
Abstract
Background
Wilms tumor is the most common pediatric renal malignancy and there is a clinical need for a molecular biomarker to assess treatment response and predict relapse. The known mutated genes in this tumor type show low mutation frequencies, whereas aberrant methylation at 11p15 is by far the most common aberration. We therefore analyzed the epigenome, rather than the genome, to identify ubiquitous tumor-specific biomarkers.
Results
Methylome analysis of matched normal kidney and Wilms tumor identifies 309 preliminary methylation variable positions which we translate into three differentially methylated regions (DMR) for use as tumor-specific biomarkers. Using two novel algorithms we show that these three DMRs are not confounded by cell type composition. We further show that these DMRs are not methylated in embryonic blastema but are intermediately methylated in Wilms tumor precursor lesions. We validate the biomarker DMRs using two independent sample sets of normal kidney and Wilms tumor and seven Wilms tumor histological subtypes, achieving 100% and 98% correct classification, respectively. As proof-of-principle for clinical utility, we successfully use biomarker DMR-2 in a pilot analysis of cell-free circulating DNA to monitor tumor response during treatment in ten patients.
Conclusions
These findings define the most common methylated regions in Wilms tumor known to date which are not associated with their embryonic origin or precursor stage. We show that this tumor-specific methylated DNA is released into the blood circulation where it can be detected non-invasively showing potential for clinical utility.
Similar content being viewed by others


Background
Wilms tumour (WT) is the most common paediatric renal cancer with a prevalence of one in 10,000 children [1]. In Europe, most patients receive four weeks of pre-operative chemotherapy prior to complete or partial nephrectomy, followed by tumour stage and histology-dependent post-operative treatment [2]. Although overall survival rates are good, there is a clinical need for a biomarker to evaluate patient response to chemotherapy and improve prediction of relapse.
Circulating cell-free DNA (cfDNA) isolated from blood has been used to assess tumour burden in other cancers [3]–[6]. In WT, the few genes that are recurrently mutated show low mutation frequencies; WTX (18%) [7], CTNNB1 (15%) [8] and WT1 (12%) [8] and do not account for the majority of WTs. However, epimutation affecting the IGF2/H19 locus at 11p15.5 is much more common, (69%) [8]. Additional genes and regions known to be affected by methylation in WT include GLIPR1[9], imprinted genes NNAT[10] and the WT1-antisense region [11], various satellite regions [12],[13], HACE1[14], RASSF1A[15], P16 and the protocadherin cluster at 5q31 [16]. Consequently, we concluded that interrogation of the methylome rather than the genome may be more likely to reveal ubiquitous tumour-specific biomarkers. Therefore, we performed genome-wide methylome analysis of matched WT and surrounding normal kidney (NK) to identify WT-specific sites of methylation which we then assessed in cfDNA for use as WT biomarkers.
Results
To identify tumour-specific methylation variable positions (MVPs), as previously defined [17], we derived methylation levels (β; 0 = unmethylated to 1 = methylated) for 462,537 CpG sites at single base-pair resolution using the Illumina Infinium HumanMethylation450 platform and performed linear modelling to compare 22 matched pairs of NK and WT (full clinical details in Additional file 1: Table S1). We identified 309 MVPs of genome-wide significance (P < 5x10−8; Figure 1). Due to the matched study design there was no need to adjust for age, race or gender and we can exclude the possibility of MVPs being confounded by genetic polymorphism(s). We then applied the novel pipeline Lasso, recently developed for analysis of Illumina 450 k data, which considers the local CpG density to group MVPs into functionally more relevant differentially methylated regions (DMRs) [18]. Using this method, we identified 3 DMRs which were hypermethylated in WT with respect to NK (Table 1).

Identification of differentially methylated loci with genome-wide significance. Manhattan plot showing –log10 p-values for all CpGs (462,537) generated by linear modelling of normal kidney (NK) against Wilms tumour (WT). CpGs with genome-wide significance (n = 309) appear above the red line. DMRs 1 and 2 on chromosome 6 and DMR-3 on chromosome 11 are shown in green.
To assess possible confounding due to differential cell type composition, we carried out two analyses. First, we conducted histological analysis confirming our samples to be composed of the expected major cell types, consisting of 95% epithelia and 5% stroma in NKs while WTs showed varying proportions of immature stroma, epithelia and blastema (Additional file 2: Figure S1). Second, we used a recently published algorithm, RefFreeEWAS [19] which corrects p-values based on estimated cell type contributions. Performing linear modelling using this algorithm, we identified 7,272 CpGs with genome-wide significance (P < 5x10−8) of which 937 had Δβ > 0.3 and were therefore considered cell-composition corrected MVPs. Of these, 766 were hyperMVPs and 171 were hypoMVPs in WT with respect to NK (Additional file 3: Table S2). For hyperMVPs in particular, we saw a striking positive enrichment for location within CpG islands (+18% compared to background). There were 483 CpG islands targeted by aberrant methylation in total (Additional file 4: Table S3) with a varying number of corrected MVPs per island. The greatest enrichment of MVPs occurred in two CpG islands on chr6 (CpG:56 and CpG:42) with 13 and 11 MVPs respectively which overlapped with DMRs 1 and 2. Using a threshold of ≥3 MVPs per DMR, we then mapped the cell-type corrected MVPs onto the DMRs identified with Lasso, confirming that all three DMRs were not confounded by cell composition effects. We therefore continued our analysis focussing on DMRs 1–3.
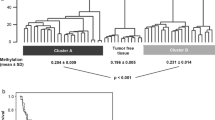
Aggregating DNA methylation across DMRs 1–3 (βmean), we found that WT had significantly greater levels of methylation with respect to NK for the discovery dataset as well as an independent dataset of 12 pairs (P = 3.85x10−17 and P = 9.26x10−10 respectively, 2-tailed t-test, Figure 2a and b). Furthermore, βmean was consistently high across an independent cohort of fresh frozen WT (n = 86; Additional file 2: Table S4) encompassing the seven post-chemotherapy WT histological subtypes classified into two risk groups, as defined by the International Society of Paediatric Oncology [20] (SIOP; Figure 2c). Within this cohort, a significant difference was seen between WT risk groups (P = 0.0024, 2-tailed T-test) with more elevated methylation levels observed in high risk WT (average βmean = 0.87 vs. 0.78). Based on methylation levels in the discovery dataset, a support vector machine (SVM; using R package e1071) correctly classified 100% and 98% samples within each replication set respectively indicating the discriminative potential of these DMRs as biomarkers. Clinical details for both of these cohorts can be found in Additional file 1: Table S1.

Methylation levels for DMRs 1–3 significantly distinguish normal kidney from Wilms tumour and whole blood. (a) Aggregated methylation levels across DMRs 1–3 significantly separate normal kidney (blue) and Wilms tumour (pink) in the test set (n = 22 pairs, P = 3.85x10−17) and in (b) the replication set (n = 12 pairs, P = 1.47x10−9). (c) Methylation levels were high in an independent dataset of WT including high risk (dark green, n = 25) and intermediate risk histological subtypes (light green, n = 61). High risk WT showed significantly higher methylation levels than intermediate risk WT (P = 0.0024). (d) Whole blood (n = 411) shows low methylation levels (red).
DMRs 1–2 are located within the extended major histocompatibility complex (MHC) region [21]. Although the MHC is highly polymorphic [22], our matched study design controlled for any genetic heterogeneity, ensuring that the observed signal was not confounded by copy number or other DNA sequence variation. We validated methylation levels for these DMRs using bisulfite-sequencing (Additional file 2: Figure S3 and Table S5) and confirmed the absence of C➔T mutation. MHC cluster hypermethylation and reciprocal loss of gene expression is common across cancers as a mechanism to evade immunosurveillance and increase oncogenic potential [23]–[26]. To further explore the association between DMR methylation and tumourigenesis, we extracted DNA from three specimens of human embryonic kidney (EK, gestational age = 22, 22 and 23 weeks) and separately microdissected embryonic blastema (EB, n = 3; the predicted WT cell-of-origin). Bisulfite sequencing of DMRs 1 and 2 showed average β-values of 0.007 for EK and 0.12 for EB. Furthermore, analysis of methylation levels in 20 cases with matched WT precursor lesions termed nephrogenic rests (NR) showed intermediate methylation levels (Additional file 2: Figure S2). Put together, this data suggests that sequential increase in methylation levels is associated with transformation of embryonic precursor cells towards a malignant phenotype.
As all DMRs were methylated in WT in comparison to NK, we predicted that levels of methylated DNA in the circulation may increase with tumour burden. When assessing levels of a methylated blood biomarker, varying proportions of leukocyte subpopulations can alter the overall methylation signal giving false yield [27]. Therefore, to assess the potential influence that blood populations may have on our dataset, we examined publically available methylation signatures that define normal peripheral blood subgroups [28] as well as methylation levels of normal whole blood (extracted using Marmal-aid [29]; n = 411). We found that DMRs 1–3 did not overlap with any blood-related methylation signature, and that normal blood methylation levels for DMRs 1–3 were extremely low (average βm = 0.12, Figure 2d). Therefore, we concluded that the WT-specific hypermethylated DMRs were not detected as a result of shifts in leukocyte populations in chemotherapy-treated tissue. Hence, we explored the potential of DMRs 1–3 as tumour-specific blood biomarkers, as they should be detectable above a low background and should not be confounded by shifts in leukocyte populations in the circulation.
To test the potential efficacy of these biomarkers, we performed bisulfite-sequencing of DMR-2 on cfDNA isolated from serum samples taken from 10 children with WT at diagnosis, during pre-operative chemotherapy and following nephrectomy as well as four cancer-free age-matched control serum samples (Additional file 2: Table S6). The entire sequenced region (chr6:32,116,940-32,117,259) spanned 319 bp and included 44 CpGs, many of which showed either no variation or extreme variation in methylation between samples. Therefore, to identify CpGs that showed consistent methylation at one time-point and differential methylation between time-points, we grouped samples within each time-point and performed an ANOVA test. From this, we selected a subset of CpGs (n = 14) that showed differential methylation between at least one pair of groups, irrespective of directionality of methylation changes, avoiding those with very low variance. Aggregating the percentage of methylated cfDNA across these 14 CpGs (%Mmean) showed that control samples had the lowest levels of methylated cfDNA (13.4) and that children with WT had relatively higher levels taken at diagnosis before treatment starts (14.5). There was then a substantial and significant increase in the level of methylated cfDNA taken after the pre-operative chemotherapy phase (19.9) that persisted into the immediate post-operative period (19.2; Figure 3). This proof-of-principle experiment establishes DMR-2 as a potential blood-based biomarker for WT.

Levels of methylation in serum cfDNA during WT treatment. Levels of %Mmean (for 14 CpGs within DMR-2) show significant differences between controls and pre-op samples (p = 0.010), controls and post-op samples (p = 0.017), pre-chemotherapy and pre-op (p = 0.028) and pre-chemotherapy and post-op (p = 0.047; all by 2-tailed T-test). Data shows increasing levels of methylated cfDNA associated with WT necrosis. Level of significance is shown by * (<0.05) or ** (<0.01) with horizontal lines indicating the respective comparison).
Discussion
This study has identified three differentially methylated regions (DMRs) with genome-wide significance in Wilms tumour (WT) that are present in nearly all WT (118 out of a total of 120 WTs examined) and are not mediated by cell type composition. In comparison to previous mutational studies, our epigenetic biomarker is far more ubiquitous and even supersedes 11p15 epimutation (in ~70% WT) as the most common biomarker in WT. Therefore, our results suggest that analysis of an epigenetic mark in cell-free DNA could be more successful than analysis of tumour-specific somatic mutations which are much less common. Furthermore, our study revealed a significant difference in methylation levels between intermediate and high risk WT which could be of diagnostic use and suggest a use for novel therapeutic approaches to high risk WT.
Although the NK samples do not contain blastema but WT samples do, our correction for cell type composition compensates for these effects allowing for identification of three true DMRs that are not mediated by cell mixture effects. Methylation analysis of DMRs 1–2 in embryonic kidney and embryonic blastema showed that these regions are not methylated in the presumed WT cell-of-origin. Analysis of nephrogenic rests showed that the precursor lesion had intermediate levels of methylation at the same regions which were then further methylated in the associated tumours (Additional file 2: Figure S2). We therefore concluded that increased methylation at these loci is associated with tumourigenic transformation and is not simply a manifestation of the embryonic origin of these tumours. Furthermore, as 84/86 WTs were correctly classified by methylation status in our independent cohort, which included tumours stratified into all 7 histological subtypes, and consistent high levels of methylation were seen in the test set of tumours with detailed assessment of their variable cellular composition (Additional file 2: Figure S1), we can conclude that all cells constituting the tumour contributed to the observed gain of methylation.
Our data suggests that WT release methylated DMR-2 cfDNA (M-cfDNA) following exposure to chemotherapy. Supporting this, a higher proportion of M-cfDNA (%Mmean of 23.6 compared to 17.3) was observed in serum sampled after pre-operative chemotherapy in patients with the regressive subtype of WT, defined by >2/3 necrosis in the nephrectomy specimen, compared to patients with other WT subtypes that show less than 2/3 necrosis in response to chemotherapy [20]. In this small ‘proof of principle’ cases series, we noted that post-operative M-cfDNA levels remained high in the immediate post-operative period, sampled at day 4 up to 24 days post-surgery. This short term persistence may be due to the fact that serum cfDNA can retain interaction with nucleosome proteins which protects the DNA from degradation rendering it relatively stable [30]. This may also explain our ability to sequence such a long fragment (319 bp). A full post-surgery time course is required to assess this rate of degradation. Interestingly, one patient showed post-operative M-cfDNA levels 1.3-fold greater than the group mean; a 22% increase post-surgery. We hypothesise that this post-surgery increase in M-cfDNA may be due to residual tumour within the patient and indeed, three months later, bone metastasis was detected.
Conclusions
In conclusion, we have defined the first epigenetic biomarker for the analysis of circulating cfDNA in Wilms tumour patients. We show that this may be useful to improve the accuracy of determining tumour response during pre-operative chemotherapy and predicting the histological risk group. This could allow appropriate modification of treatment prior to planned nephrectomy, particularly important for surgical planning in bilateral WT, where maximising tumour response to allow partial nephrectomy is the goal. Due to the low relapse rate in WT, a much larger, prospectively collected sample series of patients is required to demonstrate clinical utility as a prognostic biomarker for relapse-free survival. Based on these ‘proof of principle’ findings, a European multi-centre clinical trial with appropriate sampling is planned to rigorously test whether analysis of this epigenetic biomarker would improve the accuracy of prediction of relapse in all cases.
Methods
Sample selection
Use of patient samples in this study was conducted with appropriate parental written consent and ethical approval granted by the NHS London Bridge Research Ethics Committee (ref: 12/LO/0101) with experiments performed in compliance with the Helsinki Declaration. Patients were enrolled in the UK either into the International Society of Paediatric Oncology (SIOP) Wilms Tumour 2001 Clinical Trial and Study or the Improving Patient Outcomes for Renal Tumours of Childhood (IMPORT) study, with appropriate parental written consent. Blood serum samples from age-matched controls without cancer were taken from Great Ormond Street Hospital Department of Chemical Pathology with parental written consent. Marmal-aid v1.2.1 [31] was used to extract publically available methylation data annotated as disease = ‘Healthy’ and tissue = blood (n = 411). Embryonic kidney was obtained from fetal post-mortem examinations carried out at the Fetal Pathology Unit, University Hospital of Wales with written parental consent.
DNA extraction
As NR can only be identified by pathological review of haematoxylin and eosin (H&E) stained formalin-fixed paraffin embedded (FFPE) sections, 3 μm H&E sections from post-nephrectomy FFPE blocks were studied by two independent paediatric pathologists who marked out regions of normal kidney (NK) nephrogenic rest (NR) and Wilms tumour (WT). A total of 22 matched trios (NK, NR and WT) and 12 matched pairs of NK and WT (90 samples) were microdissected by cutting multiple 5 μm sections and removing the desired region with a scalpel. Embryonic blastema (EB) was also microdissected from 5 μm FFPE sections following a master H&E section as a guide. Tissue was taken from the whole section without microdissection to extract DNA from whole embryonic kidney (EK). Fresh frozen (FF) tissue (n = 86 samples) was taken from 83 patient nephrectomies that were classified according to centralised SIOP pathology review including stromal (n = 15), epithelial (n = 10), blastemal (n = 11), mixed (n = 23), diffuse anaplastic (n = 14), focal anaplasia (n = 2) or regressive (n = 11) type. DNA was extracted from FFPE and FF tissue using the DNeasy Blood and Tissue Kit (QIAGEN), however manufacturer’s instructions were modified for FFPE DNA: samples were heated to 90 °C for 1 hour post-incubation at 56 °C and incubated at 70 °C for 10 minutes with buffer AL. For cfDNA analysis DNA was extracted from pre-chemotherapy (n = 5), pre-operative (n = 8) or post-operative (n = 8) patient serum and age-matched cancer-free control serum (n = 7) using the QIAamp Circulating Nucleic Acid kit (QIAGEN).
Genome-wide methylation analysis
DNA extracted from FFPE (n = 90) was first treated using the REPLIg FFPE kit (QIAGEN) [32]. Both FF (n = 86) and treated FFPE DNA was then bisulfite-converted using the EZ DNA Methylation Kit (Zymo Research) and interrogated using the Illumina 450 k platform. Two FFPE NR samples failed stringent quality control metrics and were excluded. For all analyses, open source software packages implemented in R [33] or Bioconductor [34] were used as indicated. Raw data was filtered to exclude samples with detection p-value <0.01 and normalised using subset within quantile normalisation (SWAN) using the Bioconductor R package ChAMP version 2.14 [18],[35]. For initial MVP detection the normalised data matrix for 22 pairs (NK and WT) was included. Bayesian framework linear modelling using the Bioconductor R package Limma [36] version 3.20.4 [37] was performed to find sites of differential methylation that varied between NK and WT pairs and that were common across patients which avoids false positives from patient-specific SNPs or age effects. To this model, the TREAT function was applied to adjust p-values based on the Δβ-value (>0.1) [38] which were further adjusted to correct for multiple testing [39]. The DMR-lasso algorithm in the ChAMP package was then used to find differentially methylated regions (DMRs) [18] with settings adjusted to include only CpGs that reach genome-wide significance (p < 5x10−8). To the same model, we applied the RefFreeEWAS algorithm [19] which uses single value decomposition to estimate the number of cell types contributing to overall histology. The algorithm then deconvoluted the β-values based on the estimated number of cell types (d = 3) and a design matrix specifying patient pairs and sample histology, and generated bootstrap-derived CpG-specific p-values and covariates that correspond to a ‘true’ methylation signal with no cell mixture effects.
After confirming our three DMRs were not due to cell composition effects, the DMR values were compared to levels in a replication dataset of 12 pairs, the independent set of 86 FF WT and the matched NRs for 20 cases. Sample classification (by SVM) was performed using the 22 pairs as a training set and 12 pairs and 86 WT as separate test sets using R CRAN package e1071 [40].
Assessment of DMR methylation by bisulfite-sequencing
Of the three DMRs, we chose to validate methylation levels for DMRs 1–2 by sequencing both bisulfite-converted and normal DNA from 9 NK and WT pairs. We performed the same experiment to assess methylation levels in EK and EB. Bisulfite reads for DMR-1 showed poor coverage with only 6/18 validation samples giving sufficient reads (Additional file 2: Table S5). We therefore chose to focus on DMR-2 for detection in cfDNA from 28 serum samples. For cfDNA analysis, 5/21 patient (3 pre-op, 2 post-op) and 3/7 control samples failed to generate sufficient sequence reads for analysis.
Primers were designed (Additional file 2: Table S7) using Primer 3 [41] and MethPrimer [42] and optimised using commercial DNA. DNA for bisulfite-sequencing was converted using the EZ DNA methylation kit (Zymo Research). Library preparation PCRs were performed using NEBNext (New England Biolabs) and KAPA HiFi Uracil + (KAPA Biosystems). Products were cleaned using magnetic beads (Beckman Coulter) and quantified using Picogreen reagents. Sample-specific tags were added prior to sequencing using the Illumina Mi-Seq. Raw bisulfite-converted paired-end reads were mapped to human genome build hg19 with Bismark v0.9.0 [43] using Bowtie 2 [44] as the aligner. Methylated and unmethylated base counts were generated with the bismark_methylation_extractor utility and exported as BedGraph files for further analysis and display in IGV [45]. Aligned BAM files were sorted and indexed with SAMtools [46] for assessment of the regions of interest in IGV. The number of C reads divided by total reads per CpG site was then calculated to discern the % level of methylation per sample.
To generate allele counts for the full sequence of DMR-2, we used the ANGSD package [47]. ANOVA was performed in R using Bioconductor package Limma to make all possible contrasts between groups. CpGs were selected for further analysis if the Limma Toptable moderated F score >1 indicating that any of the contrasts between groups were non-zero and if group variance for that CpG was >1.
Data access
450 k methylation data described in this study are available from GEO [48] with accession ID GSE59157.
Additional files
Abbreviations
- cfDNA:
-
cell-free DNA
- DMR:
-
Differentially methylated region
- EB:
-
Embryonic blastema
- EK:
-
Embryonic kidney
- FFPE:
-
Formalin fixed paraffin embedded
- IMPORT:
-
Improving population outcomes for children with renal tumours
- M-cfDNA:
-
Methylated cell-free DNA
- NK:
-
Normal kidney
- NR:
-
Nephrogenic rest
- PCR:
-
Polymerase chain reaction
- SIOP:
-
International society of paediatric oncology
- WT:
-
Wilms tumour
References
Breslow N, Olshan A, Beckwith JB, Green DM: Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993, 21: 172-
Lemerle J, Voute PA, Tournade MF, Rodary C, Delemarre JF, Sarrazin D, Burgers JM, Sandstedt B, Mildenberger H, Carli M: Effectiveness of preoperative chemotherapy in Wilms’ tumor: results of an International Society of Paediatric Oncology (SIOP) clinical trial. J Clin Oncol. 1983, 1: 604-published online EpubOctober 1, 1983 (
Gautschi O, Bigosch C, Huegli B, Jermann M, Marx A, Chassé E, Ratschiller D, Weder W, Joerger M, Betticher DC, Stahel RA, Ziegler A: Circulating deoxyribonucleic acid as prognostic marker in non–small-cell lung cancer patients undergoing chemotherapy. J Clin Oncol. 2004, 22: 4157-published online EpubOctober 15, 2004 (10.1200/jco.2004.11.123)
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA: Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008, 14: 985-http://www.nature.com/nm/journal/v14/n9/suppinfo/nm.1789_S1.html, [http://www.nature.com/nm/journal/v14/n9/suppinfo/nm.1789_S1.html]
Ramirez JL, Rosell R, Taron M, Sanchez-Ronco M, Alberola V, de las Peñas R, Sanchez JM, Moran T, Camps C, Massuti B, Sanchez JJ, Salazar F, Catot S: 14-3-3σ Methylation in pretreatment serum circulating DNA of cisplatin-plus-gemcitabine-treated advanced Non–small-cell lung cancer patients predicts survival: the Spanish Lung Cancer Group. J Clin Oncol. 2005, 23: 9105-published online EpubDecember 20, 2005 (10.1200/jco.2005.02.2905)
Mori T, O’Day SJ, Umetani N, Martinez SR, Kitago M, Koyanagi K, Kuo C, Takeshima T-L, Milford R, Wang H-J, Vu VD, Nguyen SL, Hoon DSB: Predictive utility of circulating methylated DNA in serum of melanoma patients receiving biochemotherapy. J Clin Oncol. 2005, 23: 9351-published online EpubDecember 20, 2005 (10.1200/jco.2005.02.9876)
Ruteshouser EC, Robinson SM, Huff V: Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008, 47: 461-published online EpubJun (10.1002/gcc.20553)
Scott RH, Murray A, Baskcomb L, Turnbull C, Loveday C, Al-Saadi R, Williams R, Breatnach F, Gerrard M, Hale J, Kohler J, Lapunzina P, Levitt GA, Picton S, Pizer B, Ronghe MD, Traunecker H, Williams D, Kelsey A, Vujanic GM, Sebire NJ, Grundy P, Stiller CA, Pritchard-Jones K, Douglas J, Rahman N: Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget. 2012, 3: 327-published online EpubMar (
Chilukamarri L, Hancock AL, Malik S, Zabkiewicz J, Baker JA, Greenhough A, Dallosso AR, Huang TH, Royer-Pokora B, Brown KW, Malik K: Hypomethylation and aberrant expression of the glioma pathogenesis-related 1 gene in Wilms tumors. Neoplasia. 2007, 9: 970-published online EpubNov (
Hubertus J, Zitzmann F, Trippel F, Muller-Hocker J, Stehr M, von Schweinitz D, Kappler R: Selective methylation of CpGs at regulatory binding sites controls NNAT expression in Wilms tumors. PLoS One. 2013, 8: e67605-10.1371/journal.pone.0067605
Malik K, Salpekar A, Hancock A, Moorwood K, Jackson S, Charles A, Brown KW: Identification of differential methylation of the WT1 antisense regulatory region and relaxation of imprinting in Wilms’ tumor. Cancer Res. 2000, 60: 2356-published online EpubMay 5, 2000 (
Ehrlich M, Hopkins NE, Jiang G, Dome JS, Yu MC, Woods CB, Tomlinson GE, Chintagumpala M, Champagne M, Dillerg L, Parham DM, Sawyer J: Satellite DNA hypomethylation in karyotyped Wilms tumors. Cancer Genet Cytogenet. 2003, 141: 97-published online EpubMar (
Ludgate JL, Le Mee G, Fukuzawa R, Rodger EJ, Weeks RJ, Reeve AE, Morison IM: Global demethylation in loss of imprinting subtype of wilms tumor. Genes Chromosomes Cancer. 2012, ᅟ: ᅟ-published online EpubOct 17 (10.1002/gcc.22017)
Zhang L, Anglesio MS, O’Sullivan M, Zhang F, Yang G, Sarao R, Mai PN, Cronin S, Hara H, Melnyk N, Li L, Wada T, Liu PP, Farrar J, Arceci RJ, Sorensen PH, Penninger JM: The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med. 2007, 13: 1060-published online EpubSep (10.1038/nm1621)
Wagner KJ, Cooper WN, Grundy RG, Caldwell G, Jones C, Wadey RB, Morton D, Schofield PN, Reik W, Latif F, Maher ER: Frequent RASSF1A tumour suppressor gene promoter methylation in Wilms’ tumour and colorectal cancer. Oncogene. 2002, 21: 7277-published online EpubOct 17 (10.1038/sj.onc.1205922)
Dallosso AR, Hancock AL, Szemes M, Moorwood K, Chilukamarri L, Tsai HH, Sarkar A, Barasch J, Vuononvirta R, Jones C, Pritchard-Jones K, Royer-Pokora B, Lee SB, Owen C, Malik S, Feng Y, Frank M, Ward A, Brown KW, Malik K: Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms’ tumor. PLoS Genet. 2009, 5: e1000745-published online EpubNov (10.1371/journal.pgen.1000745)
Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, Daunay A, Busato F, Mein CA, Manfras B, Dias KR, Bell CG, Tost J, Boehm BO, Beck S, Leslie RD: Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7: e1002300-published online EpubSep (10.1371/journal.pgen.1002300)
Morris T, Butcher L, Feber A, Teschendorff A, Chakravarthy A, Wojdacz T, Beck S: 450k Chip Analysis Methylation Pipeline (ChAMP). Bioinformatics. 2013, ᅟ: ᅟ-published online EpubDecember 12, 2013 (10.1093/bioinformatics/btt684)
Houseman EA, Molitor J, Marsit CJ: Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014, 30: 1431-published online EpubMay 15 (10.1093/bioinformatics/btu029)
Vujanic GM, Sandstedt B, Harms D, Kelsey A, Leuschner I, de Kraker J: Revised International Society of Paediatric Oncology (SIOP) working classification of renal tumors of childhood. Med Pediatr Oncol. 2002, 38: 79-published online EpubFeb
Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, Lush MJ, Povey S, Talbot CC, Wright MW, Wain HM, Trowsdale J, Ziegler A, Beck S: Gene map of the extended human MHC. Nat Rev Genet. 2004, 5: 889-published online EpubDec (10.1038/nrg1489)
de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, Morrison J, Richardson A, Walsh EC, Gao X, Galver L, Hart J, Hafler DA, Pericak-Vance M, Todd JA, Daly MJ, Trowsdale J, Wijmenga C, Vyse TJ, Beck S, Murray SS, Carrington M, Gregory S, Deloukas P, Rioux JD: A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006, 38: 1166-published online EpubOct (10.1038/ng1885)
Garrido C, Paco L, Romero I, Berruguilla E, Stefansky J, Collado A, Algarra I, Garrido F, Garcia-Lora AM: MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion, and intrinsic tumorigenicity of melanoma cells. Carcinogenesis. 2012, ᅟ: ᅟ-published online Epub January 4, 2012 (10.1093/carcin/bgr318)
Doyle A, Martin WJ, Funa K, Gazdar A, Carney D, Martin SE, Linnoila I, Cuttitta F, Mulshine J, Bunn P, Minna J: Markedly decreased expression of class I histocompatibility antigens, protein, and mRNA in human small-cell lung cancer. J Exp Med. 1985, 161: 1135-published online EpubMay 1 (
Jäger E, Ringhoffer M, Altmannsberger M, Arand M, Karbach J, Jäger D, Oesch F, Knuth A: Immunoselection in vivo: Independent loss of MHC class I and melanocyte differentiation antigen expression in metastatic melanoma. Int J Cancer. 1997, 71: 142-10.1002/(sici)1097-0215(19970410)71:2<142::aid-ijc3>3.0.co;2-0
Degenhardt Y, Huang J, Greshock J, Horiates G, Nathanson K, Yang X, Herlyn M, Weber B: Distinct MHC gene expression patterns during progression of melanoma. Gene Chromosome Cancer. 2010, 49: 144-published online EpubFeb (10.1002/gcc.20728)
Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir G, Smith AV, Gudnason V: Heterogeneity in white blood cells Has potential to confound DNA methylation measurements. PLoS One. 2012, 7: e46705-10.1371/journal.pone.0046705)
Koestler DC, Marsit CJ, Christensen BC, Accomando W, Langevin SM, Houseman EA, Nelson HH, Karagas MR, Wiencke JK, Kelsey KT: Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol Biomarkers Prev. 2012, 21: 1293-published online EpubAugust 1, 2012 (10.1158/1055-9965.epi-12-0361)
Lowe R, Rakyan VK: Marmal-aid--a database for Infinium HumanMethylation450. BMC Bioinformatics. 2013, 14: 359-10.1186/1471-2105-14-359
Rumore PM, Steinman CR: Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J Clin Investig. 1990, 86: 69-published online EpubJul (10.1172/JCI114716)
Marmal-aid website. [http://marmal-aid.org/]
Thirlwell C, Eymard M, Feber A, Teschendorff A, Pearce K, Lechner M, Widschwendter M, Beck S: Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the Illumina Infinium HumanMethylation27 BeadChip. Methods. 2010, 52: 248-published online EpubNov (10.1016/j.ymeth.2010.04.012)
R: A language and environment for statistical computing. [www.r-project.org/]
Bioconductor website. [www.bioconductor.org/]
Bioconductor ChAMP package website. [http://www.bioconductor.org/packages/release/bioc/html/ChAMP.html]
Smyth GK: Limma. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Edited by: Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S. 2005, Springer, New York, 397-420.
Bioconductor Limma package website. [http://www.bioconductor.org/packages/release/bioc/html/limma.html]
McCarthy DJ, Smyth GK: Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009, 25: 765-published online EpubMarch 15, 2009 (10.1093/bioinformatics/btp053)
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B (Methodological). 1995, 57: 589-10.2307/2346101
e1071 package website. [http://cran.r-project.org/web/packages/e1071/index.html]
Koressaar T, Remm M: Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007, 23: 1289-1291.
Li LC, Dahiya R: MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002, 18: 1427-1431.
Bismark website. [http://www.bioinformatics.babraham.ac.uk/projects/bismark/]
Bowtie 2 website. [http://bowtie-bio.sourceforge.net/bowtie2/]
Robinson JT, Thorvaldsdottir K, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP: Integrative genomics viewer. Nat Biotechnol. 2011, 29: 24-26.
SAMtools website. [http://samtools.sourceforge.net/]
ANGSD website. [http://popgen.dk/wiki/index.php/ANGSD]
Gene Expression Omnibus (GEO) website. [http://www.ncbi.nlm.nih.gov/geo/]
Acknowledgements
J.C was funded by the UCL Grand Challenges Scheme and the Olivia Hodson fund. The Pritchard-Jones laboratory was funded by Cancer Research UK (C1188/A4614), Great Ormond Street Hospital (GOSH) Children’s Charity and Children with Cancer (11MH16). NJS and KPJ are part supported by the NIHR GOSH UCL Biomedical Research Centre. The Beck laboratory was funded by the Wellcome Trust (99148), the UCL Biomedical Research Centre (BRC84/CN/SB/5984), a Royal Society Wolfson Research Merit Award (WM100023), IMI-JU OncoTrack (115234) and EU-FP7 projects IDEAL (259679), EPIGENESYS (257082) and BLUEPRINT (282510).
The authors would like to thank all the investigators at the contributing Children’s Cancer and Leukaemia Group (CCLG) treatment centres and the CCLG tissue bank, which is funded by Cancer Research UK, for provision of samples. We thank Tasnim Chagtai and Becky West for processing of frozen tumour samples, Tessa Kasia, Lucy Bailey and Darren Edwards of the Great Ormond Street Hospital Translational Cancer Research Team for collection of tissue samples, the Department of Chemical Pathology at Great Ormond Street Hospital including Helen Aitkenhead, Principal Clinical Scientist, and Daley Aofolaju, Chief Biomedical Scientist for providing control serum samples. We thank Kerra Pearce and Tony Brooks from UCL Genomics for their help with processing the Illumina 450 k arrays and the Illumina sequencing.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing financial interests.
Authors’ contributions
J.C. performed DNA extraction from FFPE tissue and serum samples and carried out library preparation for bisulfite sequencing. N.J.S, S.P and W.M. marked out regions of NK, NR and WT for DNA extraction from FFPE tissue. G.V. and N.J.S are the National Reference Panel pathologists for the UK cases in the SIOP WT 2001 and IMPORT trials. G.V also supplied embryonic fetal kidney specimens. J.C. performed bioinformatics analyses with help from R.D.W. LMB provided analysis tools. M.A-G contributed data from a cohort of 86 fresh frozen tumours. MW co-ordinates the clinical study (“IMPORT”) that collects serial blood samples of patients with WT during therapy. S.B. and K.P-J conceived the study. J.C. wrote the manuscript with input from all authors. All authors approved the final version.
Electronic supplementary material
13059_2014_434_MOESM1_ESM.xlsx
Additional file 1: Table S1: Clinical information for the discovery cohort (n=22), validation set 1 (n=12) and validation set 2 (n=86). (XLSX 10 KB)
13059_2014_434_MOESM2_ESM.docx
Additional file 2: Figure S1: Quantification of cell proportions in each micro-dissected Wilms tumour section used for DNA extraction in the discovery cohort. Figure S2: WT precursor lesions show intermediate methylation at significant DMRs. Figure S3: Comparison of methylation values assessed by 450k array and bisulfite sequencing. Table S4: Fresh frozen WT (n = 86) classified by overall tumour histology with average methylation β-values across all significant DMR CpGs. Table S5: Validation of 450k methylation signal by bisulfite-sequencing. Table S6: Clinical information on patients from which cfDNA was isolated. Table S7: List of primers used. (DOCX 128 KB)
13059_2014_434_MOESM4_ESM.xlsx
Additional file 4: Table S3: A list of the CpG islands (n=483) that contained significantly hypermethylated or hypomethylated cell-corrected MVPs, with the respective number of CpGs per island. (XLSX 25 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Charlton, J., Williams, R.D., Weeks, M. et al. Methylome analysis identifies a Wilms tumor epigenetic biomarker detectable in blood. Genome Biol 15, 434 (2014). https://doi.org/10.1186/s13059-014-0434-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-014-0434-y