteosarcoma). A summary of important liquid biopsy-derived biomarkers and references to the relevant studies, per tumor type, are provided in Table 1.
Liquid biopsies in defined pediatric solid tumors
Neuroblastoma
Neuroblastoma (NB) is the most common extracranial childhood tumor, accounting for roughly 8% of childhood malignancies42,43,44. NB originates from neural crest progenitor cells, and can establish anywhere along the sympathetic nervous system with the majority arising in the adrenal glands45. NB is highly heterogeneous, and clinical symptoms and presentation can vary from a benign mass to substantially metastasized disease45. NB is classified into low, intermediate, and high-risk (approx. 50%) groups based on several criteria46. These include age at diagnosis, tumor stage and differentiation, tumor histology and associated DNA ploidy, MYCN status, and chromosome 1p and 11q copy number status42,47,48. The median age of diagnosis is 18 months, with 40% of patients being diagnosed at infancy and 90% at <10 years of age49. The 5-year survival rate is highly dependent on the risk group, varying from >90% in the low-risk group to 40–50% for patients in the high-risk group50. Treatment depends on disease stage and includes simple observation, surgical resection, chemotherapy, radiation therapy, myeloablative chemotherapy with autologous stem cell reinfusion, and immunotherapy45.
NB is one of the few pediatric cancers where biomarkers are routinely used for diagnosis, prognostication, and therapeutic monitoring, including urine and serum-derived markers (neuron-specific enolase, lactate dehydrogenase, vanillylmandelic acid, and homovanillic acid)47. Nonetheless, these biomarkers lack sensitivity and specificity for accurate prognostication42. Also, NB diagnosis, risk-classification, therapeutic monitoring, and relapse detection still mostly relies on invasive tumor and bone marrow biopsies and imaging methods43. Over the last two decades, liquid biopsies (notably blood plasma and bone marrow) have increasingly been investigated as an alternative source for potential NB biomarkers (Fig. 2b and Table 1). ctDNA is the most investigated analyte (Supplementary Table 2). Furthermore, CTCs have been analyzed using detection of nucleosomes, mRNA, DNA, cfRNA (including miRNA), and EVs.51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123. Many of the studies investigating these analytes in NB have already been thoroughly reviewed elsewhere3,4,8,31,34,35,42,43,47,50,124,125,126,127,128,129,130,131,132,133,134,135,136,137. Therefore, we will focus on the most recent development for ctDNA analysis and implementation here.
ctDNA (being the tumor fraction of cfDNA) constitutes of 90–150 bp long fragments that are actively (in EVs) or passively (by necrosis or apoptosis) released from malignant cells [12]. cfDNA is rapidly cleared from circulation (half-life of minutes to 2.5 hours) making it an interesting biomarker for real-time tumor analysis [11]. NB displays particularly high ctDNA levels at diagnosis compared to other (pediatric and adult) cancer types including Wilms tumor, rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, melanoma, renal cell carcinoma, hepatocellular carcinoma, medulloblastoma, glioma, and prostate, ovarian, colorectal, breast, thyroid, head and neck, pancreatic ductal, bladder, and gastroesophageal cancer31,47,138,139,140. Also, significant correlations among cfDNA concentration and tumor volume has been observed in NB patients, as well as higher cfDNA levels in metastatic NB versus localized NB141,140. Multiple studies demonstrated the suitability of plasma cfDNA content as a biomarker for NB risk-stratification, tumor load, monitoring of treatment response, and detection of MRD and relapse (Supplementary Table 2). Also, recent technological advances significantly facilitated ctDNA detection and analysis4. Novel methods such as droplet digital PCR (ddPCR) now allow rapid and cost-effective screening for multiple genetic targets despite small sample volumes and low ctDNA concentrations, as well as offering absolute quantification42,142. Important genetic NB targets that are detectable using ctDNA include (but are not limited to) gene amplifications (MYCN and ALK), chromosomal copy number changes (1p and 11q loss, and 17q gain), and epigenetic aberrations (RASSF1A and DCR2 hypermethylation)43,143. A comprehensive overview of studies investigating ctDNA analysis in NB is provided in Supplementary Table 2. For example, Peitz et al.142 demonstrated two multiplex ddPCR protocols allowing reliable quantification of MYCN and ALK copy numbers in NB patients using cfDNA and matched tissue samples. Similarly, Shirai et al.144 combined MYCN copy number and loss of 11q heterozygosity for accurate diagnosis in NB patients using plasma-derived cfDNA with ddPCR. Then, Gelineau et al.145 recently described a case-series where cfDNA contributed to the successful diagnosis and prognostication in NB patients using ddPCR, qPCR, and reduced representation bisulphite sequencing of cell-free DNA (cfRRBS). Finally, van Zogchel et al.146 used targeted locus amplification and ddPCR to identify patient-specific targets in the cfDNA of NB patients. These markers tracked tumor burden, decreased during induction therapy, disappeared at complete remission, and re-appeared at relapse. Some markers were detectable in cfDNA before relapse detection by imaging or standard bone marrow evaluation, demonstrating suitability for monitoring NB burden, disease progression, MRD detection, and early relapse prediction. Next to ddPCR, chromosomal micro-arrays can also be used for the identification of patient-specific copy number profiles and intra-tumoral heterogeneity42. For example, López-Carrasco et al.147 showed improved detection of intra-tumor heterogeneity in NB patients when combining tumor biopsy and ctDNA analysis versus tumor biopsies alone, therefore providing more accurate genomic diagnosis, prognosis, and therapy options.
The emergence of advanced sequencing techniques now allows for targeted or genome-wide screening and sensitive detection of patient-specific genetic aberrations4. For example, Van Paemel et al.148 accurately diagnosed 67 NB cases out of 128 pediatric cancer patients by CNA profiling using shallow whole genome sequencing (sWGS) on paired plasma cfDNA and tissue DNA samples. Also, Cahn et al.149 performed molecular profiling of 324 genes in 12 NB patients for the identification of actionable targets using a commercially available platform (Foundation One Liquid CDx). They concluded that especially in refractory or relapsed NB patients, liquid biopsies are very suitable for obtaining molecular tumor information for guiding therapeutic decisions in a clinically relevant timeframe.
NB pathogenesis is also correlated with epigenetic aberrations, providing additional targets for NB detection in ctDNA43. As with many cancers, RASSF1A is often hypermethylated in NB150. van Zogchel et al.141 found hypermethylated RASSF1A in plasma of 41 of 42 patients with metastatic NB. Levels decreased during therapy and recurred at relapse, demonstrating its potential value as a prognostic biomarker after diagnosis. Next, enzymatic methyl-sequencing (EM-seq) emerged as a new method for methylome analysis outperforming the classic bisulphite sequencing method151. Two studies recently demonstrated the feasibility of EM-seq for whole methylome analysis from ctDNA in NB patients152,153. For example, Trinidad et al.152 revealed prognostic actionable targets in high-risk 11q deleted NB and a DNA-methylation biomarker profile for disease monitoring using very low input cfDNA.
Taken together, conducted studies including the examples described above demonstrate the clinical potential of liquid biopsies (notably ctDNA) for implementation in NB, including diagnosis, tumor burden evaluation, treatment efficacy, intra-tumor heterogeneity, and the detection of MRD and relapse (Supplementary Table 2). However, several obstacles need to be tackled before clinical implementation can be realized including biomarker validation in prospective multi-center trials with harmonized and standardized protocols from sample collection to final data analysis8,31,125,154. An elaboration of these considerations is given in the ‘challenges and requirements for clinical implementation’ chapter. Importantly, liquid biopsy sampling and analyses are included in the ongoing SIOPEN-HR-NBL2 clinical trial (NCT04221035)155. Furthermore, Liquid biopsies will be used to facilitate clinical decision making in the EU-funded international MONALISA trial (EU- Grant Agreement number 101137028).
Renal tumors
Renal tumors comprise 5% of all pediatric cancers156. Important subtypes include Wilms tumor (WT)/ nephroblastoma, clear cell sarcoma of the kidney (CCSK), malignant rhabdoid tumor of the kidney (MRTK), pediatric renal cell carcinoma (RCC), and congenital mesoblastic nephroma (CMN)157,158,159,160. These subtypes display high heterogeneity in histology, genetic aberrations, and malignant potential157. WT is the most common type comprising 80-90% of cases, and hence will be the focus here161,162,163.
WT is an embryonal cancer containing undifferentiated blastemal, stromal, and epithelial elements, similar as during fetal development164. It is one of the most common malignant childhood tumors with roughly 95% occurring in patients below the age of 10165,166. The Children’s Oncology Group (COG) and The International Society of Pediatric Oncology-Renal Tumor Study Group (SIOP-RTSG) protocols comprise the two major directions of WT management, which mostly differ in surgery timing and consequent stratification for chemotherapy and radiotherapy,167,168. Outcomes for WT patients have significantly improved in recent decades with a five-year survival rate from 75% in 1975-1979 to 90% in 2003–2009169,170. Nonetheless, approximately 15% of patients eventually relapse with an OS rate of 50%171. These relapses occur mainly in the (stage and histology-based) intermediate-risk subgroups, and are often not recognized172. Also, although fine needle biopsies are considered rather safe with respect to tumor rupture risk, strict criteria for such diagnostic procedures are currently being used4,31,173. Consequently, a lack of histological confirmation could potentially lead to misdiagnosis and hence suboptimal treatment, especially in non-HR WT patients135. Finally, WT displays a high degree of intra-tumor genetic heterogeneity174,175. Hence, liquid biopsies could aid in renal tumor diagnosis, subtype identification, treatment guidance, risk-stratification, assessment of intra-tumor heterogeneity, and early relapse detection161,165,174. A comprehensive overview of studies involving analysis of liquid biopsy-based biomarkers in pediatric renal tumors is depicted in Supplementary Table 3.
Among pediatric solid tumors, WT presents with relatively high ctDNA concentrations, further demonstrating the potential for liquid biopsy application139,141,140. For example, Ruas et al.140 and Klega et al.139 found the second highest (following NB) plasma cfDNA levels in WT patients versus Ewing sarcoma, rhabdomyosarcoma, leiomyosarcoma, osteosarcoma, and benign teratoma patients. Next, van Zogchel et al.141 measured significantly higher ctDNA levels in WT (and NB) patients versus pediatric controls, while this difference was not significant in patients with rhabdomyosarcoma, lymphoma, or CNS tumors. Also, despite its genetic heterogeneity, WT displays several recurrent aberrations161,165. COG identified 1p loss, 1q gain, and 16q loss, as adverse prognostic factors, which have now been included for risk-stratification in the USA176,177. Also, 1q gain and 11p15 loss have been associated with relapse and death in WT, making these CNAs interesting targets for early relapse detection165. Indeed, several studies investigated plasma or serum-derived ctDNA for WT biomarkers including CNAs (1q, 7q, and MYCN gain, and loss of 1p, 7p, 11p15, and 16q), but also somatic mutations (including TP53, MYCN, WT1, and CTNNB1) and genomic methylation (Supplementary Table 3). A recent report from the COG prospective AREN0533 trial by Madanat-Harjuoja et al.178 used NGS approaches for ctDNA detection patients with stage III or IV WT. They detected ctDNA in serum of 41/50 patients and observed a trend toward worse event-free survival (EFS) for patients with detectable ctDNA (80% versus 100%). In contrast, none of the patients with undetectable ctDNA in serum relapsed and these all survived. Finally, they identified several CNAs (1q, 1p, and 16q) and single nucleotide variants (SNVs) in ctDNA of which some were not detectable in tumor biopsies178. Concurrently, van Paemel et al.148 identified several chromosomal aberrations (including the prognostic marker 1q gain) in cfDNA of WT patients that were not detectable in matched tissue DNA. Therefore, ctDNA analysis could potentially identify subclonal copy-numbers that might be missed when profiling a single tumor tissue at a specific site161. Still, other studies indicated that some genetic aberrations (CNAs and SNVs) in tumor biopsies were not detectable in corresponding cfDNA in some WT patients, suggesting the need for larger cohort studies that include harmonized methods for ctDNA isolation and detection for biomarker validation179,180.
Next to ctDNA alternative peripheral blood-derived analytes have been identified as potential WT biomarkers (Supplementary Table 3). For example, He et al.181 measured elevated levels of XIST, a lncRNA, in 49 WT patients using microarrays and RT-qPCR, and significantly correlated XIST upregulation with tumor staging and shorter survival time. Next, Ludwig et al.182 demonstrated a serum miRNA panel (most notably miR-100-50 and miR-130b-3p) that differentiated 43 WT patients from 13 healthy controls with an accuracy, sensitivity, and specificity of 79.6%, 69.2%, and 90.0%, respectively. Similarly, Schmitt et al.183 measured an elevated panel miRNA signature in 43 WT patients versus 19 healthy controls, but did not observe a significant difference in signature between patients prior and after chemotherapy. An elaborate description and explanation of miRNAs is provided in the germ cell tumor chapter.
Urine is increasingly being investigated as an alternative liquid biopsy matrix for renal tumor detection184,185,186. Especially renal tumor proximity, facilitation for serial-sampling, and the non-invasive nature make urine an interesting alternative27,180. Indeed, there have been several efforts investigating urine-based biomarkers in pediatric renal tumors (Supplementary Table 3). Early studies in the 90 s showed elevated levels of hyaluronidase, hyaluronic acid, and basic fibroblast growth factor in urine of WT patients187,188,189. More recently, Ortiz et al.190 identified multiple urinary protein biomarkers in 139 pediatric patients with different types of pediatric renal tumors. Particularly, high urinary prohibitin concentrations at diagnosis were significantly associated with relapse in WT patients. Also, the earlier described COG study by Madanat-Harjuoja et al.178 detected ctDNA in urine of WT patients, however, in a significantly smaller sample group versus matched serum samples (13/50 versus 41/50). Hence, more sensitive assays or larger sample volumes might be required for urine-based liquid biopsies.
Taken together, ctDNA detection, subsequent genetic targets, and other analytes are promising markers for WT risk-stratification and prognostication but require validation in larger cohorts178. Also, there is limited data available on serially collected liquid biopsy samples for monitoring treatment response and follow-up161. Finally, no biomarkers for reliable differentiation among different pediatric renal tumor subtypes have been identified so far. This is important to prevent misdiagnosis and suboptimal treatment for patients that are being treated for WT without histological confirmation3,31,135. Peripheral blood and urine liquid biopsy sampling, and subsequent ctDNA, miRNA, and proteomic analyses are therefore pursued in the ongoing SIOP-RTSG-2016 UMBRELLA clinical trial158.
Germ cell tumors
Germ cell tumors (GCTs) arise from primordial germ cells and derivatives during embryogenesis. Most GCTs develop in the gonads or along the midline structures of the body, but other locations including the central nervous system are common as well191,192. Pediatric GCTs are classified in two types depending on age group and histology. Prepubertal type I GCTs are mostly present in children aged 0–4 and may present as yolk sac tumor (YST), (generally) benign teratoma, or mixtures of these elements193. Within this group, GCTs are generally rare representing only 3% of all cancers5. Post-pubertal type II GCTs commonly arise from puberty to early adulthood and are represented by a wider range of histologies and subtypes (germinomatous GCTs and non-germinomatous GCTs), almost always being malignant192,193. There, pediatric GCTs are more common (accounting for 13% of all tumors for adolescents aged 15–19)5.
The serum tumor markers beta-human chorionic gonadotropin (β-hCG), alpha-fetoprotein (AFP), and lactate dehydrogenase (LDH) are clinical standards for GCT diagnosis and follow-up194. β-hCG and AFP are commonly linked to choriocarcinoma and (type I and II) YST, respectively194,195. In contrast, LDH is the least specific and rather used as indicator of cell turnover and burden of disease194. Generally, the lack of specificity and low sensitivity of these serum markers limits utility for histological subtype discrimination and relapse detection196,197,198. Therefore, there is a need for alternative biomarkers allowing more sensitive and specific GCT detection regardless of age, (malignant) subtype, and anatomical location without elevation in benign-GCTs and non-GCT tumors199. miRNAs are a promising alternative to conventional serum markers200. These small non-coding RNA molecules of 20-24 bases are involved in many biological pathways within multicellular organisms201. Being regulators of cell proliferation and differentiation, they influence numerous cancer-relevant processes such as proliferation, cell cycle control, apoptosis, cell differentiation, migration, and metabolism201. There, dependent of their mRNA target(s), they can function either as oncogenes or tumor suppressors197. Also, miRNA molecules are sensitive, specific, stable, and require low-cost equipment for isolation and detection in blood (serum and plasma), making them interesting liquid biopsy biomarkers for cancer detection and pathogenesis200.
The significance of miRNAs as alternative GCT biomarkers has been emerging in the last two decades200,202,203,204,205. Voorhoeve et al.204 were the first to demonstrate this by identifying miR-372 and miR-373 as potential oncogenes in testicular GCTs (TGCTs) through intervention with the p53 pathway in cell lines. Following a high throughput screening of 156 miRNAs in human GCT material, involvement of miRNA 371-373 clusters in type II malignant GCTs (MGCTs) was confirmed206,207. Later, Palmer et al.208 demonstrated overexpression of miR-371-373 and miR-302-367 clusters in all MGCTs, regardless of age, site, and histological subtype. Following demonstrated isolation and detection from blood199,209,210,211, this miRNA panel has hence been shown to be a highly sensitive and specific liquid biopsy based approach for MGCT detection200,202,212.
Murray et al.209 were the first to document involvement of liquid biopsy-derived miRNAs in pediatric GCTs. Using a multiplex RT-qPCR approach, they measured elevated miRNA levels of the 371-373 and 302 clusters in a 4-year-old boy with a YST at diagnosis and baseline levels during clinical follow-up, with parallel AFP kinetics. Subsequently and using exclusive pediatric cohorts, they demonstrated suitability of specific miRNAs (particularly miR-371a-3p) for pediatric MGCT diagnosis at different anatomical locations (intra and extracranial) and subtypes (choriocarcinoma, YST, and mixed MGCTs (MMGCTs))196,213,214. These miRNAs could also detect MGCT relapse while AFP and β-hCG levels were still below threshold levels for diagnosis213,214. Finally, they allowed discrimination of intracranial MGCT (iMGCT) from intracranial non-GCT tumors196. Concurrently, Schonberger et al.215 recently demonstrated suitability of these miRNA clusters for iMGCT diagnosis as well. Finally, Saliyeva et al.216 recently measured elevated levels of miR-302/367 and 371-373 clusters in 20 pediatric patients with various MGCTs. There, miR-371,372,373,367, and 302d levels were significantly higher in GCT patients (various subtypes) versus controls. miRNA levels of the 371-373 cluster were highest in patients with pure dysgerminoma versus non-germinomatous GCTs. MiR-367 levels were elevated regardless of morphological GCT type, but highest in patients with stage III-IV disease216. A comprehensive overview of studies involving liquid biopsy analysis in pediatric GCT patients is depicted in Supplementary Table 4.
The studies discussed above support the use of the miR-371-373, 302, and 367 clusters as sensitive and specific liquid biopsy biomarkers for pediatric GCT diagnosis and monitoring. However, validation in large pediatric cohorts is required as the provided studies only included small patient groups since most liquid biopsy-derived miRNAs studies have focused on post-pubertal type II TGCTs197,213,214. This is especially relevant for prepubertal type I GCTs, which represent a distinct disease versus type II GCTs that seem histologically and biologically similar among adolescents and adults193. Two currently running clinical trials that include liquid biopsies and focus on pediatric GCTs include the MAKEI-V and MAGIC trials217,218.
While miRNAs from the specified clusters have shown to be involved in all MGCTs, they fail to identify the teratoma subtype (both type I and II)202,208,219. Teratomas consist of a disordered mixture of differentiated cell types possibly from all three somatic germ cell layers. They are generally benign (type I), since their formation is the result of improper developmental differentiation rather than via an oncogenetic driver193. Nonetheless, they are therapeutically relevant as they are generally resistant to chemotherapy and surgical excision is the only therapeutic option. Therefore, they need to be distinguished from MGCTs where classical serum markers are generally not useful for confirming this GCT subtype219. Lobo et al.219 combined miR-371a-3p and hypermethylated RASSF1A detection, of which the latter is a common feature in several solid malignancies. This combined liquid biopsy panel allowed for 100% sensitivity in detecting diverse (young) adult TGCT subtypes, including (type II) teratoma. Then, Nappi et al.220 suggested the combination of miR-371a-3p and miR-375-5p to be highly accurate for (type II) teratoma detection. However, the level of informativity of miR-375 has been questioned in other papers216,221,222,223,224,225. Future studies should include large pediatric cohorts with type I prepubertal GCTs to investigate whether this combined panel also allows for prepubertal teratoma detection.
Sarcomas
Osteosarcoma
Osteosarcoma is the most common bone tumor, primarily affecting children, teens, and young adults, and makes up 2% of cancers in children aged 0–14, and 3% in teens aged 15–195,226,227. It arises from a malignant transformation of mesenchymal cells producing osteoid and/ or immature bone, and can present in any bone of the body with the most common sites being the metaphyses of long bones (distal femur, proximal tibia, and the proximal humerus)228,229. Standard treatment of patients with high-grade osteosarcoma (majority of patients) involves multiagent induction chemotherapy followed by surgical resection for local tumor control with focal therapy directed to metastatic sites (10–20% of patients have metastasis at the time of initial diagnosis)34,226. Osteosarcoma incidence has been relatively stable over the last 40–50 years with a reduction in mortality rates, primarily due to the introduction of multiagent chemotherapy226. Nonetheless, metastasis and chemoresistance are still a serious problem230. Metastases are detected in roughly 10–40% of osteosarcoma patients (higher percentage in low and middle-income countries), and patients with metastasized disease have a 5-year survival rate below 30%230.
Next to the advantages of liquid biopsies addressed before, implementation within bone tumors has additional value since bone tumor biopsies can be dangerous (e.g., when in proximity to the spinal cord) and difficult to collect231. Here, we will focus on the most described liquid biopsy-based analytes for osteosarcoma, being ctDNA, CTCs, miRNAs, and lncRNAs. A comprehensive overview of liquid biopsy-based analytes (including less-studied examples) and involved studies is provided in Supplementary Table 5.
Osteosarcoma displays high genomic complexity, instability and heterogeneity, resulting in a high number of SVs that can cause fusion genes, alternative splicing, CNAs, and gene expression dysregulations31,130,228,231,232,233. This genomic heterogeneity and lack of recurrent genetic aberrations might limit the implementation of ctDNA targeted assays such as ddPCR within osteosarcoma versus other pediatric tumors3,234. Nonetheless, several studies detected ctDNA in peripheral blood of osteosarcoma patients at diagnosis and during follow-up, and demonstrated a correlation between ctDNA levels, treatment response, and survival (Supplementary Table 5). Most studies utilized NGS approaches for identification of the complex SVs associated with osteosarcoma139,148,232,235,236,237,238. For example, Shulman et al.238 performed ultra low pass-WGS (ULP-WGS) for ctDNA detection in 72 patients with primary localized osteosarcoma. Although statistically insignificant, ctDNA was detectable at diagnosis in 41/72 of these patients, and increased ctDNA levels were correlated with increased risk of death. DNA methylation is an alternative marker for ctDNA detection that could be particularly interesting for osteosarcoma as methylation changes are often retained through tumor evolution, and are only moderately confounded by tumor genetic heterogeneity239. Lyskjær et al.234 employed methylation-specific ddPCR for the discovery and validation of methylation-based biomarkers for ctDNA detection in 72 osteosarcoma patients. They detected ctDNA in 50/72 or 29/72 of pre-operative plasma samples (based on one or two marker thresholds). Also, ctDNA was detected in 5/17 post-operative plasma samples, which in four cases was associated with or preceded relapse. Both pre-operative cfDNA levels and ctDNA detection independently correlated significantly with OS234. Finally, van Paemel et al.236 used cfRRBS for methylome profiling for the correct diagnosis of 4/4 osteosarcoma cases within a pool of 60 samples derived from patients harboring different solid tumors.
As tumor recurrence and metastasis are the leading causes of death in osteosarcoma patients, there is a particular need for biomarkers assessing tumor clonal evolution, prediction of treatment resistance, and metastatic potential and/ or early relapse detection240,241. CTCs can be an additional sensitive target for this purpose. CTCs circulate freely in the bloodstream with genetic resemblance to their tumor origin3. They constitute a very small fraction of cells in the bloodstream and require validated methods for detection, enrichment, and enumeration3. CTC sampling provides live information regarding disseminated tumor cell heterogeneity, and has already been implemented in clinical practice for metastatic tumor prognosis of several adult tumors such as colorectal, breast, and prostate cancer231,242. CTCs have been described as potential predictive and prognostic biomarkers for osteosarcoma metastasis240,241,243,244,245. Dai et al.243 recently investigated CTCs using the Canpatrol CTC detection platform in 50 osteosarcoma patients (23 children and 27 adults, 34 with localized disease and 16 with metastatic disease). They detected CTCs in 43/50 patients and counts were significantly higher in Enneking Stage III (regional or distant metastasis) patients versus IIB (no metastasis) patients243,246. Also, CTC detection with positive Insulin-like growth factor mRNA-binding protein 3 signal best diagnosed osteosarcoma metastasis (87.5% sensitivity, 82.4% specificity). Finally, serial monitoring in one patient suggested that total CTCs were associated with disease progression243. Future studies focusing on CTC detection and implementation in osteosarcoma should include large cohorts involving serial sampling during treatment and follow-up. This will allow for dynamic CTC monitoring to assess chemotherapy response and timely detection of recurrence or metastasis, as well as investigate relationships among CTC detection and survival240,243.
The potential role of miRNAs and lncRNAs as biomarkers for osteosarcoma has been extensively studied over the last 10 years, and several studies showed promising diagnostic and prognostic performance (Supplementary Table 5). For example, Xie et al.247 found significantly higher circulating miR-26a-5p levels in serum of 243 osteosarcoma patients versus 96 healthy controls. Patients with high miR-26a-5p levels also had poorer OS versus those with low levels, and multivariate analysis showed miR-26a-5p to be an independent osteosarcoma risk factor247. Most other (older) miRNA and lncRNA studies for osteosarcoma have already been extensively reviewed elsewhere34,129,137,228,231,248,249,250,251,252,253,254,255,256. Importantly, despite the extensive number of publications and promising performance of these RNAs in osteosarcoma, there has been little overlap in candidates and consistency in results among studies. Also, several investigated biomarkers are not specific for osteosarcoma (such as miR-21 and miR-20a)250. Finally, most studies were performed in Asia (mostly China), thereby limiting the translation of biomarker performance for osteosarcoma to other countries and regions255. This, again, demonstrates the need for large-scale, prospective, international multi-center clinical trials. Due to osteosarcoma heterogeneity, several methods and targets should be included in such trials including ctDNA quantification and subsequent broad sequencing approaches, CTC enumeration and profiling, and RNA (miRNA and lncRNA) profiling. Combining these different analyses hopefully enables development of a sensitive and specific biomarker panel for osteosarcoma.
Ewing sarcoma
Ewing sarcoma (EWS) is a malignant tumor predominantly occurring in bone (mostly pelvis, femur, tibia, and ribs), and in soft tissues (thoracic wall, gluteal muscle, pleural cavities, and cervical muscles)257. Although EWS origin is a matter of debate, it is thought to involve neural crest cells and/ or mesenchymal stem cells.258,259,260. Despite being rare, EWS is the second most common bone tumor in children, adolescents, and young adults (following osteosarcoma) comprising roughly 2% of childhood malignancies44,257,261. Peak incidence occurs around 15 years of age, and occurrence in older adults is very rare257,260. EWS is highly aggressive and survival rate varies from 70-80% for patients with localized disease to approximately 30% for those with metastasis257. Standard of care consists of a multimodal treatment regimen including multi-agent chemotherapy, surgical resection, and radiotherapy231.
EWS is characterized by chromosomal translocations yielding fusion genes and corresponding fusion proteins that promote tumorigenesis257,262. These fusion proteins contain a member of the FET family of proteins (most commonly) being RNA-binding proteins involved in transcription and splicing, together with different members of the E26 transformation-specific (ETS) family of transcription factors, which are involved in cell proliferation, differentiation, cell-cycle control, angiogenesis, and apoptosis257,261. 90% of EWS cases are driven by a t(11;22)(q24;q12) chromosomal translocation, leading to the EWSR1-FLI1 fusion gene154,257,261,262,263. EWSR1-ERG is the next most common fusion complex (5-10%) and other EWSR1 gene fusions (with ETV1, ETV5, or FEV1) are rare (<1% of total cases)261. Similarly, some variant fusions between other FET gene families (FUS and TAF15) with ETS family members have been described257. Apart from these fusion complexes, EWS is a relatively genetically silent cancer with few recurrent mutations or CNAs31.
Fusion genes such as EWSR1-FLI1 provide a clinically relevant liquid biopsy-based target for EWS detection and monitoring231. Initial efforts investigating this utilized RT-PCR approaches for fusion gene detection in CTC-derived RNA in EWS patients264,265,266,267,268,269. While fusion transcripts were indeed detectable, the clinical significance and prognostic value of these findings were inconclusive among studies270. During the last 20 years, focus shifted from CTC detection to ctDNA analysis. Technological advances such as ddPCR allowing accurate nucleic acid detection and analysis recently rekindled interest in liquid biopsy implementation in EWS270. Indeed, over the last 7 years, many novel studies started investigating peripheral blood or bone marrow derived EWSR1 fusion genes as biomarker for pediatric EWS (Supplementary Table 6). For example, Krumbholz et al.271 evaluated the predictive and prognostic value of ctDNA in 102 EWS patients using EWSR1 fusion primers and probes for sensitive and specific ctDNA detection in plasma by ddPCR. Pretreatment ctDNA copy numbers were correlated with EFS and OS, and ctDNA levels decreased below limit of detection (LOD) after two blocks of induction chemotherapy. Also, persistence of ctDNA after chemotherapy was a strong predictor of poor outcomes and ctDNA levels correlated well with clinical risk factors (tumor size, osseous or pelvic location, metastasis, and age at diagnosis) suggesting suitability for early prediction of treatment response and outcome271. Next, Schmidkonz et al.272 used ddPCR for ctDNA detection based on individual EWSR1-FLI1 and EWSR1-ERG fusion sequences. They found a significant correlation between ctDNA signals and tomography signaling (18F-FDG-PET/CT), for diagnosis, treatment response, and relapse detection in 20 EWS patients.
Next to ddPCR approaches, sequencing-based methods also enabled ctDNA detection and analysis including identification of precise fusion breakpoints and novel DNA rearrangements (Supplementary Table 6). For example, Shah et al.237 used a deep sequencing approach for common translocation detection in plasma from patients with common sarcomas including EWS. They found that ctDNA levels correlated with metastatic status and clinical response, and quantified significant ctDNA levels several months before relapse was clinically detected by imaging. More recently, Christodoulou et al.9 demonstrated a hybridization-based capture panel targeting EWSR1 fusions that correctly identified 10/12 EWS patients within a pool of 73 pediatric solid tumor patients. Finally, Seidel et al.273 used WGS for the identification of exact EWS-FLI1 fusion gene breakpoints followed by ddPCR for specific ctDNA detection. They showed that ctDNA detection in peripheral blood of 6 pediatric EWS patients allows for real-time monitoring of MRD activity, although patients displayed high variability in ctDNA signal among individuals.
Next to ctDNA, EWSR1 fusion genes can also be detected in ctRNA and small extracellular vesicles (sEVs) using ddPCR and RT-qPCR274,275,276. However, a comparison by Bodlak et al.276 suggested that ctRNA signals seem to be lower and statistically less reliable versus ctDNA detection. A comprehensive overview of relevant studies focusing on fusion gene detection in liquid biopsies for pediatric EWS is depicted in Supplementary Table 6.
Taken together, common EWSR1 fusion gene detection provides a clinically relevant genetic biomarker panel for EWS detection. The studies mentioned here demonstrate the potential for several clinical applications including EWS diagnosis, early risk-stratification and prognosis, monitoring of therapy and clonal evolution, follow-up, MRD detection, and relapse prediction (Supplementary Table 6). The frequency of EWSR1 fusion gene occurrence in EWS supports potential clinical use for most patients130. Also, experimental studies suggest that EWS tumor cells are dependent on such fusion genes reducing the risk for target loss by clonal evolution during treatment31,262,277,278. Hence, ctDNA quantification and analysis using ddPCR and deep NGS approaches targeting the characteristic fusion genes of EWS should be the main focus of future large cohort studies for validation of these potential EWS-specific liquid biopsy-based biomarkers279. Given the poor prognosis of EWS patients with metastatic or recurrent disease, risk-stratification is a particularly important target for improving outcome. Using ddPCR for fusion gene detection requires design of patient-specific primer-probe sets, which takes time (approx. 2 weeks)271. Hence, such assays must be developed in a clinically relevant timeframe for informing risk-stratification. After development, assay implementation for follow-up can be done in a faster timeframe271. The international INTER-EWING1 and German iEuroEwing studies are examples of EWS clinical trials that include liquid biopsy analyses280,281.
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is the most common soft tissue tumor in children accounting for roughly 4.5% of all pediatric cancer cases282,283. It is the third most common extracranial solid childhood tumor following NB and WT, and most cases are diagnosed in children below the age of 6283,284. The most common sites of primary disease include the head and neck region, the genitourinary tract, and the extremities283. RMS treatment generally involves systemic treatment with intensive chemotherapy, and local surgery and/or radiotherapy285. Development and refinement of multimodal treatment regimens significantly improved pediatric RMS survival282. Nonetheless, survival rate depends heavily on the extent of disease at initial diagnosis, and survival for patients with metastatic or relapsed disease remains poor286,287. There are two RMS subtypes, being fusion gene-positive RMS (FP-RMS) and fusion gene-negative RMS (FN-RMS), depending on presence of PAX3-FOXO1 or PAX7-FOXO1 (hereafter referred to as PAX3/7-FOXO1) fusion genes caused by t(2;13)(q35;14) and t(1;13)(p36;q14) chromosomal translocations, respectively2,34,129,286,288. These characteristic fusion genes and subsequently fusion proteins drive the malignant phenotypes of FP-RMS tumors286. FP-RMS has alveolar histology, appearing histologically similar to pulmonary parenchyma129. In contrast to FP-RMS, the genetic landscape of FN-RMS is heterogeneous and does not involve specific chimeric genes285. FN-RMS tumors often include epigenetic modifications, various CNAs (e.g., chromosomal 2, 8, 12, 13 gain and 11p15.5 loss), or gene translocations/ mutations (e.g., TFCP2, VGLL2, MYOD1, NCOA2, DICER1)286,289,290,291. FN-RMS generally has embryonal histology, being composed of cells resembling immature skeletal myoblasts, although FN-RMS cases that display alveolar histology also exist129,292. Patients with FP-RMS have a worse prognosis than those with FN-RMS2. Consequently, FOXO1 status has been identified as prognostic risk factor for RMS by the COG and The European pediatric Soft Tissue Sarcoma Group (EpSSG)293,294,295. Historically, RMS main types were discriminated based on histology, being alveolar RMS (ARMS) (20-25% of RMS cases) and embryonal RMS (ERMS) (70-75% of cases), instead of FP-RMS and FN-RMS2,282. Many of the studies reviewed here still adhered to this classical differentiation. As such, we use this terminology from here on as well.
Early studies in the 2000s that investigated liquid biopsy-derived biomarkers for RMS mostly focused on RNA-based MYOD1, MYOG, and α/γACHR transcripts122,296,297,298,299 (Fig. 2b and Supplementary Table 7). During the last five years, liquid biopsies in RMS have regained attention. The PAX3/7-FOXO1 translocations are very well-suited as targets for detection of liquid biopsy-based biomarkers. Indeed, these fusion genes have demonstrated their potential in several liquid biopsy targets including ctDNA, CTC-derived mRNA, and cfRNA (Supplementary Table 7). To illustrate the potential of these fusion genes for RMS detection, Klega et al.139 used targeted sequencing and WGS for the detection of PAX3-FOXO1 rearrangements in plasma-derived cfDNA from 7 ARMS patients. ctDNA changes corresponded with therapy response, with ctDNA often becoming undetectable at the start of the second chemotherapy cycle. Given that ERMS is the most frequent RMS type, there is an urgent need for biomarkers to identify and monitor this subtype in liquid biopsies. However, considering the heterogeneous genetic landscape of ERMS, this has been quite challenging so far 286. A comprehensive overview of studies focusing on liquid biopsy identification in pediatric RMS is indicated in Supplementary Table 7. Here, we will focus on recent studies that included the largest patient populations.
RMS-derived ctDNA can be detected by targeting tumor specific CNAs, patient-specific translocation breakpoints, CNVs, and/ or SNVs with different methods (Supplementary Table 7). For example, Ruhen et al.300 utilized ddPCR, panel sequencing, and whole exome sequencing (WES) for ctDNA detection in 28 RMS patients. ctDNA was detected in 21/25 pre-treatment plasma samples, and ctDNA levels at diagnosis were significantly higher in patients with unfavorable tumor sites, positive nodal status, and metastasis. Also, in serial plasma samples, ctDNA level fluctuations corresponded to treatment response300. More recently, Abbou et al.301 used ULP-WGS and targeted sequencing for ctDNA detection in 124 RMS patients. ctDNA was detectable in 40 cases, and patients with detectable ctDNA at diagnosis had significantly worse outcomes versus patients without detectable ctDNA (OS, 33.3% versus 83.2% and 39.2% versus 75%, depending on the subgroup)301. Next, Lak et al.302 employed ddPCR, shWGS, and cfRRBS for ctDNA analysis including methylated RASSF1A detection in 57 RMS patients. ctDNA was detected in 39/57 RMS patients at diagnosis, and most samples scored negative after the first course of chemotherapy. This transition is consistent with an earlier described study by Klega et al.139 Methylated RASSF1A was specifically detected in 21 of 57 patients and its presence significantly correlated with poor outcome (5-year EFS of 46.2% versus 84.9%). This prognostic effect of methylated RASSF1A was most apparent in patients with metastatic disease. Also, cfRRBS allowed for correct classification of the ERMS subtype in 21 out of 25 samples302. The potential of cfRRBS for pediatric solid tumor type classification has already been described earlier by van Paemel et al.236. There, they correctly classified RMS in 14 out of 17 cases (within a pool of 60 samples from a pediatric population with different solid tumors). ERMS and ARMS subclass identification was correct in 13 out of 14 cases.
Next to ctDNA, other analytes including CTCs, mRNA, miRNAs, exosomes, circulating protein, and monocytes have been suggested as liquid biopsy-based biomarkers for RMS (Supplementary Table 7). For example, Poli et al.303 measured significantly higher IGFBP2 protein and corresponding autoantibodies in 114 RMS patients versus healthy controls. IGFBP2 protein also identified metastatic patients with worse EFS, and both IGFBP2 and antibodies negatively correlated with OS303. Then, Lak et al.289 developed an RNA panel for detection of RMS-specific mRNA from the cellular fraction of blood and bone marrow, using multiplexed RT-qPCR in 99 RMS patients. Metastatic disease, detected by mRNA significantly outperformed standard of care testing, and the five-year OS was 54.8% for 33/99 RNA-positive patients versus 93.7% for 66/99 RNA-negative patients. When MRNA and methylated RASSF1A were combined in paired samples, the five-year OS was 100% versus 36% for double negative and double positive samples, respectively302. Finally, Urla et al.304 investigated expression of TKTL1 and Apo10 epitopes on monocytes in 29 RMS patients. Both epitopes scored positive in 96.5% of RMS patients and negative in the control population.
Taken together, the studies indicated in Supplementary Table 7 including those mentioned above demonstrate the potential of liquid biopsies for RMS diagnosis and risk-stratification, subtype identification, therapy monitoring, and outcome prediction. Given that survival in patients with metastatic or relapsed RMS is low, there is a particular need for more sensitive prognostic monitoring tools for response monitoring and relapse surveillance286,300,301,305. While several RMS studies involved serial liquid biopsy sampling including relapse samples, with some explicitly elaborating on relapse detection prior to confirmation using standard methods, population cohorts remain scarce146,289,300,302,306,307,308,309. Future trials should focus on detection of PAX3/7-FOXO1 fusion genes using ddPCR and deep targeted NGS approaches. For PAX3/7-FOXO1 negative patients, broader sequencing approaches should be applied for identification of patient-specific SVs. Two currently running large-scale international trials (FaR-RMS and ARST1431) are including liquid biopsy studies and might offer further insight in the clinical and predictive value of these biomarkers310,311.
NRSTS
Non-rhabdomyosarcoma soft tissue sarcomas (NRSTS) constitute a heterogenous group of malignant extraskeletal mesenchymal tumors312. NRSTS includes over 35 distinct histologies, many of which are exceedingly rare313. Incidence of pediatric NRSTS types vary by age, as some typically occur in small children (e.g., infantile fibrosarcoma and malignant rhabdoid tumors), others in adolescents and young adults (e.g., synovial sarcoma (SS)), and some being more common in adults and rare in children (e.g., liposarcoma and leiomyosarcoma)314,315. Generally, NRSTS survival for patients with metastatic disease remains low316,317. For example, a report from the COG investigating pediatric SS (the most common NRSTS type) stated an OS of 97.7% and 84.9–89.5% for low and intermediate-risk patients, respectively317. In contrast, patients with metastatic disease (high-risk) only had an OS of 12.5%. In addition, a recent study from the European pediatric Soft tissue sarcoma Study Group reported a 3-year EFS and OS of 15.4% and 34.9%, respectively, for pediatric patients suffering from metastatic NRSTS318. This highlights the clinical value for liquid biopsy approaches for MRD detection as early prediction for metastatic disease319.
Gene fusions have been described as driver events in many NRSTS, of which most are recurrent in the same histologic subtype and, therefore, may serve as potential targets for liquid biopsy-based detection320. Indeed, some studies utilized this approach for SS (SS18-SSX fusions) and desmoplastic small round cell tumor (DSRCT) (EWSR1-WT1 fusions) detection237,268,309,321,322,323. Nonetheless, studies investigating liquid biopsy-based biomarkers for pediatric NRSTS are limited, as most have focused on adult NRSTS patients306,321,322,323,324,325,326,327,328,329,330,331,332. Of note, while the molecular events driving these tumors may be the same in pediatric and adult patients, biology and prognosis often differs for reasons not yet elucidated, indicating that findings from adult patients cannot easily be translated to pediatric patients333. The few examples focusing on pediatric cases are briefly discussed here and indicated in Supplementary Table 8.
Athale et al.268 detected EWSR1-WT1 fusion transcripts in peripheral blood (but not in bone marrow) in two out of three pediatric patients with DSRCT at diagnosis using RT-PCR. More recently, Cahn et al.149 used a commercially available NGS tool for detection of molecular abnormalities in plasma derived cfDNA of 45 pediatric patients including six that were suffering from NRSTS (five subtypes). They detected an EWSR1 fusion and PIK3CA mutation in one DSRCT patient and no aberrations in the other five patients. Next, Colletti et al.334 investigated the exosomal miRNA profile in plasma samples of three adolescents suffering from DSRCT using RT-qPCR, and identified 55 miRNAs that were significantly modulated compared to healthy controls, proposing their panel as promising biomarkers for characterizing disease status in DSRCT patients. Finally, Christodoulou et al.9 used targeted sequencing and LP-WGS for detection of CNAs and gene fusions in plasma-derived cfDNA in 73 patients including two with SS and two with malignant peripheral nerve sheath tumor (MPNST). CNAs at diagnosis were detected in one out of two SS and one out of two MPNST patients. In addition, they identified two mutations in one out of two MPNST patients. In the other studies provided in Supplementary Table 8, either no liquid biopsy signal was detected or no discussion on NRSTS results was given due to the focus being on other pediatric tumor types140,237,309,335,336.
The lack of studies focused on liquid biopsies for pediatric NRSTS, together with the rarity and heterogeneity of individual NRSTS types, stresses the need for multi-center cooperative initiatives to enable prospective clinical trials with sufficiently large population cohorts. MyKIDS is an upcoming clinical trial with the aim of molecular profiling of NRSTS in children, adolescents, and young adults337. This trial will include a work package for investigation of ctDNA-derived biomarkers at primary diagnosis, during treatment, follow-up, and at relapse or progressive disease, using different methods (DNA methylation profiling, WES, copy number profiling, and assessment of patient specific breakpoints)337.
Liver tumors
Primary pediatric liver tumors are rare, accounting for approximately 1% of all malignancies in children below 18338. Hepatoblastoma (HB) is the most common type (37%) followed by hepatocellular carcinoma (HCC, 21%) and very rare hepatocellular malignant neoplasm-not otherwise specified (HEM-NOS) tumors339,340. HB mostly affects children below the age of three, and although uncommon, incidence has been increasing recently, likely due to increased survival of premature infants339,341. In contrast to HB, pediatric HCC occurs mostly in children aged ≥10 496. Also, etiological predisposition of pediatric HCC is different from adult HCC suggesting a possibly different pathogenesis342. Identification of liver tumor type is essential for treatment strategy as HB is very sensitive to chemotherapy while HCC responds poorly and management relies more on a surgical approach338. Treatment plans are individualized and based on patient age, histopathological and imaging information, and serum AFP levels343. Patients are considered cured when tumor lesions are no longer visible by imaging (including lung metastases) and/ or all (residual) tumors are completely removed surgically, and serum AFP levels have dropped back to normal343.
Pediatric liver cancer diagnosis and monitoring relies on serum AFP measurements, tumor biopsies, and imaging techniques343. However, AFP measurements can be misleading as not all liver tumors secrete AFP (more often the case for HCC)339,344. Also, infants (premature babies in particular) can show high physiological AFP levels343. In addition, discrimination among liver cancer subtypes still mostly relies on invasive tumor biopsies345. Finally, repeated imaging procedures can increase future oncologic risk343. Therefore, liquid biopsies could complement existing methods and provide a valuable tool for liver cancer diagnosis, therapy response monitoring, follow-up, and MRD detection3. A comprehensive overview of studies involving liquid biopsy-based biomarkers in pediatric liver cancer patients is depicted in Supplementary Table 9.
miRNAs have been described as potential liquid biopsy-based biomarkers for pediatric HB106,129,346,347. Firstly, Murray et al.106 demonstrated suitability of miR-122-5p, miR-483-30, and miR-205-5p for differentiating HB diagnosis from NB (including non-HB liver involved tumors). Next, Liu et al.346 suggested miR-21 as a diagnostic and prognostic biomarker in HB patients. They also showed superiority of miR-21 versus serum AFP levels for HB diagnosis and indicated miRNA-21 as an independent predictor of EFS for HB patients346. Finally, Jiao et al.347 suggested miRNA-34a, miRNA-34b, and miRNA-34c as diagnostic and prognostic biomarkers for HB. Although this miRNA panel was not superior versus serum AFP for HB diagnosis, it did show superior prognosis prediction versus other risk factors (AFP levels, metastasis, vascular invasion, and advanced disease stages)347.
Next to miRNAs, other liquid biopsies-based biomarkers have been described for implementation in pediatric liver tumors (Supplementary Table 9). Firstly, protein-based biomarkers can be used for identification of tumor-associated markers and therapeutic targets348. Zhao et al.349 used SELDI-TOF-MS as a proteomic screening tool for non-inflammatory protein markers for pediatric HB. They revealed significantly reduced expression of apolipoprotein A-1 (Apo A-I) in HB patients versus healthy controls, suggesting it as a biomarker for early HB diagnosis349. Additionally, cfDNA could provide specific genetic biomarkers for pediatric liver cancer4,350. Despite the low mutational burden of HB (as in most pediatric cancer types), roughly 50-90% of HB patients harbors recurrent mutations in the CTNNB1 gene341. CTNNB1 detection in with plasma-derived ctDNA from patients with localized HB has been demonstrated by Kurihara et al.235 and Kahana-Edwin et al.350 using NGS and ddPCR approaches. By serial sampling along the course of therapy and follow-up, CTNNB1 could provide a sensitive mechanism for monitoring tumor dynamics and treatment response350. Finally, CTC sampling from liquid biopsies provides live information regarding disseminated tumor cell heterogeneity and response to therapy351. Recently, Espinoza et al.343 (article in preprint) demonstrated a CTC quantification method for pediatric patients suffering from HB and HCC (including the fibrolamellar carcinoma subtype) based on indocyanine green (ICG) staining followed by fluorescence microscopy and flow cytometry. ICG accumulation was specific for HB and HCC cells versus controls (non-malignant liver cells, non-liver tumor cells, and non-malignant cells). Also, CTC burden mirrored patients’ responses to therapy thus providing potential method for outcome prediction343.
Taken together, the studies discussed provide different potential liquid biopsy-based biomarkers for implementation in pediatric liver cancer oncology, including miRNAs, a protein, ctDNA, and CTCs (Supplementary Table 9). However, patient cohorts in most studies were very low, and there is no overlap in target identification among studies (except for CTNNB1 mutations). Hence, there is an initial need for more screening studies focusing on biomarker identification8. Next, all studies (except for343) only included HB patients. This makes sense given HB is the most frequent type of pediatric liver cancer339. Nonetheless, given the different treatment regimens for HB and HCC, accurate diagnosis between both types and hence inclusion of HCC patients in future studies is clinically important338. The morphological overlap between HB and HCC can make current histological diagnosis difficult338. Therefore, it is desired that specific liquid biopsy-based biomarkers could complement current methods for distinction among both malignancies3.
Summary of liquid biopsy-based biomarkers
Within the pediatric solid tumor type selection of this review, most literature has been published on NB. There, most studies were based on detection of characteristic genetic aberrations in plasma-derived ctDNA, and targets with high potential for clinical implementation were identified (Supplementary Table 2). Pediatric renal tumors display genetic heterogeneity and hence most studies utilized NGS approaches for biomarker identification in WT based on recurrent CNAs and mutations (Supplementary Table 3). Due to renal tumor localization, urine provides an additional interesting liquid biopsy matrix, but current methods lack sensitivity for proper implementation. miRNAs of the 371-373, 302, 367 clusters appear overexpressed in all MGCTs, with the exception of teratoma, and hence provide interesting targets (Supplementary Table 4). Nonetheless, validation in prepubertal type I GCTs is required. Osteosarcoma is genetically heterogeneous, and several liquid biopsy-based analytes have been investigated, including complex SVs, miRNAs, lncRNAs, and CTCs (Supplementary Table 5). Still, there is very little overlap in consistent targets among studies. EWS and ARMS are characterized by gene fusions that provide relevant targets for detection (EWSR1-FLI1 and PAX3/7-FOXO1, respectively), while ERMS does not display such fusions and characteristic biomarkers including patient specific SVs are less established (Supplementary Tables 6 and 7 for EWS and (A/E)RMS, respectively). Few pediatric studies focused on NRSTS and liver cancer patients, and more exploratory studies are needed for proper target identification (Supplementary Tables 8 and 9, respectively). Characteristic fusions for NRSTS (such as SS18-SSX for SS and EWSR1-WT1 for DSRCT), and CTNNB1 mutations for HB provide potential starting points.
Clinical applications and future directions
Based on published studies and future trials, clinical implementation of liquid biopsies in pediatric solid tumors is anticipated for several applications. Firstly, in a potential cancer screening setting for early diagnosis. However, this seems dubious for the general pediatric population and will likely only be relevant for specific risk groups, such as children with genetic predisposition. More relevant is risk-stratification following diagnosis and subsequent guidance of treatment management, which has already demonstrated clinical benefit in adults352,353. For many pediatric cancers, outcome differs significantly among risk groups, and several risk factors detectable in liquid biopsies have been discussed in our review354. Hence, in the context of personalized medicine and minimizing treatment-related morbidity, liquid biopsy-informed risk-stratification could guide appropriate treatment intensification or de-escalation. Serial sampling during treatment periods could similarly aid such treatment decisions. In pediatric acute lymphoblastic leukemia patients, chemotherapy was reduced successfully without affecting survival based on bone marrow-informed MRD detection throughout chemotherapy courses in a clinical setting355,356. Similarly, intensified therapy significantly improved outcome for patients with intermediate and high MRD levels. We discussed several pediatric studies for different solid tumor types that demonstrated significantly reductions in liquid biopsy-based biomarker signals during the course of therapy. This urges for implementation in clinical studies for informing patient-specific treatment plans as well. Next, by mitigating tumor heterogeneity, liquid biopsy sampling allows molecular profiling, monitoring of clonal evolution, and hence identification of therapy-resistant subclones, as demonstrated in adult and pediatric studies357,358. Deep sequencing approaches on ctDNA material could allow spatiotemporal genomics and gene expression profiling for identification of relevant cancer-associated genetic targets and pathways, and enable opportunities for targeted treatment3. Indeed, several liquid biopsy-based biomarkers mentioned in this review are eligible for targeted treatment, including ALK mutations for NB and RMS, and EWSR1-FLI1 fusions for EWS359. Finally, liquid biopsy serial sampling during follow-up could monitor MRD levels, predict outcome, and inform decisions regarding need and frequency for adjuvant therapy. Relapse is a prognostic factor for poor outcome and is the primary cause for cancer related death in children4,354. In different adults cancers, ctDNA and/or cfDNA detection during follow-up has already been shown to outperform imaging and standard biomarkers for disease recurrence and relapse detection, with subsequent adjuvant therapy leading to improved outcome360,361,362,363,364. While we discussed some examples indicating liquid biopsy suitability for MRD monitoring versus conventional methods during follow-up, dedicated studies are lacking237. The upcoming MONALISA trial mentioned earlier aims to investigate liquid biopsy efficacy during follow-up versus standard methods in a clinical setting for NB patients.
In practice, during the screening and diagnosis phase, NGS approaches could complement standard of care for diagnostic molecular profiling of tumor material. There, sequencing strategies would involve targeted deep approaches in pediatric tumors with recurrent genetic or epigenetic aberrations (e.g., NB, EWS, ARMS, NRSTS, and HB) or more shallow but wider approaches for tumors with genetic heterogeneity (e.g., renal tumors, osteosarcoma, and ERMS), focusing on targets as displayed in Table 1. This genetic profiling should inform risk-stratification and subsequent treatment selection, and guide development of cost-effective and highly sensitive patient-specific ddPCR assays for the treatment and follow-up phases. During treatment, serial sampling should inform treatment efficacy and hence guide subsequent intensification or de-escalation, and monitoring of clonal evolution and therapy resistance. If the latter is observed, treatment should be changed accordingly. During follow-up, surveillance using these ddPCR assays as well as CTC detection methods can be used for MRD screening. If detected, broad NGS approaches should be utilized again for new diagnostic molecular profiling of (potentially treatment resistant) clones, before new treatment is initiated. Still, design of patient-specific ddPCR assays might not always be possible (e.g., due to absence of an appropriate chromosomal region, presence of the sequence in the normal genome, or little ctDNA (being more relevant for sarcomas)) or alternative analytes to ctDNA might be more informative4,139,141,146,140. Important examples include miRNA clusters (for MGCTs and osteosarcoma), CTC analysis including derived mRNA material (for NB, osteosarcoma, EWS, and RMS), and lncRNAs (for osteosarcoma) (Supplementary Tables 2–9). In such cases, assessment of preferably single and otherwise panels of simple and routinely applicable, specific, sensitive, robust, accurate, and cost-effective biomarkers using relevant platforms (e.g., CTC identification and analysis techniques, RNA seq, and miRNA profiling), depending on tumor type, will then provide most value for implementation in pediatric oncology137,365.
Finally, liquid biopsies could aid in identification of clonal hematopoiesis (CH). CH involves accumulation of somatic mutations in hematopoietic stem cells leading to clonal expansions of genetically distinct blood cell subpopulations without evidence of dysplasia, neoplasm, or cytopenia8,366. Although initially considered an age-related phenomenon, a recent study by Hagiwara et al.367 found significantly higher (15% vs 8.5%) mutations in CH-related genes versus community controls in 2860 long-term survivors of pediatric cancer with a mean follow-up time of 23.5 years. CH in survivors is associated with exposures to hazards (alkalyting agents, radiation, and bleomycin), and is now recognized as a risk for developing cardiovascular disease and hematological malignancies367,368. This urges the need for monitoring to investigate long-term effects of therapy-induced CH in pediatric cancer survivors. CH detection using liquid biopsies has already been suggested for different adult cancers369,370,371,372. In children, however, VAFs of CH remain very low and currently undetectable in many liquid biopsy assays8. Future efforts exploring this should be wary to avoid mix-up of signals derived from tumors and from CH, for example by targeted sequencing of paired cfDNA and white blood cells for correct identification of corresponding genetic aberrations373.
Taken together, methods and targets of choice inherently depend on tumor type and the clinical question at hand, most commonly being either diagnostic or prognostic. Importantly, regardless of the application or tumor type, liquid biopsy validation studies will ultimately need to demonstrate improved outcome and/ or quality of life versus standard methods in order to serve purpose in clinical oncology.
Challenges and requirements for clinical implementation
Liquid biopsies are increasingly being investigated as non-invasive clinical biomarkers in pediatric solid tumors (Fig. 2a, b). Still, in contrast to adult oncology, liquid biopsies have not yet been implemented in daily clinical practice for several reasons. Firstly, clinical implementation requires biomarker validation in large-scale prospective trials with comparison to the standard of care at predefined timepoints4. Considering sampling volume restrictions and given that many pediatric solid tumor subtypes are rare, pediatric liquid biopsy material is precious, and availability is limited8,31,47,125. This complicates validation of findings in independent cohorts. In order to overcome these limitations, there is a need for multi-institutional collaboration with defined targets and methods in clinical trials, frequent sampling, and open access to published data31,125,135. Such trials are starting to emerge and examples were presented in the main text, per tumor type.
Next, biomarker validation in prospective clinical trials requires standardization and harmonization of pre-analytical conditions and protocols374. Pre-analytical parameters of a liquid biopsy workflow involve sampling, processing (e.g., analyte isolation or purification), and storage. For ctDNA, for example, yield, stability, and contamination with genomic DNA are affected by collection tube choice (e.g., serum versus plasma collection) and agitation, centrifugation force and speed, time between sampling and storage, storage temperature, freeze-thaw cycles, isolation methods, and quantification374. Standardized sampling conditions within and among international trials should decrease variation. Fortunately, recommendations on these conditions are emerging in recent studies24,374,375.
Clinical implementation of liquid biopsies also requires consistent, specific, sensitive, quick, and cost-effective methods for biomarker detection and analysis, including interpretation. Generally, relevant methods include single target approaches (e.g., PCR-based), panel approaches (targeted sequencing and arrays) and broad strategies (comprehensive sequencing)8,40,154. Naturally, methods of preference depend on the tumor type being investigated, involved biomarkers, and which information is required at that time. For ctDNA detection and analysis, ddPCR and NGS-based approaches are currently the most commonly used methods. ddPCR is sensitive, relatively cheap, quick, and suitable for detecting recurrent genetic aberrations such as mutations and SVs (CNAs, fusions, and translocations) with variant allele fractions (VAFs) as low as 0.001%8,125,146,374. Still, patients often display specific DNA breakpoints in the case of SVs, therefore requiring development of a unique assay per patient with existing knowledge of the respective genetic aberrations4,125. In contrast, NGS approaches allow genomic analysis without previous knowledge of tumor genetics, and can be used for investigation of individual genes, selected regions, or the entire genome40,374. NGS, however, is costly and hence requires balancing among sequencing breadth (proportion of genome being sequenced) and depth (number of times a nucleotide is read during sequencing). Shallow WGS approaches, for example, can detect CNAs in samples with a high fraction of ctDNA but lack sensitivity for the detection of SNVs, INDELs, and translocations. Conversely, while deep sequencing panels can identify such aberrations, they are generally restricted to targeted regions8,125,146,376. Deep WGS is currently still too cost-prohibitive for routinary practice125. Fortunately, costs are still decreasing making this method potentially feasible in the future.
ctDNA-based biomarker detection raises several challenges. Firstly, depending on cancer type, disease staging, and inherently ctDNA secretion, target scarcity among the pool of normal cfDNA fragments within peripheral blood may mask a positive signal despite ctDNA being present in the patient140,141,377. In pediatrics, this issue is compounded by blood volume restrictions. The maximum amount of blood that can be drawn per moment depends on the child’s age, weight, and medical condition, but is typically 0.8–0.9 mL/ kg of body weight (corresponding to 1% of total blood volume) as recommended by the European Union (most often less is taken)378. Also, an assay’s LOD is constrained by the number of unique molecules being present in the sample. This is primarily a concern when using sequencing assays for ctDNA detection. Sequencing to a depth greater than unique genome equivalents in the sample results in duplicate reads without improvement of the actual LOD8. Current efforts to mitigate these challenges include improved method sensitivity, enrichment protocols promoting yield and stability, application of sequencing error suppression algorithms, identification and removal of mutations from CH, and deep sequencing efforts with implementation of unique molecular identifiers (UMIs)4,377,379. UMIs tag original DNA molecules as distinct identities and therefore allow quantification of thoroughly unique molecules present in the sample379. Hence, these UMIs improve characterization of rare genetic alterations, reduce background signal, and maintain high positive predictive value and sensitivity379. Finally, several different units of measurement for ctDNA are currently used, such as ng/mL, haploid genome equivalents (copies)/ mL, fraction of total cfDNA (VAF), and copies/ mL4. Harmonization and guideline development will allow for better interpretation and comparisons of ctDNA analyses. Importantly, the failure rate of ctDNA-based biomarker detection and causes (e.g., too low input ctDNA, ctDNA being too fragmented, or VAF below method LOD) are currently not monitored well in pediatric studies investigating liquid biopsies. Hence, future studies should track this to elucidate their impact and clinical relevance.
Final remarks
In this review, we provide a comprehensive overview of the literature on liquid biopsies in pediatric solid tumors and elaborate on recent developments. Especially ctDNA, but also CTCs (including derived mRNA material) and miRNAs, are the most commonly investigated liquid biopsy analytes, and could serve valuable purposes including cancer diagnosis, risk-stratification, prognostication, monitoring of therapy response and clonal evolution, MRD detection, and premature detection of relapse, as indicated by many studies (Table 1, Supplementary Tables 2–9). Taken together, liquid biopsy-derived biomarkers hold tremendous potential for application in pediatric solid tumors with the field emerging rapidly, especially during the last decade (Fig. 2a, b). Still, validation in large-scale prospective international trials with defined biomarker targets, frequent sampling, standardization of pre-analytical conditions, and defined analytical techniques is required before liquid biopsies can be implemented in the routine care for pediatric oncology patients.
Data availability
All data generated and analyzed during this study are included in this published article (and its supplementary information files).
References
Tomasetti, C., Li, L. & Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355, 1330–1334 (2017).
WHO Classification of Tumours Editorial Board. Paediatric Tumours. Lyon (France): International Agency for Research on Cancer, 5th edn, Vol. 7 WHO classification of tumours series. https://publications.iarc.fr/608 (WHO, 2022).
Weiser, D. A. et al. Progress toward liquid biopsies in pediatric solid tumors. Cancer Metastasis Rev. 38, 553–571 (2019).
Doculara, L., Trahair, T. N., Bayat, N. & Lock, R. B. Circulating tumor DNA in pediatric cancer. Front Mol. Biosci. 9, 885597 (2022).
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48 (2023).
Kattner, P. et al. Compare and contrast: pediatric cancer versus adult malignancies. Cancer Metastasis Rev. 38, 673–682 (2019).
Grossi, M. Management and long-term complications of pediatric cancer. Pediatr. Clin. North Am. 45, 1637–1658 (1998).
Sundby, R. T., Pan, A. & Shern, J. F. Liquid biopsies in pediatric oncology: opportunities and obstacles. Curr. Opin. Pediatr. 34, 39–47 (2022).
Christodoulou, E. et al. Combined low-pass whole genome and targeted sequencing in liquid biopsies for pediatric solid tumors. NPJ Precis. Oncol. 7, 21 (2023).
Sweet-Cordero, E. A. & Biegel, J. A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 363, 1170–1175 (2019).
Grobner, S. N. et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018).
Parsa, N. Environmental factors inducing human cancers. Iran. J. Public Health 41, 1–9 (2012).
Wu, S., Zhu, W., Thompson, P. & Hannun, Y. A. Evaluating intrinsic and non-intrinsic cancer risk factors. Nat. Commun. 9, 3490 (2018).
Doll, R. & Peto, R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 66, 1191–1308 (1981).
Zhang, J. et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 373, 2336–2346 (2015).
Bakhuizen, J. J. et al. Assessment of cancer predisposition syndromes in a national cohort of children with a neoplasm. JAMA Netw. Open 6, e2254157 (2023).
Miller, K. D. et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 69, 363–385 (2019).
Bryant, R. Managing side effects of childhood cancer treatment. J. Pediatr. Nurs. 18, 113–125 (2003).
Basak, D., Arrighi, S., Darwiche, Y. & Deb, S. Comparison of anticancer drug toxicities: paradigm shift in adverse effect profile. Life (Basel) 12, 48 (2021).
Lone, S. N. et al. Liquid biopsy: a step closer to transform diagnosis, prognosis and future of cancer treatments. Mol. Cancer 21, 79 (2022).
Weiser, D. A., Kaste, S. C., Siegel, M. J. & Adamson, P. C. Imaging in childhood cancer: a Society for Pediatric Radiology and Children’s Oncology Group Joint Task Force report. Pediatr. Blood Cancer 60, 1253–1260 (2013).
Stankunaite, R. et al. Liquid biopsy for children with central nervous system tumours: clinical integration and technical considerations. Front Pediatr. 10, 957944 (2022).
Anderson, N. M. & Simon, M. C. The tumor microenvironment. Curr. Biol. 30, R921–R925 (2020).
Kahana-Edwin, S., Cain, L. E. & Karpelowsky, J. Roadmap to liquid biopsy biobanking from pediatric cancers-challenges and opportunities. Biopreserv. Biobank 19, 124–129 (2021).
Madlener, S. & Gojo, J. Liquid biomarkers for pediatric brain tumors: biological features, advantages and perspectives. J. Pers. Med. 10, 254 (2020).
Werner, B., Warton, K. & Ford, C. E. Transcending blood-opportunities for alternate liquid biopsies in oncology. Cancers (Basel) 14, 1309 (2022).
Peng, M., Chen, C., Hulbert, A., Brock, M. V. & Yu, F. Non-blood circulating tumor DNA detection in cancer. Oncotarget 8, 69162–69173 (2017).
McEwen, A. E., Leary, S. E. S. & Lockwood, C. M. Beyond the blood: CSF-derived cfDNA for diagnosis and characterization of CNS tumors. Front Cell Dev. Biol. 8, 45 (2020).
Miller, A. M. & Karajannis, M. A. Current role and future potential of CSF ctDNA for the diagnosis and clinical management of pediatric central nervous system tumors. J. Natl. Compr. Canc. Netw. 20, 1363–1369 (2022).
Liu, A. P., Northcott, P. A., Robinson, G. W. & Gajjar, A. Circulating tumor DNA profiling for childhood brain tumors: technical challenges and evidence for utility. Lab Invest. 102, 134–142 (2022).
Andersson, D., Fagman, H., Dalin, M. G. & Stahlberg, A. Circulating cell-free tumor DNA analysis in pediatric cancers. Mol. Asp. Med. 72, 100819 (2020).
Nikanjam, M., Kato, S. & Kurzrock, R. Liquid biopsy: current technology and clinical applications. J. Hematol. Oncol. 15, 131 (2022).
Alix-Panabieres, C. & Pantel, K. Liquid biopsy: from discovery to clinical application. Cancer Discov. 11, 858–873 (2021).
Liu, F., Xiong, Q. W., Wang, J. H. & Peng, W. X. Roles of lncRNAs in childhood cancer: current landscape and future perspectives. Front Oncol. 13, 1060107 (2023).
Varkey, J. & Nicolaides, T. Tumor-educated platelets: a review of current and potential applications in solid tumors. Cureus 13, e19189 (2021).
Narayan, P. et al. State of the science and future directions for liquid biopsies in drug development. Oncologist 25, 730–732 (2020).
Sato, Y. Clinical utility of liquid biopsy-based companion diagnostics in the non-small-cell lung cancer treatment. Explor. Target Antitumor Ther. 3, 630–642 (2022).
Cisneros-Villanueva, M. et al. Cell-free DNA analysis in current cancer clinical trials: a review. Br. J. Cancer 126, 391–400 (2022).
Ignatiadis, M., Sledge, G. W. & Jeffrey, S. S. Liquid biopsy enters the clinic—implementation issues and future challenges. Nat. Rev. Clin. Oncol. 18, 297–312 (2021).
Alexandrou, G. et al. The evolution of affordable technologies in liquid biopsy diagnostics: the key to clinical implementation. Cancers (Basel) 15, 5434 (2023).
Archer™ LIQUIDPlex™ Universal Solid Tumor panel, https://www.idtdna.com/pages/products/next-generation-sequencing/archer-ngs-assay-solutions/solid-tumor-research/archer-liquidplex-universal-panel (2024).
Yang, R., Zheng, S. & Dong, R. Circulating tumor cells in neuroblastoma: current status and future perspectives. Cancer Med. 12, 7–19 (2023).
Wei, M., Ye, M., Dong, K. & Dong, R. Circulating tumor DNA in neuroblastoma. Pediatr. Blood Cancer 67, e28311 (2020).
Kaatsch, P. Epidemiology of childhood cancer. Cancer Treat. Rev. 36, 277–285 (2010).
Mahapatra, S., Challagundla, K. B. Neuroblastoma. [Updated 2023 Jul 10]. In StatPearls [Internet]. Treasure Island (FL) (StatPearls Publishing 2024). Available from: https://www.ncbi.nlm.nih.gov/books/NBK448111/
Krystal, J. & Foster, J. H. Treatment of high-risk neuroblastoma. Children (Basel) 10, 1302 (2023).
Trigg, R. M., Shaw, J. A. & Turner, S. D. Opportunities and challenges of circulating biomarkers in neuroblastoma. Open Biol. 9, 190056 (2019).
Cohn, S. L. et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J. Clin. Oncol. 27, 289–297 (2009).
Matthay, K. K. et al. Neuroblastoma. Nat. Rev. Dis. Prim. 2, 16078 (2016).
Lerone, M. et al. Molecular genetics in neuroblastoma prognosis. Children (Basel) 8, 456 (2021).
Ma, J. et al. Exosomal hsa-miR199a-3p promotes proliferation and migration in neuroblastoma. Front Oncol. 9, 459 (2019).
Lak, N. S. M. et al. Cell-free RNA from plasma in patients with neuroblastoma: exploring the technical and clinical potential. Cancers (Basel) 15, 2108 (2023).
Abbasi, M. R. et al. Bone marrows from neuroblastoma patients: an excellent source for tumor genome analyses. Mol. Oncol. 9, 545–554 (2015).
Batth, I. S. et al. Cell surface vimentin-positive circulating tumor cell-based relapse prediction in a long-term longitudinal study of postremission neuroblastoma patients. Int. J. Cancer 147, 3550–3559 (2020).
Burchill, S. A. et al. Circulating neuroblastoma cells detected by reverse transcriptase polymerase chain reaction for tyrosine hydroxylase mRNA are an independent poor prognostic indicator in stage 4 neuroblastoma in children over 1 year. J. Clin. Oncol. 19, 1795–1801 (2001).
Liu, X. et al. Circulating tumor cells detection in neuroblastoma patients by EpCAM-independent enrichment and immunostaining-fluorescence in situ hybridization. EBioMedicine 35, 244–250 (2018).
Kuroda, T., Saeki, M., Nakano, M. & Mizutani, S. Clinical application of minimal residual neuroblastoma cell detection by reverse transcriptase-polymerase chain reaction. J. Pediatr. Surg. 32, 69–72 (1997).
Lee, N. H. et al. Clinical significance of tyrosine hydroxylase mRNA transcripts in peripheral blood at diagnosis in patients with neuroblastoma. Cancer Res. Treat. 48, 1399–1407 (2016).
Moss, T. J. et al. Clonogenicity of circulating neuroblastoma cells: implications regarding peripheral blood stem cell transplantation. Blood 83, 3085–3089 (1994).
Merugu, S. et al. Detection of circulating and disseminated neuroblastoma cells using the imagestream flow cytometer for use as predictive and pharmacodynamic biomarkers. Clin. Cancer Res. 26, 122–134 (2020).
Moss, T. J. & Sanders, D. G. Detection of neuroblastoma cells in blood. J. Clin. Oncol. 8, 736–740 (1990).
Gao, Y., Li, G., Zhang, X., Xu, Q. & Zheng, B. Detection of neuroblastoma cells in blood by reverse transcriptase-polymerase chain reaction. Chin. Med. J. (Engl.) 110, 341–345 (1997).
Miyajima, Y., Kato, K., Numata, S., Kudo, K. & Horibe, K. Detection of neuroblastoma cells in bone marrow and peripheral blood at diagnosis by the reverse transcriptase-polymerase chain reaction for tyrosine hydroxylase mRNA. Cancer 75, 2757–2761 (1995).
Corrias, M. V. et al. Detection of neuroblastoma cells in bone marrow and peripheral blood by different techniques: accuracy and relationship with clinical features of patients. Clin. Cancer Res. 10, 7978–7985 (2004).
Yanagisawa, T. Y. et al. Detection of the PGP9.5 and tyrosine hydroxylase mRNAs for minimal residual neuroblastoma cells in bone marrow and peripheral blood. Tohoku J. Exp. Med. 184, 229–240 (1998).
Pagani, A. et al. Detection procedures for neuroblastoma cells metastatic to blood and bone marrow: blinded comparison of chromogranin A heminested reverse transcription polymerase chain reaction to tyrosine hydroxylase nested reverse transcription polymerase chain reaction and to anti-GD2 immunocytology. Diagn. Mol. Pathol. 11, 98–106 (2002).
Oltra, S. et al. The doublecortin gene, a new molecular marker to detect minimal residual disease in neuroblastoma. Diagn. Mol. Pathol. 14, 53–57 (2005).
Burchill, S. A., Bradbury, F. M., Selby, P. & Lewis, I. J. Early clinical evaluation of neuroblastoma cell detection by reverse transcriptase-polymerase chain reaction (RT-PCR) for tyrosine hydroxylase mRNA. Eur. J. Cancer 31a, 553–556 (1995).
Hirase, S. et al. Early detection of tumor relapse/regrowth by consecutive minimal residual disease monitoring in high-risk neuroblastoma patients. Oncol. Lett. 12, 1119–1123 (2016).
Corrias, M. V. et al. Event-free survival of infants and toddlers enrolled in the HR-NBL-1/SIOPEN trial is associated with the level of neuroblastoma mRNAs at diagnosis. Pediatr. Blood Cancer 65, e27052 (2018).
Cheung, I. Y., Feng, Y., Gerald, W. & Cheung, N. K. Exploiting gene expression profiling to identify novel minimal residual disease markers of neuroblastoma. Clin. Cancer Res. 14, 7020–7027 (2008).
Marachelian, A. et al. Expression of five neuroblastoma genes in bone marrow or blood of patients with relapsed/refractory neuroblastoma provides a new biomarker for disease and prognosis. Clin. Cancer Res. 23, 5374–5383 (2017).
Faulkner, L. B. et al. High-sensitivity immunocytologic analysis of neuroblastoma cells in paired blood and marrow samples. J. Hematother. 7, 361–366 (1998).
Burchill, S. A., Lewis, I. J. & Selby, P. Improved methods using the reverse transcriptase polymerase chain reaction to detect tumour cells. Br. J. Cancer 79, 971–977 (1999).
Thwin, K. K. M. et al. Level of seven neuroblastoma-associated mRNAs detected by droplet digital PCR is associated with tumor relapse/regrowth of high-risk neuroblastoma patients. J. Mol. Diagn. 22, 236–246 (2020).
Uemura, S. et al. Limited correlation between tumor markers and minimal residual disease detected by seven neuroblastoma-associated mRNAs in high-risk neuroblastoma patients. Mol. Clin. Oncol. 15, 137 (2021).
Cheung, I. Y., Sahota, A. & Cheung, N. K. Measuring circulating neuroblastoma cells by quantitative reverse transcriptase-polymerase chain reaction analysis. Cancer 101, 2303–2308 (2004).
Yanez, Y. et al. Minimal disease detection in peripheral blood and bone marrow from patients with non-metastatic neuroblastoma. J. Cancer Res. Clin. Oncol. 137, 1263–1272 (2011).
Oltra, S. et al. Minimal residual disease in neuroblastoma: to GAGE or not to GAGE. Oncol. Res. 14, 291–295 (2004).
Bozzi, F. et al. Molecular detection of dopamine decarboxylase expression by means of reverse transcriptase and polymerase chain reaction in bone marrow and peripheral blood: utility as a tumor marker for neuroblastoma. Diagn. Mol. Pathol. 13, 135–143 (2004).
Cheung, I. Y. & Cheung, N. K. Molecular detection of GAGE expression in peripheral blood and bone marrow: utility as a tumor marker for neuroblastoma. Clin. Cancer Res. 3, 821–826 (1997).
Trager, C. et al. mRNAs of tyrosine hydroxylase and dopa decarboxylase but not of GD2 synthase are specific for neuroblastoma minimal disease and predicts outcome for children with high-risk disease when measured at diagnosis. Int. J. Cancer 123, 2849–2855 (2008).
Burchill, S. A., Bradbury, F. M., Smith, B., Lewis, I. J. & Selby, P. Neuroblastoma cell detection by reverse transcriptase-polymerase chain reaction (RT-PCR) for tyrosine hydroxylase mRNA. Int. J. Cancer 57, 671–675 (1994).
Lanino, E., Melodia, A., Casalaro, A. & Cornaglia-Ferraris, P. Neuroblastoma cells circulate in peripheral blood. Pediatr. Hematol. Oncol. 6, 193–195 (1989).
van Wezel, E. M. et al. Neuroblastoma messenger RNA is frequently detected in bone marrow at diagnosis of localised neuroblastoma patients. Eur. J. Cancer 54, 149–158 (2016).
Viprey, V. F. et al. Neuroblastoma mRNAs predict outcome in children with stage 4 neuroblastoma: a European HR-NBL1/SIOPEN study. J. Clin. Oncol. 32, 1074–1083 (2014).
Swerts, K. et al. Potential application of ELAVL4 real-time quantitative reverse transcription-PCR for detection of disseminated neuroblastoma cells. Clin. Chem. 52, 438–445 (2006).
Parareda, A. et al. Prognostic impact of the detection of microcirculating tumor cells by a real-time RT-PCR assay of tyrosine hydroxylase in patients with advanced neuroblastoma. Oncol. Rep. 14, 1021–1027 (2005).
Kuroda, T. et al. Prognostic significance of circulating tumor cells and bone marrow micrometastasis in advanced neuroblastoma. J. Pediatr. Surg. 43, 2182–2185 (2008).
Träger, C. et al. Quantitative analysis of tyrosine hydroxylase mRNA for sensitive detection of neuroblastoma cells in blood and bone marrow. Clin. Chem. 49, 104–112 (2003).
Seeger, R. C. et al. Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children’s Cancer Group Study. J. Clin. Oncol. 18, 4067–4076 (2000).
Lambooy, L. H. et al. Real-time analysis of tyrosine hydroxylase gene expression: a sensitive and semiquantitative marker for minimal residual disease detection of neuroblastoma. Clin. Cancer Res. 9, 812–819 (2003).
Mattano, L. A. Jr., Moss, T. J. & Emerson, S. G. Sensitive detection of rare circulating neuroblastoma cells by the reverse transcriptase-polymerase chain reaction. Cancer Res. 52, 4701–4705 (1992).
Miyajima, Y. et al. Sequential detection of tumor cells in the peripheral blood and bone marrow of patients with stage IV neuroblastoma by the reverse transcription-polymerase chain reaction for tyrosine hydroxylase mRNA. Cancer 77, 1214–1219 (1996).
Sanders, D. G., Wiley, F. M. & Moss, T. J. Serial immunocytologic analysis of blood for tumor cells in two patients with neuroblastoma. Cancer 67, 1423–1427 (1991).
Tchirkov, A. et al. Significance of molecular quantification of minimal residual disease in metastatic neuroblastoma. J. Hematother. Stem Cell Res 12, 435–442 (2003).
Kuroda, T. et al. Surgical treatment of neuroblastoma with micrometastasis. J. Pediatr. Surg. 35, 1638–1642 (2000).
Yanez, Y. et al. TH and DCX mRNAs in peripheral blood and bone marrow predict outcome in metastatic neuroblastoma patients. J. Cancer Res. Clin. Oncol. 142, 573–580 (2016).
Kuroda, T. et al. Tumor cell dynamics and metastasis in advanced neuroblastoma. Pediatr. Surg. Int. 21, 859–863 (2005).
Stutterheim, J. et al. Detecting minimal residual disease in neuroblastoma: the superiority of a panel of real-time quantitative PCR markers. Clin. Chem. 55, 1316–1326 (2009).
Hartomo, T. B. et al. Minimal residual disease monitoring in neuroblastoma patients based on the expression of a set of real-time RT-PCR markers in tumor-initiating cells. Oncol. Rep. 29, 1629–1636 (2013).
Corrias, M. V. et al. Multiple target molecular monitoring of bone marrow and peripheral blood samples from patients with localized neuroblastoma and healthy donors. Pediatr. Blood Cancer 58, 43–49 (2012).
Viprey, V. F. et al. Standardisation of operating procedures for the detection of minimal disease by QRT-PCR in children with neuroblastoma: quality assurance on behalf of SIOPEN-R-NET. Eur. J. Cancer 43, 341–350 (2007).
Kojima, M. et al. Single-cell next-generation sequencing of circulating tumor cells in patients with neuroblastoma. Cancer Sci. 114, 1616–1624 (2023).
Stagno, M. J. et al. Epitope detection in monocytes (EDIM) for liquid biopsy including identification of GD2 in childhood neuroblastoma-a pilot study. Br. J. Cancer 127, 1324–1331 (2022).
Murray, M. J. et al. Solid tumors of childhood display specific serum microRNA profiles. Cancer Epidemiol. Biomark. Prev. 24, 350–360 (2015).
Zeka, F. et al. Circulating microRNA biomarkers for metastatic disease in neuroblastoma patients. JCI Insight 3, 1–13 (2018).
Morini, M. et al. Exosomal microRNAs from longitudinal liquid biopsies for the prediction of response to induction chemotherapy in high-risk neuroblastoma patients: a proof of concept SIOPEN study. Cancers (Basel). 11, 1476 (2019).
Ikematsu, S. et al. Correlation of elevated level of blood midkine with poor prognostic factors of human neuroblastomas. Br. J. Cancer 88, 1522–1526 (2003).
Cheung, I. Y. & Cheung, N. K. Detection of microscopic disease: comparing histology, immunocytology, and RT-PCR of tyrosine hydroxylase, GAGE, and MAGE. Med. Pediatr. Oncol. 36, 210–212 (2001).
Cheung, I. Y., Barber, D. & Cheung, N. K. Detection of microscopic neuroblastoma in marrow by histology, immunocytology, and reverse transcription-PCR of multiple molecular markers. Clin. Cancer Res. 4, 2801–2805 (1998).
Hoon, D. S. et al. Ganglioside GM2/GD2 synthetase mRNA is a marker for detection of infrequent neuroblastoma cells in bone marrow. Am. J. Pathol. 159, 493–500 (2001).
Stutterheim, J. et al. Methylated RASSF1a is the first specific DNA marker for minimal residual disease testing in neuroblastoma. Clin. Cancer Res 18, 808–814 (2012).
Vasudevan, S. A. et al. Neuroblastoma-derived secretory protein messenger RNA levels correlate with high-risk neuroblastoma. J. Pediatr. Surg. 42, 148–152 (2007).
Cheung, I. Y., Lo Piccolo, M. S., Kushner, B. H., Kramer, K. & Cheung, N. K. Quantitation of GD2 synthase mRNA by real-time reverse transcriptase polymerase chain reaction: clinical utility in evaluating adjuvant therapy in neuroblastoma. J. Clin. Oncol. 21, 1087–1093 (2003).
Cheung, I. Y., Lo Piccolo, M. S., Collins, N., Kushner, B. H. & Cheung, N. K. Quantitation of GD2 synthase mRNA by real-time reverse transcription-polymerase chain reaction: utility in bone marrow purging of neuroblastoma by anti-GD2 antibody 3F8. Cancer 94, 3042–3048 (2002).
Cheung, I. Y. & Cheung, N. K. Quantitation of marrow disease in neuroblastoma by real-time reverse transcription-PCR. Clin. Cancer Res. 7, 1698–1705 (2001).
Pession, A. et al. Real-time RT-PCR of tyrosine hydroxylase to detect bone marrow involvement in advanced neuroblastoma. Oncol. Rep. 10, 357–362 (2003).
Cheung, I. Y., Vickers, A. & Cheung, N. K. Sialyltransferase STX (ST8SiaII): a novel molecular marker of metastatic neuroblastoma. Int. J. Cancer 119, 152–156 (2006).
van Zogchel, L. M. J. et al. Specific and sensitive detection of neuroblastoma mRNA markers by multiplex RT-qPCR. Cancers (Basel). 13, 150 (2021).
Stutterheim, J. et al. Stability of PCR targets for monitoring minimal residual disease in neuroblastoma. J. Mol. Diagn. 14, 168–175 (2012).
Gattenloehner, S. et al. A comparison of MyoD1 and fetal acetylcholine receptor expression in childhood tumors and normal tissues: implications for the molecular diagnosis of minimal disease in rhabdomyosarcomas. J. Mol. Diagn. 1, 23–31 (1999).
Subhash, V. V. et al. Whole-genome sequencing facilitates patient-specific quantitative PCR-based minimal residual disease monitoring in acute lymphoblastic leukaemia, neuroblastoma and Ewing sarcoma. Br. J. Cancer 126, 482–491 (2022).
Zhenjian, Z., Lin, Lei, Miao, Lei, Li, Meng & He, Jing Advances in liquid biopsy in neuroblastoma. Fundam. Res. 2, 903–917 (2022).
Abbou, S. D., Shulman, D. S., DuBois, S. G. & Crompton, B. D. Assessment of circulating tumor DNA in pediatric solid tumors: the promise of liquid biopsies. Pediatr. Blood Cancer 66, e27595 (2019).
Kojima, M. & Hiyama, E. Circulating tumor cells and tumor progression, metastasis, and poor prognosis in patients with neuroblastoma. Anticancer Res. 43, 4327–4331 (2023).
Trigg, R. M., Turner, S. D., Shaw, J. A. & Jahangiri, L. Diagnostic accuracy of circulating-free DNA for the determination of MYCN amplification status in advanced-stage neuroblastoma: a systematic review and meta-analysis. Br. J. Cancer 122, 1077–1084 (2020).
Uemura, S. et al. Dynamics of minimal residual disease in neuroblastoma patients. Front Oncol. 9, 455 (2019).
Galardi, A. et al. Exosomal miRNAs in pediatric cancers. Int. J. Mol. Sci. 20, 4600 (2019).
Vellichirammal, N. N., Chaturvedi, N. K., Joshi, S. S., Coulter, D. W. & Guda, C. Fusion genes as biomarkers in pediatric cancers: a review of the current state and applicability in diagnostics and personalized therapy. Cancer Lett. 499, 24–38 (2021).
Gholamin, S. et al. GD2-targeted immunotherapy and potential value of circulating microRNAs in neuroblastoma. J. Cell Physiol. 233, 866–879 (2018).
Segura, M. F. et al. Methodological advances in the discovery of novel neuroblastoma therapeutics. Expert Opin. Drug Discov. 17, 167–179 (2022).
Andreeva, N., Usman, N. & Druy, A. MicroRNAs as prospective biomarkers, therapeutic targets and pharmaceuticals in neuroblastoma. Mol. Biol. Rep. 50, 1895–1912 (2023).
Galardi, A. et al. MicroRNAs in neuroblastoma: biomarkers with therapeutic potential. Curr. Med Chem. 25, 584–600 (2018).
Van Paemel, R. et al. The pitfalls and promise of liquid biopsies for diagnosing and treating solid tumors in children: a review. Eur. J. Pediatr. 179, 191–202 (2020).
Bhavsar, S. P. Recent advances in the roles of exosomal microRNAs in neuroblastoma. Front Oncol. 12, 1091847 (2022).
de Carvalho, I. N., de Freitas, R. M. & Vargas, F. R. Translating microRNAs into biomarkers: What is new for pediatric cancer? Med. Oncol. 33, 49 (2016).
Bettegowda, C. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra224 (2014).
Klega, K. et al. Detection of somatic structural variants enables quantification and characterization of circulating tumor DNA in children with solid tumors. JCO Precis. Oncol. 2018, PO.17.00285 (2018).
Ruas, J. S. et al. Somatic copy number alteration in circulating tumor DNA for monitoring of pediatric patients with cancer. Biomedicines 11, 1082 (2023).
van Zogchel, L. M. J. et al. Novel circulating hypermethylated RASSF1A ddPCR for liquid biopsies in patients with pediatric solid tumors. JCO Precis. Oncol. 5, PO.21.00130 (2021).
Peitz, C. et al. Multiplexed quantification of four neuroblastoma DNA targets in a single droplet digital PCR reaction. J. Mol. Diagn. 22, 1309–1323 (2020).
Bosse, K. R. & Maris, J. M. Advances in the translational genomics of neuroblastoma: from improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 122, 20–33 (2016).
Shirai, R. et al. Quantitative assessment of copy number alterations by liquid biopsy for neuroblastoma. Genes Chromosomes Cancer 61, 662–669 (2022).
Gelineau, N. U. et al. Case series on clinical applications of liquid biopsy in pediatric solid tumors: towards improved diagnostics and disease monitoring. Front Oncol. 13, 1209150 (2023).
van Zogchel, L. M. J. et al. Targeted locus amplification to develop robust patient-specific assays for liquid biopsies in pediatric solid tumors. Front Oncol. 13, 1124737 (2023).
Lopez-Carrasco, A. et al. Intra-tumour genetic heterogeneity and prognosis in high-risk neuroblastoma. Cancers (Basel) 13, 5173 (2021).
Van Paemel, R. et al. The feasibility of using liquid biopsies as a complementary assay for copy number aberration profiling in routinely collected paediatric cancer patient samples. Eur. J. Cancer 160, 12–23 (2022).
Cahn, F. et al. Blood-derived liquid biopsies using foundation One((R)) liquid CDx for children and adolescents with high-risk malignancies: a monocentric experience. Cancers (Basel) 14, 2774 (2022).
Michalowski, M. B. et al. Methylation of tumor-suppressor genes in neuroblastoma: The RASSF1A gene is almost always methylated in primary tumors. Pediatr. Blood Cancer 50, 29–32 (2008).
Vaisvila, R. et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 31, 1280–1289 (2021).
Trinidad, E. M. et al. Liquidhope: methylome and genomic profiling from very limited quantities of plasma-derived DNA. Brief. Bioinform. 24, bbac575 (2023).
Trinidad, E. M. et al. Evaluation of circulating tumor DNA by electropherogram analysis and methylome profiling in high-risk neuroblastomas. Front Oncol. 13, 1037342 (2023).
van der Laan, P., van Houdt, W. J., van den Broek, D., Steeghs, N. & van der Graaf, W. T. A. Liquid biopsies in sarcoma clinical practice: Where Do We Stand? Biomedicines 9, 1315 (2021).
High-Risk Neuroblastoma Study 2 of SIOP-Europa-Neuroblastoma (SIOPEN) (HR-NBL2), https://clinicaltrials.gov/study/NCT04221035 (2023).
Nakata, K., Colombet, M., Stiller, C. A., Pritchard-Jones, K. & Steliarova-Foucher, E. Incidence of childhood renal tumours: an international population-based study. Int J. Cancer 147, 3313–3327 (2020).
Ooms, A. et al. Renal tumors of childhood-a histopathologic pattern-based diagnostic approach. Cancers (Basel). 12, 729 (2020).
van den Heuvel-Eibrink, M. M. et al. Position paper: rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat. Rev. Urol. 14, 743–752 (2017).
van der Beek, J. N. et al. Characteristics and outcome of children with renal cell carcinoma: a narrative review. Cancers (Basel). 12, 1776 (2020).
Gooskens, S. L. et al. Clear cell sarcoma of the kidney: a review. Eur. J. Cancer 48, 2219–2226 (2012).
Walz, A. L. et al. Tumor biology, biomarkers, and liquid biopsy in pediatric renal tumors. Pediatr. Blood Cancer 70, e30130 (2023).
Schulpen, M. et al. Incidence and survival of paediatric renal tumours in the Netherlands between 1990 and 2014. Eur. J. Cancer 175, 282–290 (2022).
Roy, P. et al. Characteristics and outcome of children with renal tumors in the Netherlands: the first five-year’s experience of national centralization. PLoS One 17, e0261729 (2022).
Charlton, J., Pavasovic, V. & Pritchard-Jones, K. Biomarkers to detect Wilms tumors in pediatric patients: where are we now? Future Oncol. 11, 2221–2234, (2015).
Zheng, H., Liu, J., Pan, X. & Cui, X. Biomarkers for patients with Wilms tumor: a review. Front Oncol. 13, 1137346 (2023).
Huszno, J., Starzyczny-Słota, D., Jaworska, M. & Nowara, E. Adult Wilms’ tumor—diagnosis and current therapy. Cent. Eur. J. Urol. 66, 39–44 (2013).
Jain, J., Sutton, K. S. & Hong, A. L. Progress update in pediatric renal tumors. Curr. Oncol. Rep. 23, 33 (2021).
Wang, J., Li, M., Tang, D., Gu, W., Mao, J., & Shu, Q. Current treatment for Wilms tumor: COG and SIOP standards. World J. Pediatr. Surg. 2, e000038 (2019).
Pater, L. et al. Wilms tumor. Pediatr. Blood Cancer 68, e28257 (2021).
Hol, J. A. et al. Prognostic significance of age in 5631 patients with Wilms tumour prospectively registered in International Society of Paediatric Oncology (SIOP) 93-01 and 2001. PLoS One 14, e0221373 (2019).
Zekri, W., Yacoub, D. M., Ibrahim, A. & Madney, Y. Relapsed Wilms’ tumor in pediatric patients: challenges in low- to middle-income countries-a single-center experience. J. Egypt Natl. Canc. Inst. 32, 21 (2020).
Groenendijk, A. et al. Prognostic factors for Wilms tumor recurrence: a review of the literature. Cancers (Basel). 13, 3142 (2021).
Jackson, T. J. et al. How we approach paediatric renal tumour core needle biopsy in the setting of preoperative chemotherapy: a review from the SIOP Renal Tumour Study Group. Pediatr. Blood Cancer 69, e29702 (2022).
de Sa Pereira, B. M. et al. Intra-tumor genetic heterogeneity in Wilms tumor samples. Rev. Assoc. Med Bras. (1992) 65, 1496–1501 (2019).
Gadd, S. et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat. Genet 49, 1487–1494 (2017).
Grundy, P. E. et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J. Clin. Oncol. 23, 7312–7321 (2005).
Gratias, E. J. et al. Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children’s Oncology Group. Cancer 119, 3887–3894 (2013).
Madanat-Harjuoja, L. M. et al. Circulating tumor DNA as a biomarker in patients with stage III and IV Wilms tumor: analysis from a children’s oncology group trial, AREN0533. J. Clin. Oncol. 40, 3047–3056 (2022).
Jiménez, I. et al. Circulating tumor DNA analysis enables molecular characterization of pediatric renal tumors at diagnosis. Int. J. Cancer 144, 68–79 (2019).
Miguez, A. C. K. et al. Assessment of somatic mutations in urine and plasma of Wilms tumor patients. Cancer Med. 9, 5948–5959 (2020).
He, X. et al. Long non-coding RNA XIST promotes Wilms tumor progression through the miR-194-5p/YAP axis. Cancer Manag. Res. 13, 3171–3180 (2021).
Ludwig, N. et al. Circulating serum miRNAs as potential biomarkers for nephroblastoma. Pediatr. Blood Cancer 62, 1360–1367 (2015).
Schmitt, J. et al. Treatment-independent miRNA signature in blood of Wilms tumor patients. BMC Genom. 13, 379 (2012).
Li, M. et al. Liquid biopsy at the frontier in renal cell carcinoma: recent analysis of techniques and clinical application. Mol. Cancer 22, 37 (2023).
Salfer, B., Li, F., Wong, D. T. W. & Zhang, L. Urinary cell-free DNA in liquid biopsy and cancer management. Clin. Chem. 68, 1493–1501 (2022).
Oshi, M. et al. Urine as a source of liquid biopsy for cancer. Cancers (Basel). 13, 2652 (2021).
Lin, R. Y., Argenta, P. A., Sullivan, K. M. & Adzick, N. S. Diagnostic and prognostic role of basic fibroblast growth factor in Wilms’ tumor patients. Clin. Cancer Res. 1, 327–331, (1995).
Stern, M., Longaker, M. T., Adzick, N. S., Harrison, M. R. & Stern, R. Hyaluronidase levels in urine from Wilms’ tumor patients. J. Natl. Cancer Inst. 83, 1569–1574, (1991).
Lin, R. Y., Argenta, P. A., Sullivan, K. M., Stern, R. & Adzick, N. S. Urinary hyaluronic acid is a Wilms’ tumor marker. J. Pediatr. Surg. 30, 304–308 (1995).
Ortiz, M. V. et al. Prohibitin is a prognostic marker and therapeutic target to block chemotherapy resistance in Wilms’ tumor. JCI Insight 4, e127098 (2019).
Weil, B. R. & Billmire, D. F. Management of germ cell tumors in pediatric patients. Surg. Oncol. Clin. N. Am. 30, 325–338 (2021).
Oosterhuis, J. W. & Looijenga, L. H. J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 19, 522–537 (2019).
Pierce, J. L., Frazier, A. L. & Amatruda, J. F. Pediatric germ cell tumors: a developmental perspective. Adv. Urol. 2018, 9059382 (2018).
Egan, J. & Salari, K. Biomarkers in testicular cancer: classic tumor markers and beyond. Urol. Clin. North Am. 50, 133–143 (2023).
Kattuoa Ml, Dunton, C. J. Yolk Sac Tumors. In StatPearls [Internet]. Treasure Island (FL) (StatPearls Publishing, 2024). Available from: https://www.ncbi.nlm.nih.gov/books/NBK563163/
Murray, M. J. et al. Circulating microRNAs as biomarkers to assist the management of the malignant germ-cell-tumour subtype choriocarcinoma. Transl. Oncol. 14, 100904 (2021).
Lobo, J., Leao, R., Jeronimo, C. & Henrique, R. Liquid biopsies in the clinical management of germ cell tumor patients: state-of-the-art and future directions. Int. J. Mol. Sci. 22, 2654 (2021).
Nicholson, B. D. et al. The diagnostic performance of current tumour markers in surveillance for recurrent testicular cancer: A diagnostic test accuracy systematic review. Cancer Epidemiol. 59, 15–21 (2019).
Murray, M. J. & Coleman, N. Testicular cancer: a new generation of biomarkers for malignant germ cell tumours. Nat. Rev. Urol. 9, 298–300 (2012).
Almstrup, K. et al. Application of miRNAs in the diagnosis and monitoring of testicular germ cell tumours. Nat. Rev. Urol. 17, 201–213 (2020).
Jansson, M. D. & Lund, A. H. MicroRNA and cancer. Mol. Oncol. 6, 590–610 (2012).
Leao, R. et al. Circulating MicroRNAs, the next-generation serum biomarkers in testicular germ cell tumours: a systematic review. Eur. Urol. 80, 456–466 (2021).
Chovanec, M., Kalavska, K., Mego, M. & Cheng, L. Liquid biopsy in germ cell tumors: biology and clinical management. Expert Rev. Mol. Diagn. 20, 187–194 (2020).
Voorhoeve, P. M. et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 124, 1169–1181 (2006).
Dieckmann, K. P. et al. Serum levels of MicroRNA-371a-3p (M371 Test) as a new biomarker of testicular germ cell tumors: results of a prospective multicentric study. J. Clin. Oncol. 37, 1412–1423 (2019).
Gillis, A. J. et al. High-throughput microRNAome analysis in human germ cell tumours. J. Pathol. 213, 319–328 (2007).
Looijenga, L. H., Gillis, A. J., Stoop, H., Hersmus, R. & Oosterhuis, J. W. Relevance of microRNAs in normal and malignant development, including human testicular germ cell tumours. Int. J. Androl. 30, 304–314 (2007).
Palmer, R. D. et al. Malignant germ cell tumors display common microRNA profiles resulting in global changes in expression of messenger RNA targets. Cancer Res. 70, 2911–2923 (2010).
Murray, M. J. et al. Identification of microRNAs From the miR-371~373 and miR-302 clusters as potential serum biomarkers of malignant germ cell tumors. Am. J. Clin. Pathol. 135, 119–125 (2011).
Belge, G., Dieckmann, K. P., Spiekermann, M., Balks, T. & Bullerdiek, J. Serum levels of microRNAs miR-371-3: a novel class of serum biomarkers for testicular germ cell tumors? Eur. Urol. 61, 1068–1069, (2012).
Gillis, A. J. et al. Targeted serum miRNA (TSmiR) test for diagnosis and follow-up of (testicular) germ cell cancer patients: a proof of principle. Mol. Oncol. 7, 1083–1092 (2013).
Piao, J. et al. A multi-institutional pooled analysis demonstrates that circulating miR-371a-3p alone is sufficient for testicular malignant germ cell tumor diagnosis. Clin. Genitourin. Cancer 19, 469–479 (2021).
Murray, M. J. et al. A pipeline to quantify serum and cerebrospinal fluid microRNAs for diagnosis and detection of relapse in paediatric malignant germ-cell tumours. Br. J. Cancer 114, 151–162 (2016).
Murray, M. J. et al. Clinical utility of circulating miR-371a-3p for the management of patients with intracranial malignant germ cell tumors. Neurooncol. Adv. 2, vdaa048 (2020).
Schonberger, S. et al. MicroRNA-profiling of miR-371~373- and miR-302/367-clusters in serum and cerebrospinal fluid identify patients with intracranial germ cell tumors. J. Cancer Res. Clin. Oncol. 149, 791–802 (2023).
Saliyeva, S., Boranbayeva, R., Bulegenova, M. & Beloussov, V. Application of microRNAs in the diagnosis and monitoring of pediatric germ cell tumors: Kazakh experience. Pediatr. Hematol. Oncol. 41, 1–14 (2023).
Multicenter prospective study of a randomized comparison of carboplatin with cisplatin in extracranial malignant germ cell tumors https://www.gpoh.de/kinderkrebsinfo/content/health_professionals/clinical_trials/therapy_trials_and_registries_in_the_gpoh/makei_v/index_eng.html (2024).
Magic Consortium AGCT1531 Clinical Trail, https://magicconsortium.com/magic-research/clinical-trials/ (2021).
Lobo, J. et al. Combining hypermethylated RASSF1A detection using ddPCR with miR-371a-3p testing: an improved panel of liquid biopsy biomarkers for testicular germ cell tumor patients. Cancers (Basel). 13, 5228 (2021).
Nappi, L. et al. Integrated expression of circulating miR375 and miR371 to identify teratoma and active germ cell malignancy components in malignant germ cell tumors. Eur. Urol. 79, 16–19 (2021).
Lobo, J. et al. Identification and validation model for informative liquid biopsy-based microRNA biomarkers: insights from germ cell tumor in vitro, in vivo and patient-derived data. Cells 8, 1637 (2019).
Lafin, J. T. et al. Serum small RNA sequencing and miR-375 assay do not identify the presence of pure teratoma at postchemotherapy retroperitoneal lymph node dissection. Eur. Urol. Open Sci. 26, 83–87 (2021).
Belge, G., Grobelny, F., Matthies, C., Radtke, A. & Dieckmann, K. P. Serum level of microRNA-375-3p is not a reliable biomarker of teratoma. Vivo 34, 163–168 (2020).
Myklebust, M. P. et al. MicroRNAs in differentiation of embryoid bodies and the teratoma subtype of testicular cancer. Cancer Genom. Proteom. 19, 178–193 (2022).
Lafin, J. T. et al. Serum MicroRNA-371a-3p levels predict viable germ cell tumor in chemotherapy-naive patients undergoing retroperitoneal lymph node dissection. Eur. Urol. 77, 290–292 (2020).
Eaton, B. R. et al. Osteosarcoma. Pediatr. Blood Cancer 68, e28352 (2021).
Beird, H. C. et al. Osteosarcoma. Nat. Rev. Dis. Prim. 8, 77 (2022).
Aran, V. et al. Osteosarcoma, chondrosarcoma and Ewing sarcoma: clinical aspects, biomarker discovery and liquid biopsy. Crit. Rev. Oncol. Hematol. 162, 103340 (2021).
Ritter, J. & Bielack, S. S. Osteosarcoma. Ann. Oncol. 21, vii320–vii325 (2010).
Odri, G. A., Tchicaya-Bouanga, J., Yoon, D. J. Y. & Modrowski, D. Metastatic progression of osteosarcomas: a review of current knowledge of environmental versus oncogenic drivers. Cancers (Basel). 14, 360 (2022).
Ucci, A., Rucci, N. & Ponzetti, M. Liquid biopsies in primary and secondary bone cancers. Cancer Drug Resist 5, 541–559 (2022).
Barris, D. M. et al. Detection of circulating tumor DNA in patients with osteosarcoma. Oncotarget 9, 12695–12704 (2018).
Bao, Q. et al. Extracellular vesicle RNA sequencing reveals dramatic transcriptomic alterations between metastatic and primary osteosarcoma in a liquid biopsy approach. Ann. Surg. Oncol. 25, 2642–2651 (2018).
Lyskjær, I. et al. Osteosarcoma: novel prognostic biomarkers using circulating and cell-free tumour DNA. Eur. J. Cancer 168, 1–11 (2022).
Kurihara, S. et al. Circulating free DNA as non-invasive diagnostic biomarker for childhood solid tumors. J. Pediatr. Surg. 50, 2094–2097 (2015).
Van Paemel, R. et al. Minimally invasive classification of paediatric solid tumours using reduced representation bisulphite sequencing of cell-free DNA: a proof-of-principle study. Epigenetics 16, 196–208 (2021).
Shah, A. T. et al. A comprehensive circulating tumor DNA assay for detection of translocation and copy-number changes in pediatric sarcomas. Mol. Cancer Ther. 20, 2016–2025 (2021).
Shulman, D. S. et al. Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: a report from the Children’s Oncology Group. Br. J. Cancer 119, 615–621 (2018).
Gai, W. & Sun, K. Epigenetic biomarkers in cell-free DNA and applications in liquid biopsy. Genes (Basel) 10, 32 (2019).
Li, M. et al. Prognostic and clinicopathological significance of circulating tumor cells in osteosarcoma. J. Bone Oncol. 16, 100236 (2019).
Wu, Z. J., Tan, J. C., Qin, X., Liu, B. & Yuan, Z. C. Significance of circulating tumor cells in osteosarcoma patients treated by neoadjuvant chemotherapy and surgery. Cancer Manag. Res. 10, 3333–3339 (2018).
Sharma, S. et al. Circulating tumor cell isolation, culture, and downstream molecular analysis. Biotechnol. Adv. 36, 1063–1078 (2018).
Dai, S. et al. Association of circulating tumor cells and IMP3 expression with metastasis of osteosarcoma. Front Oncol. 13, 819357 (2023).
Zhang, H. Q. et al. [Detection and clinical significance of circulating tumor cells in osteosarcoma using immunofluorescence combined with in situ hybridization]. Zhonghua Zhong Liu Za Zhi 39, 485–489 (2017).
Zhang, H. et al. A liquid biopsy-based method for the detection and quantification of circulating tumor cells in surgical osteosarcoma patients. Int J. Oncol. 50, 1075–1086 (2017).
Jawad, M. U. & Scully, S. P. In brief: classifications in brief: enneking classification: benign and malignant tumors of the musculoskeletal system. Clin. Orthop. Relat. Res. 468, 2000–2002, (2010).
Xie, X. Y., Chen, X. M., Shi, L. & Liu, J. W. Increased expression of microRNA-26a-5p predicted a poor survival outcome in osteosarcoma patients: an observational study. Medicine (Baltimore) 100, e24765 (2021).
Raimondi, L. et al. Circulating biomarkers in osteosarcoma: new translational tools for diagnosis and treatment. Oncotarget 8, 100831–100851 (2017).
Moonmuang, S., Chaiyawat, P., Jantrapirom, S., Pruksakorn, D. & Lo Piccolo, L. Circulating long non-coding RNAs as novel potential biomarkers for osteogenic sarcoma. Cancers (Basel). 13, 4214 (2021).
Gally, T. B., Aleluia, M. M., Borges, G. F. & Kaneto, C. M. Circulating MicroRNAs as novel potential diagnostic biomarkers for osteosarcoma: a systematic review. Biomolecules 11, 1432 (2021).
Wang, Y. et al. Diagnostic and prognostic value of circulating miR-21 for cancer: a systematic review and meta-analysis. Gene 533, 389–397 (2014).
Dean, D. C., Shen, S., Hornicek, F. J. & Duan, Z. From genomics to metabolomics: emerging metastatic biomarkers in osteosarcoma. Cancer Metastasis Rev. 37, 719–731 (2018).
Liu, H. et al. MicroRNAs as a novel class of diagnostic biomarkers for the detection of osteosarcoma: a meta-analysis. Onco Targets Ther. 10, 5229–5236 (2017).
Zhang, J. et al. MicroRNAs in osteosarcoma. Clin. Chim. Acta 444, 9–17 (2015).
Gao, S. S., Wang, Y. J., Zhang, G. X. & Zhang, W. T. Potential diagnostic value of miRNAs in peripheral blood for osteosarcoma: a meta-analysis. J. Bone Oncol. 23, 100307 (2020).
Wang, S., Ma, F., Feng, Y., Liu, T. & He, S. Role of exosomal miR‑21 in the tumor microenvironment and osteosarcoma tumorigenesis and progression (Review). Int. J. Oncol. 56, 1055–1063 (2020).
Grünewald, T. G. P. et al. Ewing sarcoma. Nat. Rev. Dis. Prim. 4, 5 (2018).
Lin, P. P., Wang, Y. & Lozano, G. Mesenchymal stem cells and the origin of Ewing’s sarcoma. Sarcoma 2011, 276463 (2011).
Tu, J. et al. The histogenesis of Ewing sarcoma. Cancer Rep. Rev. 1, 1-4 (2017).
Khan, S. et al. Incidence of Ewing’s sarcoma in different age groups, their associated features, and its correlation with primary care interval. Cureus 13, e13986 (2021).
Zhang, P., Samuel, G., Crow, J., Godwin, A. K. & Zeng, Y. Molecular assessment of circulating exosomes toward liquid biopsy diagnosis of Ewing sarcoma family of tumors. Transl. Res. 201, 136–153 (2018).
Cervera, S. T. et al. Therapeutic potential of EWSR1-FLI1 inactivation by CRISPR/Cas9 in Ewing sarcoma. Cancers (Basel). 13, 3783 (2021).
Crow, J. et al. MicroRNA content of Ewing sarcoma derived extracellular vesicles leads to biomarker potential and identification of a previously undocumented EWS-FLI1 translocation. Biomark. Insights 17, 11772719221132693 (2022).
West, D. C. et al. Detection of circulating tumor cells in patients with Ewing’s sarcoma and peripheral primitive neuroectodermal tumor. J. Clin. Oncol. 15, 583–588 (1997).
Zoubek, A. et al. Predictive potential of testing for bone marrow involvement in Ewing tumor patients by RT-PCR: a preliminary evaluation. Int. J. Cancer 79, 56–60 (1998).
Fagnou, C. et al. Presence of tumor cells in bone marrow but not in blood is associated with adverse prognosis in patients with Ewing’s tumor. Société Française d’Oncologie Pédiatrique. J. Clin. Oncol. 16, 1707–1711 (1998).
de Alava, E., Lozano, M. D., Patiño, A., Sierrasesúmaga, L. & Pardo-Mindán, F. J. Ewing family tumors: potential prognostic value of reverse-transcriptase polymerase chain reaction detection of minimal residual disease in peripheral blood samples. Diagn. Mol. Pathol. 7, 152–157 (1998).
Athale, U. H. et al. Use of reverse transcriptase polymerase chain reaction for diagnosis and staging of alveolar rhabdomyosarcoma, Ewing sarcoma family of tumors, and desmoplastic small round cell tumor. J. Pediatr. Hematol. Oncol. 23, 99–104 (2001).
Schleiermacher, G. et al. Increased risk of systemic relapses associated with bone marrow micrometastasis and circulating tumor cells in localized Ewing tumor. J. Clin. Oncol. 21, 85–91 (2003).
Rizk, V. T., Walko, C. M. & Brohl, A. S. Precision medicine approaches for the management of Ewing sarcoma: current perspectives. Pharmgenom. Pers. Med 12, 9–14 (2019).
Krumbholz, M. et al. Quantification of translocation-specific ctDNA provides an integrating parameter for early assessment of treatment response and risk stratification in Ewing sarcoma. Clin. Cancer Res. 27, 5922–5930 (2021).
Schmidkonz, C. et al. Assessment of treatment responses in children and adolescents with Ewing sarcoma with metabolic tumor parameters derived from (18)F-FDG-PET/CT and circulating tumor DNA. Eur. J. Nucl. Med. Mol. Imaging 47, 1564–1575 (2020).
Seidel, M. G. et al. Clinical implementation of plasma cell-free circulating tumor DNA quantification by digital droplet PCR for the monitoring of Ewing sarcoma in children and adolescents. Front Pediatr. 10, 926405 (2022).
Allegretti, M. et al. Precision diagnostics of Ewing’s sarcoma by liquid biopsy: circulating EWS-FLI1 fusion transcripts. Ther. Adv. Med. Oncol. 10, 1758835918774337 (2018).
Samuel, G. et al. Ewing sarcoma family of tumors-derived small extracellular vesicle proteomics identify potential clinical biomarkers. Oncotarget 11, 2995–3012 (2020).
Bodlak, A. et al. Circulating plasma tumor DNA is superior to plasma tumor RNA detection in Ewing sarcoma patients: ptDNA and ptRNA in Ewing sarcoma. J. Mol. Diagn. 23, 872–881 (2021).
Tanaka, K., Iwakuma, T., Harimaya, K., Sato, H. & Iwamoto, Y. EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma and primitive neuroectodermal tumor cells. J. Clin. Invest 99, 239–247 (1997).
Erkizan, H. V. et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med 15, 750–756 (2009).
Salguero-Aranda, C., Amaral, A. T., Olmedo-Pelayo, J., Diaz-Martin, J. & Álava, E. Breakthrough technologies reshape the Ewing sarcoma molecular landscape. Cells 9 (2020).
International Euro Ewing Trial for Treatmentoptimisation in Patients with Ewing Sarcoma, https://www.gpoh.de/kinderkrebsinfo/content/fachinformationen/studienportal/onkologische_studien_und_register/ieuroewing/index_ger.html (2024).
International clinical research programme to improve outcomes in newly diagnosed Ewing sarcoma – Trial 1, https://www.isrctn.com/ISRCTN17938906 (2023).
Skapek, S. X. et al. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 5, 1 (2019).
Dasgupta, R., Fuchs, J. & Rodeberg, D. Rhabdomyosarcoma. Semin Pediatr. Surg. 25, 276–283 (2016).
Kaseb, H., Kuhn, J., Gasalberti, D. P. et al. Rhabdomyosarcoma. In StatPearls [Internet]. Treasure Island (FL) (StatPearls Publishing, 2024). Available from: https://www.ncbi.nlm.nih.gov/books/NBK507721/
Miwa, S. et al. Recent advances and challenges in the treatment of rhabdomyosarcoma. Cancers (Basel). 12, 1758 (2020).
Hettmer, S. et al. Molecular testing of rhabdomyosarcoma in clinical trials to improve risk stratification and outcome: a consensus view from European paediatric Soft tissue sarcoma Study Group, Children’s Oncology Group and Cooperative Weichteilsarkom-Studiengruppe. Eur. J. Cancer 172, 367–386 (2022).
Oberlin, O. et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J. Clin. Oncol. 26, 2384–2389 (2008).
Ouchi, K. et al. Oncogenic role of HMGA2 in fusion-negative rhabdomyosarcoma cells. Cancer Cell Int. 20, 192 (2020).
Lak, N. S. M. et al. Improving risk stratification for pediatric patients with rhabdomyosarcoma by molecular detection of disseminated disease. Clin. Cancer Res. 27, 5576–5585 (2021).
Wang, C. Childhood rhabdomyosarcoma: recent advances and prospective views. J. Dent. Res. 91, 341–350, (2012).
Leiner, J. & Le Loarer, F. The current landscape of rhabdomyosarcomas: an update. Virchows Arch. 476, 97–108 (2020).
Williamson, D. et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J. Clin. Oncol. 28, 2151–2158 (2010).
Hibbitts, E. et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children’s Oncology Group. Cancer Med 8, 6437–6448 (2019).
Gallego, S. et al. Fusion status in patients with lymph node-positive (N1) alveolar rhabdomyosarcoma is a powerful predictor of prognosis: Experience of the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG). Cancer 124, 3201–3209 (2018).
Heske, C. M. et al. Survival outcomes of patients with localized FOXO1 fusion-positive rhabdomyosarcoma treated on recent clinical trials: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer 127, 946–956 (2021).
Michelagnoli, M. P., Burchill, S. A., Cullinane, C., Selby, P. J. & Lewis, I. J. Myogenin–a more specific target for RT-PCR detection of rhabdomyosarcoma than MyoD1. Med. Pediatr. Oncol. 40, 1–8 (2003).
Gallego, S. et al. Detection of bone marrow micrometastasis and microcirculating disease in rhabdomyosarcoma by a real-time RT-PCR assay. J. Cancer Res. Clin. Oncol. 132, 356–362 (2006).
Sartori, F. et al. Results of a prospective minimal disseminated disease study in human rhabdomyosarcoma using three different molecular markers. Cancer 106, 1766–1775 (2006).
Krskova, L. et al. Detection and clinical significance of bone marrow involvement in patients with rhabdomyosarcoma. Virchows Arch. 456, 463–472 (2010).
Ruhen, O. et al. Molecular characterization of circulating tumor DNA in pediatric rhabdomyosarcoma: a feasibility study. JCO Precis. Oncol. 6, e2100534 (2022).
Abbou, S. et al. Circulating tumor DNA is prognostic in intermediate-risk rhabdomyosarcoma: a report from the children’s oncology group. J. Clin. Oncol. 41, 2382–2393 (2023).
Lak, N. S. M. et al. Cell-free DNA as a diagnostic and prognostic biomarker in pediatric rhabdomyosarcoma. JCO Precis. Oncol. 7, e2200113 (2023).
Poli, E. et al. Prognostic value of circulating IGFBP2 and related autoantibodies in children with metastatic rhabdomyosarcomas. Diagnostics (Basel) 10, 115 (2020).
Urla, C. et al. Epitope Detection in Monocytes (EDIM) as a new method of liquid biopsy in pediatric rhabdomyosarcoma. Biomedicines 10, 1812 (2022).
de Traux de Wardin, H. et al. Sequential genomic analysis using a multisample/multiplatform approach to better define rhabdomyosarcoma progression and relapse. NPJ Precis. Oncol. 7, 96 (2023).
Hayashi, M. et al. Size-based detection of sarcoma circulating tumor cells and cell clusters. Oncotarget 8, 78965–78977 (2017).
Eguchi-Ishimae, M. et al. Early detection of the PAX3-FOXO1 fusion gene in circulating tumor-derived DNA in a case of alveolar rhabdomyosarcoma. Genes Chromosomes. Cancer 58, 521–529 (2019).
Tombolan, L. et al. Clinical significance of circulating tumor cells and cell-free DNA in pediatric rhabdomyosarcoma. Mol. Oncol. 16, 2071–2085 (2022).
Stegmaier, S. et al. Fusion transcripts as liquid biopsy markers in alveolar rhabdomyosarcoma and synovial sarcoma: A report of the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr. Blood Cancer 69, e29652 (2022).
FaR-RMS: An Overarching Study for Children and Adults With Frontline and Relapsed RhabdoMyoSarcoma (FaR-RMS), https://clinicaltrials.gov/study/NCT04625907 (2023).
Combination Chemotherapy With or Without Temsirolimus in Treating Patients With Intermediate Risk Rhabdomyosarcoma, https://clinicaltrials.gov/study/NCT02567435 (2024).
Ferrari, A. et al. Paediatric non-rhabdomyosarcoma soft tissue sarcomas: the prospective NRSTS 2005 study by the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG). Lancet Child Adolesc. Health 5, 546–558 (2021).
Waxweiler, T. V. et al. Non-rhabdomyosarcoma soft tissue sarcomas in children: a surveillance, epidemiology, and end results analysis validating COG risk stratifications. Int. J. Radiat. Oncol. Biol. Phys. 92, 339–348 (2015).
Ferrari, A. et al. Pediatric non-rhabdomyosarcoma soft tissue sarcomas: standard of care and treatment recommendations from the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG). Cancer Manag Res 14, 2885–2902 (2022).
Ferrari, A. et al. Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr. Blood Cancer 57, 943–949 (2011).
Qureshi, S. S. & Bhagat, M. Non-rhabdomyosarcoma soft-tissue sarcomas in children: contemporary appraisal and experience from a single centre. J. Indian Assoc. Pediatr. Surg. 20, 165–169 (2015).
Venkatramani, R. et al. Synovial sarcoma in children, adolescents, and young adults: a report from the children’s oncology group ARST0332 study. J. Clin. Oncol. 39, 3927–3937 (2021).
Ferrari, A. et al. Metastatic adult-type non-rhabdomyosarcoma soft tissue sarcomas in children and adolescents: a cohort study from the European paediatric Soft tissue sarcoma Study Group. Cancer 129, 2542–2552 (2023).
Alix-Panabières, C., Marchetti, D. & Lang, J. E. Liquid biopsy: from concept to clinical application. Sci. Rep. 13, 21685 (2023).
Mertens, F., Antonescu, C. R. & Mitelman, F. Gene fusions in soft tissue tumors: Recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer 55, 291–310 (2016).
Shukla, N. N. et al. Plasma DNA-based molecular diagnosis, prognostication, and monitoring of patients with EWSR1 fusion-positive sarcomas. JCO Precis. Oncol. 2017, 1–11 (2017).
Uotani, K. et al. Circulating MicroRNA-92b-3p as a novel biomarker for monitoring of synovial sarcoma. Sci. Rep. 7, 14634 (2017).
Yokoo, S. et al. Liquid biopsy targeting monocarboxylate transporter 1 on the surface membrane of tumor-derived extracellular vesicles from synovial sarcoma. Cancers (Basel). 13, 1823 (2021).
Szymanski, J. J. et al. Cell-free DNA ultra-low-pass whole genome sequencing to distinguish malignant peripheral nerve sheath tumor (MPNST) from its benign precursor lesion: a cross-sectional study. PLoS Med 18, e1003734 (2021).
Wakely, P. E. Jr. Cytopathology of classic type epithelioid sarcoma: a series of 20 cases and review of the literature. J. Am. Soc. Cytopathol. 9, 126–136 (2020).
Viñal, D. et al. Prognostic value of neutrophil-to-lymphocyte ratio and other inflammatory markers in patients with high-risk soft tissue sarcomas. Clin. Transl. Oncol. 22, 1849–1856 (2020).
Que, Y. et al. Preoperative platelet-lymphocyte ratio is superior to neutrophil-lymphocyte ratio as a prognostic factor for soft-tissue sarcoma. BMC Cancer 15, 648 (2015).
Park, S. J. et al. Serum biomarkers for neurofibromatosis type 1 and early detection of malignant peripheral nerve-sheath tumors. BMC Med 11, 109 (2013).
Hummel, T. R. et al. Gene expression analysis identifies potential biomarkers of neurofibromatosis type 1 including adrenomedullin. Clin. Cancer Res. 16, 5048–5057 (2010).
Chen, S. et al. Prognostic analysis of surgically treated clear cell sarcoma: an analysis of a rare tumor from a single center. Int. J. Clin. Oncol. 24, 1605–1611 (2019).
Bottillo, I. et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J. Pathol. 217, 693–701 (2009).
Arshad, J. et al. Utility of circulating tumor DNA in the management of patients with GI stromal tumor: analysis of 243 patients. JCO Precis. Oncol. 4, 66–73 (2020).
Sultan, I. et al. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115, 3537–3547 (2009).
Colletti, M. et al. Expression profiles of exosomal miRNAs isolated from plasma of patients with desmoplastic small round cell tumor. Epigenomics 11, 489–500 (2019).
Peneder, P. et al. Multimodal analysis of cell-free DNA whole-genome sequencing for pediatric cancers with low mutational burden. Nat. Commun. 12, 3230 (2021).
Tombolan, L. et al. Pediatric sarcomas display a variable EpCAM expression in a histology-dependent manner. Transl. Oncol. 13, 100846 (2020).
MyKids: molecular profiling of non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) in children, adolescents and young adults, https://www.isrctn.com/ISRCTN12831761 (2022).
Ranganathan, S., Lopez-Terrada, D. & Alaggio, R. Hepatoblastoma and pediatric hepatocellular carcinoma: an update. Pediatr. Dev. Pathol. 23, 79–95 (2020).
Ng, K. & Mogul, D. B. Pediatric liver tumors. Clin. Liver Dis. 22, 753–772 (2018).
Geramizadeh, B., Foroughi, R. & Shojazadeh, A. Hepatocellular malignant neoplasm, not otherwise specified: a new name in liver tumors: a brief narrative review of published cases. Gastrointest. Tumors 8, 96–100 (2021).
Crippa, S. et al. Mutant CTNNB1 and histological heterogeneity define metabolic subtypes of hepatoblastoma. EMBO Mol. Med. 9, 1589–1604 (2017).
Varol, F. I. Pediatric hepatocellular carcinoma. J. Gastrointest. Cancer 51, 1169–1175 (2020).
Espinoza, A. F. et al. An indocyanine green-based liquid biopsy test for circulating tumor cells for pediatric liver cancer. bioRxiv, (2023).
Carr, B. I. et al. HCC with low- and normal-serum alpha-fetoprotein levels. Clin. Pract. (Lond.) 15, 453–464 (2018).
Rabbani, T., Bartlett, J. M. A. & Mittal, N. Liver biopsy in children. Indian Pediatr. 57, 734–740 (2020).
Liu, W., Chen, S. & Liu, B. Diagnostic and prognostic values of serum exosomal microRNA-21 in children with hepatoblastoma: a Chinese population-based study. Pediatr. Surg. Int. 32, 1059–1065 (2016).
Jiao, C., Jiao, X., Zhu, A., Ge, J. & Xu, X. Exosomal miR-34s panel as potential novel diagnostic and prognostic biomarker in patients with hepatoblastoma. J. Pediatr. Surg. 52, 618–624 (2017).
Maurya, P., Meleady, P., Dowling, P. & Clynes, M. Proteomic approaches for serum biomarker discovery in cancer. Anticancer Res. 27, 1247–1255, (2007).
Zhao, W. et al. Screening and identification of apolipoprotein A-I as a potential hepatoblastoma biomarker in children, excluding inflammatory factors. Oncol. Lett. 10, 233–239 (2015).
Kahana-Edwin, S. et al. Exploration of CTNNB1 ctDNA as a putative biomarker for hepatoblastoma. Pediatr. Blood Cancer 67, e28594 (2020).
Visal, T. H., den Hollander, P., Cristofanilli, M. & Mani, S. A. Circulating tumour cells in the -omics era: how far are we from achieving the ‘singularity’? Br. J. Cancer 127, 173–184 (2022).
Kim, E. S. et al. Potential utility of risk stratification for multicancer screening with liquid biopsy tests. NPJ Precis. Oncol. 7, 39 (2023).
Tie, J. et al. Circulating tumor DNA analysis guiding adjuvant therapy in stage II colon cancer. N. Engl. J. Med. 386, 2261–2272 (2022).
Cho, H. W. et al. Treatment outcomes in children and adolescents with relapsed or progressed solid tumors: a 20-year, single-center study. J. Korean Med Sci. 33, e260 (2018).
Pieters, R. et al. Successful therapy reduction and intensification for childhood acute lymphoblastic leukemia based on minimal residual disease monitoring: study ALL10 from the dutch childhood oncology group. J. Clin. Oncol. 34, 2591–2601 (2016).
Pieters, R. et al. Dutch ALL11 study: improved outcome for acute lymphoblastic leukemia by prolonging therapy for IKZF1 deletion and decreasing therapy for ETV6::RUNX1, down syndrome and prednisone poor responders. Blood 140, 519–520 (2022).
Chicard, M. et al. Whole-exome sequencing of cell-free DNA reveals temporo-spatial heterogeneity and identifies treatment-resistant clones in neuroblastoma. Clin. Cancer Res. 24, 939–949 (2018).
Remon, J. et al. Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Ann. Oncol. 28, 784–790 (2017).
Bertacca, I., Pegoraro, F., Tondo, A. & Favre, C. Targeted treatment of solid tumors in pediatric precision oncology. Front Oncol. 13, 1176790 (2023).
Dawson, S. J. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med 368, 1199–1209 (2013).
Tie, J. et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut 68, 663–671 (2019).
Chaudhuri, A. A. et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 7, 1394–1403 (2017).
Henriksen, T. V. V. et al. Serial circulating tumor DNA analysis to assess recurrence risk, benefit of adjuvant therapy, growth rate and early relapse detection in stage III colorectal cancer patients. J. Clin. Oncol. 39, 3540–3540 (2021).
Powles, T. et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 595, 432–437 (2021).
Karley, D., Gupta, D. & Tiwari, A. Biomarker for cancer: a great promise for future. World J. Oncol. 2, 151–157 (2011).
Novetsky Friedman, D. et al. Clonal hematopoiesis in survivors of childhood cancer. Blood Adv. 7, 4102–4106 (2023).
Hagiwara, K. et al. Dynamics of age- versus therapy-related clonal hematopoiesis in long-term survivors of pediatric cancer. Cancer Discov. 13, 844–857 (2023).
Chan, H. T., Chin, Y. M., Nakamura, Y. & Low, S. K. Clonal hematopoiesis in liquid biopsy: from biological noise to valuable clinical implications. Cancers (Basel). 12, 2277 (2020).
Huang, F. et al. Chemotherapy-associated clonal hematopoiesis mutations should be taken seriously in plasma cell-free DNA KRAS/NRAS/BRAF genotyping for metastatic colorectal cancer. Clin. Biochem 92, 46–53 (2021).
Leal, A. et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 11, 525 (2020).
Chan, H. T. et al. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol. Oncol. 14, 1719–1730 (2020).
Ococks, E. et al. Longitudinal tracking of 97 esophageal adenocarcinomas using liquid biopsy sampling. Ann. Oncol. 32, 522–532 (2021).
Croitoru, V. M. et al. Clonal hematopoiesis and liquid biopsy in gastrointestinal cancers. Front Med. (Lausanne) 8, 772166 (2021).
Greytak, S. R. et al. Harmonizing cell-free DNA collection and processing practices through evidence-based guidance. Clin. Cancer Res. 26, 3104–3109 (2020).
Almasi, M. et al. Biobanking - the first step to successful liquid biopsy experiments. Klin. Onkol. 30, 9–12 (2017).
Colomer, R. et al. Usefulness and real-world outcomes of next generation sequencing testing in patients with cancer: an observational study on the impact of selection based on clinical judgement. EClinicalMedicine 60, 102029 (2023).
Anagnostou, V. & Velculescu, V. E. Pushing the boundaries of liquid biopsies for early precision intervention. Cancer Discov. 14, 615–619 (2024).
European Union. Ethical considerations for clinical trials on medicinal products conducted with the paediatric population. Eur. J. Health Law 15, 223–250 (2008).
De Luca, G. & Dono, M. The opportunities and challenges of molecular tagging next-generation sequencing in liquid biopsy. Mol. Diagn. Ther. 25, 537–547 (2021).
Kobayashi, K. et al. Cell-free DNA oncogene copy number as a surrogate molecular biomarker in ALK/MYCN-coamplified neuroblastoma. J. Pediatr. Hematol. Oncol. 43, e165–e168 (2021).
Combaret, V. et al. Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer Res. 62, 3646–3648 (2002).
Combaret, V., Bergeron, C., Noguera, R., Iacono, I. & Puisieux, A. Circulating MYCN DNA predicts MYCN-amplification in neuroblastoma. J. Clin. Oncol. 23, 8919–8920 (2005). author reply 8920.
Ma, Y. et al. Detection of MYCN amplification in serum DNA using conventional polymerase chain reaction. J. Korean Med. Sci. 31, 1392–1396 (2016).
Kojima, M. et al. Detection of MYCN amplification using blood plasma: noninvasive therapy evaluation and prediction of prognosis in neuroblastoma. Pediatr. Surg. Int 29, 1139–1145 (2013).
Panachan, J. et al. Extracellular Vesicle-Based Method for Detecting MYCN Amplification Status of Pediatric Neuroblastoma. Cancers (Basel) 14, 2627 (2022).
Chicard, M. et al. Genomic Copy Number Profiling Using Circulating Free Tumor DNA Highlights Heterogeneity in Neuroblastoma. Clin. Cancer Res 22, 5564–5573 (2016).
Combaret, V. et al. Influence of neuroblastoma stage on serum-based detection of MYCN amplification. Pediatr. Blood Cancer 53, 329–331 (2009).
Gotoh, T. et al. Prediction of MYCN amplification in neuroblastoma using serum DNA and real-time quantitative polymerase chain reaction. J. Clin. Oncol. 23, 5205–5210 (2005).
Iehara, T. et al. A prospective evaluation of liquid biopsy for detecting MYCN amplification in neuroblastoma patients. Jpn J. Clin. Oncol. 49, 743–748 (2019).
Yagyu, S. et al. Serum-based quantification of MYCN gene amplification in young patients with neuroblastoma: potential utility as a surrogate biomarker for neuroblastoma. PLoS One 11, e0161039 (2016).
Van Roy, N. et al. Shallow whole genome sequencing on circulating cell-free DNA allows reliable noninvasive copy-number profiling in neuroblastoma patients. Clin. Cancer Res. 23, 6305–6314 (2017).
Lodrini, M. et al. Targeted analysis of cell-free circulating tumor DNA is suitable for early relapse and actionable target detection in patients with neuroblastoma. Clin. Cancer Res. 28, 1809–1820 (2022).
Lodrini, M. et al. Using droplet digital PCR to analyze MYCN and ALK copy number in plasma from patients with neuroblastoma. Oncotarget 8, 85234–85251 (2017).
Combaret, V. et al. Detection of tumor ALK status in neuroblastoma patients using peripheral blood. Cancer Med. 4, 540–550 (2015).
Kahana-Edwin, S. et al. Neuroblastoma molecular risk-stratification of DNA copy number and ALK genotyping via cell-free circulating tumor DNA profiling. Cancers (Basel) 13, 3365 (2021).
Combaret, V. et al. Determination of 17q gain in patients with neuroblastoma by analysis of circulating DNA. Pediatr. Blood Cancer 56, 757–761 (2011).
Yagyu, S. et al. Preoperative analysis of 11q loss using circulating tumor-released DNA in serum: a novel diagnostic tool for therapy stratification of neuroblastoma. Cancer Lett. 309, 185–189 (2011).
Applebaum, M. A. et al. 5-Hydroxymethylcytosine profiles in circulating cell-free DNA associate with disease burden in children with neuroblastoma. Clin. Cancer Res. 26, 1309–1317 (2020).
Lodrini, M. et al. Circulating cell-free DNA assessment in biofluids from children with neuroblastoma demonstrates feasibility and potential for minimally invasive molecular diagnostics. Cancers (Basel) 14, 2080 (2022).
Stutterheim, J. et al. The prognostic value of fast molecular response of marrow disease in patients aged over 1 year with stage 4 neuroblastoma. Eur. J. Cancer 47, 1193–1202 (2011).
Cimmino, F., Lasorsa, V. A., Vetrella, S., Iolascon, A. & Capasso, M. A targeted gene panel for circulating tumor DNA sequencing in neuroblastoma. Front Oncol. 10, 596191 (2020).
Riehl, L. et al. Targeted parallel DNA sequencing detects circulating tumor-associated variants of the mitochondrial and nuclear genomes in patients with neuroblastoma. Cancer Rep. (Hoboken) 6, e1687 (2023).
Duan, C. et al. Whole exome sequencing reveals novel somatic alterations in neuroblastoma patients with chemotherapy. Cancer Cell Int. 18, 21 (2018).
van Wezel, E. M. et al. Whole-genome sequencing identifies patient-specific DNA minimal residual disease markers in neuroblastoma. J. Mol. Diagn. 17, 43–52 (2015).
Su, Y. et al. Dynamic alterations of plasma cell free DNA in response to chemotherapy in children with neuroblastoma. Cancer Med. 8, 1558–1566 (2019).
van Zogchel, L. M. J. et al. Hypermethylated RASSF1A as circulating tumor DNA marker for disease monitoring in neuroblastoma. JCO Precis Oncol. 4, PO.19.00261 (2020).
Su, Y. et al. Increased plasma concentration of cell-free DNA precedes disease recurrence in children with high-risk neuroblastoma. BMC Cancer 20, 102 (2020).
Wang, X. et al. Plasma cell-free DNA quantification is highly correlated to tumor burden in children with neuroblastoma. Cancer Med 7, 3022–3030 (2018).
Sausen, M. et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet 45, 12–17 (2013).
Yagyu, S. et al. Circulating methylated-DCR2 gene in serum as an indicator of prognosis and therapeutic efficacy in patients with MYCN nonamplified neuroblastoma. Clin. Cancer Res. 14, 7011–7019 (2008).
Misawa, A. et al. RASSF1A hypermethylation in pretreatment serum DNA of neuroblastoma patients: a prognostic marker. Br. J. Cancer 100, 399–404 (2009).
Charlton, J. et al. Methylome analysis identifies a Wilms tumor epigenetic biomarker detectable in blood. Genome Biol. 15, 434 (2014).
Ueno-Yokohata, H. et al. Preoperative diagnosis of clear cell sarcoma of the kidney by detection of BCOR internal tandem duplication in circulating tumor DNA. Genes Chromosomes Cancer 57, 525–529 (2018).
Biderman Waberski, M. et al. Urine cell-free DNA is a biomarker for nephroblastomatosis or Wilms tumor in PIK3CA-related overgrowth spectrum (PROS). Genet Med 20, 1077–1081 (2018).
Treger, T. D. et al. Somatic TP53 mutations are detectable in circulating tumor DNA from children with anaplastic Wilms tumors. Transl. Oncol. 11, 1301–1306 (2018).
Chen, J. et al. Design of a targeted sequencing assay to detect rare mutations in circulating tumor DNA. Genet Test. Mol. Biomark. 23, 264–269 (2019).
Rossi, E. et al. Liquid biopsy in pediatric renal cancer: stage I and stage IV cases compared. Diagnostics (Basel) 10, 810 (2020).
Li, H. et al. Detection of circulating tumor cells from cryopreserved human sarcoma peripheral blood mononuclear cells. Cancer Lett. 403, 216–223 (2017).
Sittiju, P. et al. Osteosarcoma-specific genes as a diagnostic tool and clinical predictor of tumor progression. Biology (Basel) 11 (2022).
Batth, I. S. et al. Rare osteosarcoma cell subpopulation protein array and profiling using imaging mass cytometry and bioinformatics analysis. BMC Cancer 20, 715 (2020).
Satelli, A. et al. Universal marker and detection tool for human sarcoma circulating tumor cells. Cancer Res. 74, 1645–1650 (2014).
Fasanya, H. O., Dopico, P. J., Yeager, Z., Fan, Z. H. & Siemann, D. W. Using a combination of gangliosides and cell surface vimentin as surface biomarkers for isolating osteosarcoma cells in microfluidic devices. J. Bone Oncol. 28, 100357 (2021).
Li, X. et al. [Expressions of ERCC2 and ERCC4 genes in osteosarcoma and peripheral blood lymphocytes and their clinical significance]. Beijing Da Xue Xue Bao Yi Xue Ban. 39, 467–471 (2007).
Wong, I. H., Chan, A. T. & Johnson, P. J. Quantitative analysis of circulating tumor cells in peripheral blood of osteosarcoma patients using osteoblast-specific messenger RNA markers: a pilot study. Clin. Cancer Res. 6, 2183–2188, (2000).
Luo, Z., Liu, M., Zhang, H. & Xia, Y. Association of circulating miR-125b and survival in patients with osteosarcoma-A single center experience. J. Bone Oncol. 5, 167–172 (2016).
Nakka, M. et al. Biomarker significance of plasma and tumor miR-21, miR-221, and miR-106a in osteosarcoma. Oncotarget 8, 96738–96752 (2017).
Tian, Q. et al. A causal role for circulating miR-34b in osteosarcoma. Eur. J. Surg. Oncol. 40, 67–72 (2014).
Zhu, K. et al. Circular RNA hsa_circ_0000885 levels are increased in tissue and serum samples from patients with osteosarcoma. Med. Sci. Monit. 25, 1499–1505 (2019).
Ma, W. et al. Circulating miR-148a is a significant diagnostic and prognostic biomarker for patients with osteosarcoma. Tumour Biol. 35, 12467–12472 (2014).
Monterde-Cruz, L. et al. Circulating miR-215-5p and miR-642a-5p as potential biomarker for diagnosis of osteosarcoma in Mexican population. Hum. Cell 31, 292–299 (2018).
Fujiwara, T. et al. Clinical significance of circulating miR-25-3p as a novel diagnostic and prognostic biomarker in osteosarcoma. Oncotarget 8, 33375–33392 (2017).
Zhang, C., Yao, C., Li, H., Wang, G. & He, X. Combined elevation of microRNA-196a and microRNA-196b in sera predicts unfavorable prognosis in patients with osteosarcomas. Int. J. Mol. Sci. 15, 6544–6555 (2014).
Kohama, I. et al. Comprehensive serum and tissue microRNA profiling in dedifferentiated liposarcoma. Oncol. Lett. 22, 623 (2021).
Allen-Rhoades, W. et al. Cross-species identification of a plasma microRNA signature for detection, therapeutic monitoring, and prognosis in osteosarcoma. Cancer Med. 4, 977–988 (2015).
Yao, Z. S. et al. Diagnostic and prognostic implications of serum miR-101 in osteosarcoma. Cancer Biomark. 22, 127–133 (2018).
Tang, J., Zhao, H., Cai, H. & Wu, H. Diagnostic and prognostic potentials of microRNA-27a in osteosarcoma. Biomed. Pharmacother. 71, 222–226 (2015).
Zhou, L. et al. The diagnostic effect of serum miR-139-5p as an indicator in osteosarcoma. Cancer Biomark. 23, 561–567 (2018).
Wang, N. G., Wang, D. C., Tan, B. Y., Wang, F. & Yuan, Z. N. Down-regulation of microRNA152 is associated with the diagnosis and prognosis of patients with osteosarcoma. Int. J. Clin. Exp. Pathol. 8, 9314–9319 (2015).
Zhao, X. et al. Downregulation of microRNA-95-3p suppresses cell growth of osteosarcoma via CDKN1A/p21 expression. Oncol. Rep. 39, 289–297 (2018).
Wang, L., Gao, H., Gong, N. & Gong, M. Downregulation of microRNA-497 is associated with upregulation of synuclein γ in patients with osteosarcoma. Exp. Ther. Med 12, 3761–3766 (2016).
Shi, L., Xie, C., Zhu, J. & Chen, X. Downregulation of serum miR-194 predicts poor prognosis in osteosarcoma patients. Ann. Diagn. Pathol. 46, 151488 (2020).
Gong, L. et al. Exosomal miR-675 from metastatic osteosarcoma promotes cell migration and invasion by targeting CALN1. Biochem. Biophys. Res. Commun. 500, 170–176 (2018).
Xu, J. F. et al. Exosomes containing differential expression of microRNA and mRNA in osteosarcoma that can predict response to chemotherapy. Oncotarget 8, 75968–75978 (2017).
Zhang, K. et al. Extracellular vesicle-mediated delivery of miR-101 inhibits lung metastasis in osteosarcoma. Theranostics 10, 411–425 (2020).
Huang, C. et al. A four serum-miRNA panel serves as a potential diagnostic biomarker of osteosarcoma. Int. J. Clin. Oncol. 24, 976–982 (2019).
Cuscino, N. et al. Gathering novel circulating exosomal microRNA in osteosarcoma cell lines and possible implications for the disease. Cancers (Basel) 11, 1924 (2019).
Lian, F., Cui, Y., Zhou, C., Gao, K. & Wu, L. Identification of a plasma four-microRNA panel as potential noninvasive biomarker for osteosarcoma. PLoS One 10, e0121499 (2015).
Huang, C. et al. Identification of circulating miR-663a as a potential biomarker for diagnosing osteosarcoma. Pathol. Res Pract. 215, 152411 (2019).
Zhou, G. et al. Identification of miR-199a-5p in serum as noninvasive biomarkers for detecting and monitoring osteosarcoma. Tumour Biol. 36, 8845–8852 (2015).
Xu, K. et al. Identification of potential micro-messenger RNAs (miRNA-mRNA) interaction network of osteosarcoma. Bioengineered 12, 3275–3293 (2021).
Yuan, J., Chen, L., Chen, X., Sun, W. & Zhou, X. Identification of serum microRNA-21 as a biomarker for chemosensitivity and prognosis in human osteosarcoma. J. Int. Med. Res 40, 2090–2097 (2012).
Cong, C. et al. Identification of serum miR-124 as a biomarker for diagnosis and prognosis in osteosarcoma. Cancer Biomark. 21, 449–454 (2018).
Xie, L. et al. Identification of the miRNA-mRNA regulatory network of small cell osteosarcoma based on RNA-seq. Oncotarget 8, 42525–42536 (2017).
Wang, T., Ji, F., Dai, Z., Xie, Y. & Yuan, D. Increased expression of microRNA-191 as a potential serum biomarker for diagnosis and prognosis in human osteosarcoma. Cancer Biomark. 15, 543–550 (2015).
Zhang, X. et al. Influence mechanism of miRNA-144 on proliferation and apoptosis of osteosarcoma cells. Oncol. Lett. 19, 1530–1536 (2020).
Li, H. et al. MicroRNA screening identifies circulating microRNAs as potential biomarkers for osteosarcoma. Oncol. Lett. 10, 1662–1668 (2015).
Tian, Z. G. et al. MicroRNA-337-5p participates in the development and progression of osteosarcoma via ERBB, MAPK and VEGF pathways. Eur. Rev. Med Pharm. Sci. 22, 5460–5470 (2018).
Liu, W., Zhao, X., Zhang, Y. J., Fang, G. W. & Xue, Y. MicroRNA-375 as a potential serum biomarker for the diagnosis, prognosis, and chemosensitivity prediction of osteosarcoma. J. Int. Med. Res. 46, 975–983 (2018).
Xu, N., Yang, W., Liu, Y., Yan, F. & Yu, Z. MicroRNA-411 promoted the osteosarcoma progression by suppressing MTSS1 expression. Environ. Sci. Pollut. Res. Int. 25, 12064–12071 (2018).
Zhou, X., Natino, D., Zhai, X., Gao, Z. & He, X. MicroRNA‑22 inhibits the proliferation and migration, and increases the cisplatin sensitivity, of osteosarcoma cells. Mol. Med. Rep. 17, 7209–7217 (2018).
Lian, H., Zhou, Y., Sun, Z. & Liu, K. MicroRNA34a is associated with chemotherapy resistance, metastasis, recurrence, survival, and prognosis in patient with osteosarcoma. Medicine (Baltimore) 101, e30722 (2022).
Sun, Y., He, N., Dong, Y. & Jiang, C. MiR-24-BIM-Smac/DIABLO axis controls the sensitivity to doxorubicin treatment in osteosarcoma. Sci. Rep. 6, 34238 (2016).
Cao, L., Wang, J. & Wang, P. Q. MiR-326 is a diagnostic biomarker and regulates cell survival and apoptosis by targeting Bcl-2 in osteosarcoma. Biomed. Pharmacother. 84, 828–835 (2016).
Zhou, S. et al. miR-421 is a diagnostic and prognostic marker in patients with osteosarcoma. Tumour Biol. 37, 9001–9007 (2016).
Wang, S. N. et al. miR-491 inhibits osteosarcoma lung metastasis and chemoresistance by targeting αB-crystallin. Mol. Ther. 25, 2140–2149 (2017).
Pang, P. C., Shi, X. Y., Huang, W. L. & Sun, K. miR-497 as a potential serum biomarker for the diagnosis and prognosis of osteosarcoma. Eur. Rev. Med Pharm. Sci. 20, 3765–3769 (2016).
Liu, K., Sun, X., Zhang, Y., Liu, L. & Yuan, Q. MiR-598: a tumor suppressor with biomarker significance in osteosarcoma. Life Sci. 188, 141–148 (2017).
Lei, J. et al. miRNA identification by nuclease digestion in ELISA for diagnosis of osteosarcoma. Biotechnol. Appl. Biochem. 69, 1365–1372 (2022).
Dong, J. et al. miRNA-223 is a potential diagnostic and prognostic marker for osteosarcoma. J. Bone Oncol. 5, 74–79 (2016).
Zhang, C., Yao, C., Li, H., Wang, G. & He, X. Serum levels of microRNA-133b and microRNA-206 expression predict prognosis in patients with osteosarcoma. Int. J. Clin. Exp. Pathol. 7, 4194–4203 (2014).
Li, S. et al. Serum microRNA-17 functions as a prognostic biomarker in osteosarcoma. Oncol. Lett. 12, 4905–4910 (2016).
Yang, Z. et al. Serum microRNA-221 functions as a potential diagnostic and prognostic marker for patients with osteosarcoma. Biomed. Pharmacother. 75, 153–158 (2015).
Fei, D. et al. Serum miR-9 as a prognostic biomarker in patients with osteosarcoma. J. Int. Med. Res. 42, 932–937 (2014).
Wang, T., Wu, J., Liu, X. & Li, S. Serum miR-34a is a potential diagnostic and prognostic marker for osteosarcoma. Int. J. Clin. Exp. Pathol. 10, 9683–9689 (2017).
Niu, J., Sun, Y., Guo, Q., Niu, D. & Liu, B. Serum miR-95-3p is a diagnostic and prognostic marker for osteosarcoma. Springerplus 5, 1947 (2016).
Cai, H., Zhao, H., Tang, J. & Wu, H. Serum miR-195 is a diagnostic and prognostic marker for osteosarcoma. J. Surg. Res. 194, 505–510 (2015).
Liu, J. D. et al. Serum miR-300 as a diagnostic and prognostic biomarker in osteosarcoma. Oncol. Lett. 12, 3912–3918 (2016).
Li, Q., Song, S., Ni, G., Li, Y. & Wang, X. Serum miR-542-3p as a prognostic biomarker in osteosarcoma. Cancer Biomark. 21, 521–526 (2018).
Ouyang, L. et al. A three-plasma miRNA signature serves as novel biomarkers for osteosarcoma. Med. Oncol. 30, 340 (2013).
Hua, Y., Jin, Z., Zhou, F., Zhang, Y. Q. & Zhuang, Y. The expression significance of serum MiR-21 in patients with osteosarcoma and its relationship with chemosensitivity. Eur. Rev. Med Pharm. Sci. 21, 2989–2994 (2017).
Hua, J. et al. Diagnostic and prognostic values of blood microRNA-Let7A for osteosarcoma. J. Bone Oncol. 12, 65–68 (2018).
Hong, Q., Fang, J., Pang, Y. & Zheng, J. Prognostic value of the microRNA-29 family in patients with primary osteosarcomas. Med. Oncol. 31, 37 (2014).
Wen, J. J., Ma, Y. D., Yang, G. S. & Wang, G. M. Analysis of circulating long non-coding RNA UCA1 as potential biomarkers for diagnosis and prognosis of osteosarcoma. Eur. Rev. Med. Pharm. Sci. 21, 498–503 (2017).
Xia, W. K. et al. Clinical implication of long noncoding RNA 91H expression profile in osteosarcoma patients. Onco Targets Ther. 9, 4645–4652 (2016).
Chen, S., Liu, Z., Lu, S. & Hu, B. EPEL promotes the migration and invasion of osteosarcoma cells by upregulating ROCK1. Oncol. Lett. 17, 3133–3140 (2019).
Zhang, G. F., Zhou, B. S., An, X. C., An, F. M. & Li, S. H. LINC01278 is highly expressed in osteosarcoma and participates in the development of tumors by mediating the miR-134-5p/KRAS axis. Onco Targets Ther. 14, 683–695 (2021).
Zhao, B., Liu, K. & Cai, L. LINK-A lncRNA functions in the metastasis of osteosarcoma by upregulating HIF1α. Oncol. Lett. 17, 5005–5011 (2019).
Wang, Y. et al. LncRNA FAL1 is a negative prognostic biomarker and exhibits pro-oncogenic function in osteosarcoma. J. Cell Biochem 119, 8481–8489 (2018).
Cai, L. et al. The lncRNA HNF1A-AS1 is a negative prognostic factor and promotes tumorigenesis in osteosarcoma. J. Cell Mol. Med. 21, 2654–2662 (2017).
Sheng, K. & Li, Y. LncRNA TUG1 promotes the development of osteosarcoma through RUNX2. Exp. Ther. Med 18, 3002–3008 (2019).
Yang, Q., Yu, H., Yin, Q., Hu, X. & Zhang, C. lncRNA-NEF is downregulated in osteosarcoma and inhibits cancer cell migration and invasion by downregulating miRNA-21. Oncol. Lett. 17, 5403–5408 (2019).
Chen, S., Xu, X., Lu, S. & Hu, B. Long non-coding RNA HAND2-AS1 targets glucose metabolism and inhibits cancer cell proliferation in osteosarcoma. Oncol. Lett. 18, 1323–1329 (2019).
Han, F., Wang, C., Wang, Y. & Zhang, L. Long noncoding RNA ATB promotes osteosarcoma cell proliferation, migration and invasion by suppressing miR-200s. Am. J. Cancer Res. 7, 770–783 (2017).
Huo, Y. et al. MALAT1 predicts poor survival in osteosarcoma patients and promotes cell metastasis through associating with EZH2. Oncotarget 8, 46993–47006 (2017).
Song, Q. H. et al. Study on targeting relationship between miR-320b and FGD5-AS1 and its effect on biological function of osteosarcoma cells. Cancer Manag. Res. 12, 13589–13598 (2020).
Ma, B. et al. Upregulation of long non-coding RNA TUG1 correlates with poor prognosis and disease status in osteosarcoma. Tumour Biol. 37, 4445–4455 (2016).
Wang, J. et al. Circulating exosomal PD-L1 at initial diagnosis predicts outcome and survival of patients with osteosarcoma. Clin. Cancer Res. 29, 659–666 (2023).
Han, Z. et al. Integrated microfluidic-SERS for exosome biomarker profiling and osteosarcoma diagnosis. Biosens. Bioelectron. 217, 114709 (2022).
Han, Z. et al. Matrix-assisted laser desorption ionization mass spectrometry profiling of plasma exosomes evaluates osteosarcoma metastasis. iScience 24, 102906 (2021).
Han, Z. et al. SERS and MALDI-TOF MS based plasma exosome profiling for rapid detection of osteosarcoma. Analyst 146, 6496–6505 (2021).
Thomson, B., Hawkins, D., Felgenhauer, J. & Radich, J. RT-PCR evaluation of peripheral blood, bone marrow and peripheral blood stem cells in children and adolescents undergoing VACIME chemotherapy for Ewing’s sarcoma and alveolar rhabdomyosarcoma. Bone Marrow Transpl. 24, 527–533 (1999).
Yu, M., Wan, Y. F. & Zou, Q. H. Cell-free circulating mitochondrial DNA in the serum: a potential non-invasive biomarker for Ewing’s sarcoma. Arch. Med Res. 43, 389–394 (2012).
Nie, C. L., Ren, W. H., Ma, Y., Xi, J. S. & Han, B. Circulating miR-125b as a biomarker of Ewing’s sarcoma in Chinese children. Genet Mol. Res. 14, 19049–19056 (2015).
Krumbholz, M. et al. Genomic EWSR1 fusion sequence as highly sensitive and dynamic plasma tumor marker in Ewing sarcoma. Clin. Cancer Res. 22, 4356–4365 (2016).
Hayashi, M. et al. Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer 122, 3015–3023 (2016).
Lee, S. Y., Lim, S. & Cho, D. H. Personalized genomic analysis based on circulating tumor cells of extra-skeletal Ewing sarcoma of the uterus: a case report of a 16-year-old Korean female. Exp. Ther. Med 16, 1343–1349 (2018).
Benini, S. et al. Detection of circulating tumor cells in liquid biopsy from Ewing sarcoma patients. Cancer Manag. Res. 10, 49–60 (2018).
Turaga, S. M. et al. Identification of small extracellular vesicle protein biomarkers for pediatric Ewing Sarcoma. Front Mol. Biosci. 10, 1138594 (2023).
Miyachi, M. et al. Circulating muscle-specific microRNA, miR-206, as a potential diagnostic marker for rhabdomyosarcoma. Biochem Biophys. Res. Commun. 400, 89–93 (2010).
Ghamloush, F. et al. The PAX3-FOXO1 oncogene alters exosome miRNA content and leads to paracrine effects mediated by exosomal miR-486. Sci. Rep. 9, 14242 (2019).
Tombolan, L. et al. Circulating miR-26a as potential prognostic biomarkers in pediatric rhabdomyosarcoma. Front Genet 11, 606274 (2020).
Li, X., Seebacher, N. A., Hornicek, F. J., Xiao, T. & Duan, Z. Application of liquid biopsy in bone and soft tissue sarcomas: present and future. Cancer Lett. 439, 66–77 (2018).
Murray, M. J., Nicholson, J. C. & Coleman, N. Biology of childhood germ cell tumours, focussing on the significance of microRNAs. Andrology 3, 129–139 (2015).
Takami, H. & Ichimura, K. Biomarkers for risk-based treatment modifications for CNS germ cell tumors: updates on biological underpinnings, clinical trials, and future directions. Front Oncol. 12, 982608 (2022).
Fankhauser, C. D., Nuño, M. M., Murray, M. J., Frazier, L. & Bagrodia, A. Circulating MicroRNAs for detection of germ cell tumours: a narrative review. Eur. Urol. Focus 8, 660–662 (2022).
Bottani, M., Banfi, G. & Lombardi, G. Circulating miRNAs as diagnostic and prognostic biomarkers in common solid tumors: focus on lung, breast, prostate cancers, and osteosarcoma. J. Clin. Med. 8, 1661 (2019).
Jezierska, M., Gawrychowska, A. & Stefanowicz, J. Diagnostic, prognostic and predictive markers in pediatric germ cell tumors-past, present and future. Diagnostics (Basel) 12, 278 (2022).
Perut, F., Roncuzzi, L. & Baldini, N. The emerging roles of extracellular vesicles in osteosarcoma. Front Oncol. 9, 1342 (2019).
Lobo, J., Gillis, A. J. M., Jerónimo, C., Henrique, R. & Looijenga, L. H. J. Human germ cell tumors are developmental cancers: impact of epigenetics on pathobiology and clinic. Int. J. Mol. Sci. 20, 258 (2019).
Sasaki, R., Osaki, M. & Okada, F. MicroRNA-based diagnosis and treatment of metastatic human osteosarcoma. Cancers (Basel). 11, 553 (2019).
De Martino, M., Chieffi, P. & Esposito, F. miRNAs and biomarkers in testicular germ cell tumors: an update. Int. J. Mol. Sci. 22, 1380 (2021).
Chavarriaga, J. & Hamilton, R. J. miRNAs for testicular germ cell tumours: contemporary indications for diagnosis, surveillance and follow-up. Andrology 11, 628–633 (2023).
Zeuschner, P., Linxweiler, J. & Junker, K. Non-coding RNAs as biomarkers in liquid biopsies with a special emphasis on extracellular vesicles in urological malignancies. Expert Rev. Mol. Diagn. 20, 151–167 (2020).
Lakpour, N. et al. Potential biomarkers for testicular germ cell tumour: Risk assessment, diagnostic, prognostic and monitoring of recurrence. Andrologia 53, e13998 (2021).
Looijenga, L. H. J., Kao, C. S. & Idrees, M. T. Predicting gonadal germ cell cancer in people with disorders of sex development; insights from developmental biology. Int. J. Mol. Sci. 20, 5017 (2019).
Murray, M. J., Huddart, R. A. & Coleman, N. The present and future of serum diagnostic tests for testicular germ cell tumours. Nat. Rev. Urol. 13, 715–725 (2016).
Ditonno, F. et al. The role of miRNA in testicular cancer: current insights and future perspectives. Medicina (Kaunas) 59, 2033 (2023).
Shulman, D. S. & Crompton, B. D. Using liquid biopsy in the treatment of patient with OS. Adv. Exp. Med Biol. 1257, 95–105 (2020).
Acknowledgements
This study was funded by the Dutch foundation Stichting Kinderen Kankervrij (KiKa). The funder was not involved in the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: F.J. and L.L.; literature analysis and design and writing: F.J.; revisions and editing: N.L., C.J., L.K., M.M., M.v.N., J.M., M.v.d.H.E., J.Z., G.T., and L.L.; supervision: L.L.; corresponding author: L.L. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Janssen, F.W., Lak, N.S.M., Janda, C.Y. et al. A comprehensive overview of liquid biopsy applications in pediatric solid tumors. npj Precis. Onc. 8, 172 (2024). https://doi.org/10.1038/s41698-024-00657-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00657-z
- Springer Nature Limited