
Abstract
We present here the K9 lymphoma assay, a novel 31-gene targeted next-generation sequencing panel designed for genomic profiling of canine lymphoid neoplasms. Addressing the growing demand for advanced diagnostics in veterinary oncology, this assay enables sensitive identification of known and actionable mutations specific to canine lymphomas, while evaluating its prognostic potential to facilitate diagnosis and prognosis. Our analysis, spanning several B- and T-cell lymphoma histotypes, unveiled distinct mutational landscapes distinguishing tumors derived from immature versus mature lymphocytes. Clustering analysis revealed a shared genetic origin between diffuse large B-cell lymphoma and marginal zone lymphoma, aligning with findings in human lymphomas, with TRAF3 emerging as the most frequently mutated gene across B-cell lymphoma subtypes. Significantly, TP53 mutations demonstrated universal adverse prognostic implications across B-cell lymphomas. Additionally, SETD2 mutations contributed to shorter time-to-progression, underscoring the role of epigenetic dysregulation in B-cell tumors. In T-cell lymphomas, SATB1 and FBXW7 were frequently mutated, warranting further investigation in larger cohorts. Our findings advocate for tailored therapeutic approaches based on the genetic profile, impacting treatment decisions and outcomes in canine lymphoma management. This study provides pivotal insights bridging veterinary and human oncology, paving the way for comprehensive genomic diagnostics and therapeutic strategies in comparative oncology.
Similar content being viewed by others


Introduction
Canine nodal lymphomas arise from the malignant proliferation of lymphoid cells, primarily occurring within the lymph nodes, and they represent the most prevalent hematologic neoplasms in dogs1. The classification of canine lymphomas has evolved, and the World Health Organization (WHO) classification is now the widely adopted system2. The WHO classification categorizes canine lymphomas based on histologic and immunohistochemical criteria, sorting subtypes by morphologic and phenotypic features. Additionally, lymphomas are clinically divided into aggressive or indolent3.
Approximately 60–70% of canine lymphomas originate from B-cells, while the remaining cases derive from T-cells4. The most common aggressive B-cell lymphomas include diffuse large B-cell lymphoma (DLBCL), B-cell lymphoblastic lymphoma (B-LL) and Burkitt lymphoma (BL). Conversely, indolent B-cell lymphomas encompass follicular lymphoma (FL), marginal zone lymphoma (MZL), and B-cell small lymphocytic lymphoma (B-SLL). Aggressive T-cell lymphomas comprise peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) and T-cell lymphoblastic lymphoma (T-LL), while T-zone lymphoma (TZL) and T-cell small lymphocytic lymphoma (T-SLL) are among the indolent forms.
While this classification system provides a framework for understanding and categorizing canine lymphomas based on morphological characteristics, it does not entirely elucidate the heterogeneity among tumor histotypes and their sensitivity to therapies. Despite the significant advancements in treatment protocols that have improved survival rates for dogs with both aggressive and indolent lymphomas, disease progression remains the primary cause of death in affected animals3,5.
Most research in veterinary medicine, covering clinics, genetics, transcriptomics, and epigenetics, has predominantly focused on canine DLBCL. This specific subtype, comprising 50–60% of cases, is the most common lymphoma and is generally associated with a relatively poor prognosis5. Furthermore, treatment outcomes show significant variability, making prediction challenging. Gene expression profiling (GEP) and RNA sequencing studies have revealed two distinct subgroups with significantly different survival times6,7. Recurrently mutated genes identified through whole exome sequencing (WES) include TRAF3, SETD2, POT1, TP53, and FBXW78,9,10. However, limited clinical data in most studies hampered a comprehensive understanding of the significance of these genetic variants. A recent study that integrated "omics" data and clinicopathological features developed a predictive model to identify negative prognostic factors. One contributing factor is the genetic status of TP53, which identifies a subgroup of dogs that significantly benefit from adding immunotherapy to the standard CHOP-based therapy10.
In human medicine, the relevance of targeted next-generation sequencing (tNGS), directed at specific genomic regions of interest, has steadily gained importance in diagnosis, risk assessment, and in the management of patients with confirmed or suspected malignancies. Integrating these tests into several tumor practice guidelines is now common, with numerous genetic alterations serving as criteria for disease classification and treatment decisions11.
In veterinary medicine, a limited number of genetic profiling tests are available for canine cancers in the USA12,13. However, none of these tests have been tailored specifically to target the most common mutations in canine lymphomas. With the increasing availability of targeted therapies in veterinary medicine, understanding the genetic profile of each tumor histotype has become crucial for predicting treatment response. To this end, the primary objective of this study was to design and develop a targeted sequencing gene panel (named the “K9 lymphoma assay”) covering exons frequently mutated across different canine lymphoma studies. The secondary objective was to investigate the prognostic potential of this panel, paving the way for a diagnostic and prognostic clinical assay in canine B- and T-cell lymphomas.
Results
Study population and clinical characteristics of dogs with B-cell lymphoma
Overall, 205 dogs with BCL were included in the analysis (Supplementary Information and Supplementary Data 1). The most common subtype was DLBCL (57.1%), followed by MZL (18.5%), FL (10.2%), B-SLL (5.4%), BL (4.9%), and B-LL (3.9%). Almost all dogs (97.1%) underwent complete staging, revealing that most had advanced stage disease (stage V, 75.4%) and bone marrow (BM) infiltration (67.0%). Treatment varied based on the subtype, with the majority of dogs with both aggressive (60%) and indolent (60%) BCL receiving chemo-immunotherapy, while the remaining dogs primarily received chemotherapy alone.
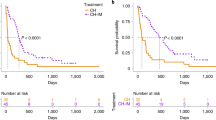
At data analysis closure, 168 (81.9%) dogs had died (142 due to lymphoma and 26 to tumor-unrelated causes), 26 (12.7%) were still alive, and 11 (5.4%) were lost to follow-up. Median time to progression (TTP) and lymphoma specific survival (LSS) were 180 days (range, 1–1116 days) and 282 days (range, 5–1321 days), respectively. By multivariate Cox proportional-hazards analysis (Supplementary Data 2), dogs treated with chemo-immunotherapy presented both longer TTP (202 days) and LSS (352 days) compared to dogs that had received chemotherapy only (median TTP = 81 days; median LSS = 230 days) (p < 0.01). Dogs with aggressive histotypes showed significantly longer median TTP and LSS when treated with chemo-immunotherapy (TTP = 202 days; LSS = 347 days) compared to those treated with chemotherapy alone (TTP = 80 days, p = 0.02; LSS = 169 days, p < 0.01) according to multivariable analysis (Supplementary Data 2).
In the group of indolent histotypes, TTP was significantly shorter in dogs showing symptoms (107 vs. 209 days, p = 0.02), having BM involvement (134 vs. 212 days, p = 0.01) and receiving chemotherapy only (168 vs. 211 days, p < 0.01). No significant associations with LSS were retrieved by multivariable analysis (Supplementary Data 2).
Among DLBCL cases, dogs treated with chemo-immunotherapy had a significantly longer LSS than those treated with chemotherapy only (343 vs. 230 days, p = 0.02) (Supplementary Data 2). Among dogs with MZL, treatment significantly impacted both TTP and LSS, with dogs treated with chemo-immunotherapy experiencing longer TTP (226 vs. 60 days, p = 0.02) and LSS (354 vs. 211 days, p = 0.05) (Supplementary Data 2). No significant associations were found for the other histotypes.
Study population and clinical characteristics of dogs with T-cell lymphoma
Overall, 52 dogs with TCL were included in the analysis (Supplementary Data 1). The most common subtype was PTCL-NOS (59.6%), followed by T-LL (13.5%), T-SLL (11.5%), TZL (11.5%), and cutaneous lymphomas (CL) (3.9%). Most dogs (75%) had advanced stage disease (stage V), with 40.4% showing bone marrow infiltration. More than half of the dogs (61.5%) were symptomatic at presentation. The majority of dogs with both aggressive (95%) and indolent (75%) TCL subtypes received chemotherapy, while the remaining dogs were treated with prednisolone alone.
At data analysis closure, 48 (92.3%) dogs had died (40 due to lymphoma and 8 to tumor-unrelated causes), 3 (5.8%) were lost to follow-up, and 1 (1.9%) was still alive. Median TTP and LSS were 86 days (range, 44–182 days) and 132 days (range, 91–179 days), respectively.
By multivariate analysis, substage (a vs. b) significantly impacted both TTP (median TTP = 307 vs. 59 days, p = 0.01) and LSS (median LSS = 362 vs. 120 days, p < 0.01). Moreover, both TTP and LSS were significantly shorter in mixed-breed dogs compared to pure dogs (median TTP = 35 vs. 131 days, respectively, p = 0.008; median LSS = 103 vs. 154 days, respectively, p = 0.015) (Supplementary Data 3).
In dogs with aggressive histotypes, the breed (mixed vs pure) significantly impacted TTP (median TTP = 35 vs. 131 days, respectively, p = 0.02) and LSS (median LSS = 76 vs. 166 days, respectively, p < 0.01) by multivariate analysis (Supplementary Data 3).
The K9 lymphoma assay is accurate and provides similar genetic data
The K9 lymphoma assay, covering a total of 196 kbp, was designed and built to interrogate 31 genes recurrently mutated in canine lymphomas8,9,10, as well as oncogenes and tumor suppressor genes mutated in other canine tumors14,15,16,17,18,19,20,21 (Table 1). An in silico validation of the panel utilizing the data of 77 dogs with DLBCL previously analyzed by WES10 demonstrated that the panel identified at least one mutation in 89.6% of cases (Supplementary Information and Supplementary Data 4 and 5). When applied to the study cases, an average sequencing depth of 702X was achieved across all tumors, with values ranging from 402X to 3,632X. On average, 77% of reads aligned to the canine genome, ranging from 54 to 85%, and there was a mean of 13% duplicate reads (range, 9–39%).
Genetic landscape of canine B-cell lymphoma
Putatively somatic variants were identified in almost all dogs with B-cell lymphoma (203/205, 99% of the cases). The full list of identified nucleotide variants is reported in Supplementary Data 6. The median number of variants per case was 6 (range, 1–13), and a total of 577 single nucleotide variants (SNVs) and 91 insertion/deletion variants (indels) in coding regions were identified. Among the SNVs, 53 were synonymous (S), while 524 were nonsynonymous (NS), with 108 determining the appearance of premature stop codons. This yielded a nonsynonymous-to-synonymous (NS:S) ratio of 9.9. Protein-coding variants were retrieved in 191/205 dogs (93.2%). Missense mutations were the most frequent variants (65.3%), followed by nonsense mutations (17%). Frameshift insertions and deletions accounted for 5.7% of mutations each, while in-frame deletions were 2.2%. Variants affecting splicing were 2%. Finally, start-loss and in-frame insertions were the least frequent, each accounting for 0.8%. The median tumor mutational burden (TMB) of the whole cohort was 15.3 mutations/Mb (range, 5.1–40.8) (Supplementary Data 7). The impact of the most frequently mutated genes on TMB was also explored. Mutations in FBXW7, TRAF3, SETD2, FAT4, DDX3X, POT1, and KMT2D were associated with higher TMB compared with wild-type genes after multiple testing correction (p adj < 0.01) (Supplementary Data 8).
Six genes (TRAF3, POT1, FBXW7, MYC, DDX3X, and SETD2) were mutated in at least 20% of the cases (Fig. 1). In line with previous reports8,9,10, TRAF3 was the most frequently altered gene (52.7%), harboring 132 putatively somatic variants in 108 dogs, with 23 dogs presenting multiple aberrations. Truncating mutations (i.e. nonsense variants, frameshift insertions, and deletions) accounted for 75% of TRAF3 mutations, while missense variants were 19.7%, all but two predicted as deleterious by Sorting Intolerant from Tolerant (SIFT) tool. In-frame insertions were 1.5%, while splicing variants were reported in 5 (4.6%) dogs (Supplementary Data 6). The TRAF-MATH domain was the most frequently hit, harboring 48% of mutations. In addition, the variant p.R340* (ENSCAFT00000028719.4) was recurrently observed (10.5% of cases) (Fig. 2a).

Oncoplot of mutated genes in B-cell lymphoma. Genes of the K9 lymphoma assay harboring putative somatic short variants (SNVs and indels) identified in B-cell lymphomas are depicted. Genes are represented in descending order according to the frequency of mutation. Mutation frequency is calculated on dogs harboring at least one protein-coding mutation in a gene of the panel. Different mutation types are identified with different colors. Clinico-pathological data including diagnosis, aggressive or indolent histotype, sex, breed, stage and substage are shown at the bottom.

Lollipop plots of the top-four mutated genes in B-cell lymphomas. Localization of putative somatic short variants (SNVs and indels) at protein level for TRAF3 (a), POT1 (b), FBXW7 (c) and MYC (d). The height of each bar represents the frequency at which each mutation occurred. Proteins’ functional domains were retrieved from UniProt database (https://www.uniprot.org/). For each protein, the longest isoform was considered.
The second most frequently mutated gene was POT1 (30.7%). Missense mutations, predicted to be deleterious by SIFT analysis, were the most prevalent aberrations (70.3%), followed by nonsense mutations (21.9%). Splicing variants were identified in four dogs (6.3%) (Supplementary data 6). Notably, 31.7% of the mutations were localized within the single-strand (ss) DNA-binding domain (Fig. 2b). FBXW7 mutations were identified in 25.8% of cases. Missense mutations accounted for 78.6% of the FBXW7 variants, with 51.2% predicted as deleterious and 48.8% as tolerated by SIFT analysis (Supplementary Data 6). Recurrent FBXW7 variants were observed, including p.S3N (n = 10), p.R552C (n = 6), p.R512C (n = 6), and p.R512H (n = 5) (Fig. 2c). The MYC gene was altered in 21.5% of dogs. Almost all MYC mutations (93.5%) were missense variants, with all but two predicted as deleterious by SIFT analysis. These variants predominantly (93.5%) mapped within exon 2 (Supplementary Data 6). The p.P72S substitution in MYC was recurrently observed (19.6%) (Fig. 2d).
DDX3X variants were retrieved in 19.5% of cases. Missense variants were the most frequent (93.6%) and were all predicted as deleterious by SIFT, while nonsense mutations and in-frame insertions were 4.3% and 2.1%, respectively. Exons 8 and 9 (ENSCAFT00000061818.1), which encode for the ATP-binding domain, were the most frequently hit (46.8% and 30%, respectively). The variants p.R277W, p.F283Y and p.D255N were reported in 10.6% of dogs each.
SETD2 gene was mutated in 19% of cases. Missense variants accounted for 44.9%, of which 72.3% were predicted as deleterious and 27.3% as tolerated by SIFT. Nonsense mutations were 26.5%, and frameshift insertions and deletions were 14.3% each. One dog presented a splicing variant. Exon 3 (ENSCAFT00000021260.4) harbored 44.9% of all retrieved variants.
Patterns of co-mutation and mutual exclusivity were also explored. TRAF3 mutations were mutually exclusive with MAP3K14 (p adj < 0.001), while TP53 mutations co-occurred with FAT4 (p adj = 0.048).
We employed hierarchical clustering to investigate variations in mutation profiles among various B-cell lymphoma histotypes, analyzing 203 samples. The results revealed two prominent clusters, distinguishing nodal lymphomas derived from immature lymphocytes from those originating from mature lymphocytes (Fig. 3). Further investigation of the primary lymphoma clusters indicated a subdivision between BL and lymphomas originating from centroblasts and marginal zone cells, such as FL, DLBCL, and MZL.

Hierarchical clustering. Hierarchical clustering was used to investigate variations in mutation profiles among present histotypes. Two resulting clusters emerge clearly from the above figure, distinguishing lymphomas originating from the BM from those originating from lymph nodes. The relevant genes for this clustering are MYC, POT1, TRAF3 and TBL1XR1.
Genetic landscape of canine T-cell lymphoma
In TCL, putatively somatic variants were identified in 48 dogs (92%) (Supplementary Data 9). The mean number of variants per case was 3.5 (range, 1–7) and a total of 72 SNVs and 3 indels in coding regions were detected. Among the SNVs, 13 were S, while 59 were NS, with 5 resulting in the gain of stop codons. This yielded a NS:S ratio of 4.5. Protein-coding variants were retrieved in 33/48 (68.8%) dogs. Missense mutations were the most frequent aberrations (85.5%), followed by nonsense mutations (9.7%), frameshift insertions (3.2%) and frameshift deletions (1.6%) (Fig. 4). The median TMB of the whole cohort was 10.2 mutations/Mb (range: 5.10–25.51) (Supplementary Data 10).

Oncoplot of mutated genes in TCL. Genes of the K9 lymphoma assay harboring putative somatic short variants (SNVs and indels) identified in TCLs are depicted. Genes are represented in descending order according to the frequency of mutation. Mutation frequency is calculated on dogs harboring at least one protein-coding mutation in one panel gene. Different mutation types are identified with different colors. Clinico-pathological data including diagnosis, aggressive or indolent histotype, sex, breed, stage and substage are shown at the bottom.
In aggressive TCL, protein-coding variants were found in 26 dogs, distributed across 17 genes. Six genes exhibited mutations in at least 10% of the cases. Mutations in SATB1, FBXW7, FAT4, and ARPC1A were found exclusively in aggressive lymphomas, except for a single SATB1 variant observed in one dog with indolent TCL (Supplementary Data 9 and Supplementary Fig. 1). The SATB1 gene had the highest mutation frequency, identified in 9 dogs (23.7%), with 6 carrying the same mutation (p.Q420R). All SATB1 variants were predicted as deleterious by SIFT. Three out of 4 dogs harboring ARPC1A alterations presented the same nonsense variant (c.A364T) that determines the appearance of a premature stop codon (p.K122*). The variant has not been reported previously. One sample (130CIM) harbored two splicing variants involving TP53 and KMT2D genes. A higher median TMB was observed in dogs carrying FBXW7 mutations compared to wild-type (10.2 vs. 5.1; p = 0.04). However, this association did not remain significant after multiple testing correction.
In indolent TCL, protein-coding variants were identified in 7 dogs (58%), affecting 13 genes. SETD2 gene was altered in 2 (17%) dogs, and one sample (156BAR) presented two different SETD2 variants, a nonsense mutation involving the exon 3 (p.Q1062*) and a missense variant involving the exon 10 (p.I1756K). Other genes in the panel were seldom mutated. (Supplementary Fig. 2).
TP53, SETD2, and TBL1XR1 mutations influence time to progression and overall survival in aggressive and indolent B-cell lymphomas
The impact of the most frequently mutated genes on TTP and LSS was examined in the entire BCL population and separately for aggressive and indolent histotypes. Both TP53 and SETD2 mutations were associated with significantly shorter TTP and LSS. Dogs with TP53 mutations had a median TTP of 53 days and a median LSS of 108 days, compared to 202 days and 347 days, respectively, for those with wild-type TP53 (p < 0.01). Similarly, dogs with SETD2 mutations had a median TTP of 123 days, compared to 194 days for those with wild-type SETD2 (p < 0.01).
In aggressive BCLs, TP53 and SETD2 mutations were associated with significantly shorter TTP and LSS, while TBL1XR1 mutations were correlated with longer TTP and LSS. Dogs with TP53 mutations had a median TTP of 52 days and a median LSS of 113 days, compared to 202 days and 343 days, respectively, for those with wild-type TP53 (p = 0.01). Similarly, SETD2 mutations were linked to a shorter TTP (130 days vs. 202 days, p = 0.02) and LSS (176 days vs. 343 days, p = 0.02) compared to wild-type SETD2. Conversely, patients with TBL1XR1 mutations had a longer median TTP (252 days vs. 171 days, p = 0.01) and LSS (368 days vs. 257 days, p = 0.02) compared to those with wild-type TBL1XR1, based on multivariate analysis.
In indolent BCLs, TP53 mutations were the only genetic alteration found to be predictive of a shorter TTP and LSS. Dogs with TP53 mutations had a significantly shorter median TTP (78 days vs. 188 days, p < 0.01) and LSS (105 days vs. 359 days, p < 0.01) compared to those with wild-type TP53.
No significant associations were found after multiple testing corrections.
A regression tree model was developed to provide a more comprehensive significant understanding of the clinico-pathologic features and mutations identified in canine BCL. The model used TP53 genetic status, treatment, and sex as variables to divide the BCL cohort into seven nodes, each with a different prognosis. Nodes 1 and 2 were associated with a good prognosis (survival 100 days longer than the cohort median), nodes 3 and 4 with a fair prognosis (survival around the cohort median), and nodes 5, 6, and 7 with a poor prognosis. The median survival for dogs with a good, fair, and poor prognosis was 575 days, 354 days, and 118 days, respectively (p < 0.0001) (Fig. 5).

Regression tree model (all B-cell lymphomas). All relevant features were processed through a regression tree model (displayed above). The prognostic relevance of TP53 mutations and treatment are immediately visible. Furthermore, the interplay between TRAF3 status and indolent diagnosis, along with the correlation between clinical stages and chemo-immunotherapy, show the benefit of this approach in uncovering masked factors strongly affecting the prognosis.
TP53 and SETD2 mutations influence time to progression and overall survival in DLBCL
In DLBCL, SETD2 mutations were associated with shorter TTP (101 days vs. 202 days, p = 0.02) and LSS (175 days vs. 345 days) compared to wild-type SETD2. Additionally, TP53 mutations were predictive of shorter LSS (117 days vs. 343 days, p = 0.04) by multivariable analysis. A regression tree decision model applied to the DLBCL dataset revealed that treatment, TP53 status, sex, MYC, POT1, and FBXW7 status were crucial in identifying seven distinct DLBCL clusters for LSS. When analyzing TTP, SETD2 and KMT2D also played a significant role, resulting in seven DLBCL clusters (Fig. 6).

Regression tree model (DLBCL). To provide a deeper understanding of DLBCL malignancies, we built the regression tree model shown above. It revealed that treatment choice emerges as the most relevant prognostic factor. Additionally, the other features of the tree primarily pertain to gene status, displaying the importance and complexity of genetic variations.
KMT2D mutations are associated with bone marrow infiltration in aggressive T-cell lymphomas
In aggressive TCL, dogs with KMT2D mutations had a higher likelihood of bone marrow (BM) infiltration compared to those with wild-type KMT2D (80% vs. 29%, p = 0.02). However, this association did not maintain statistical significance after multiple testing corrections. In both indolent and aggressive TCLs, no associations were found between gene mutational status and LSS or TTP.
Discussion
Here, we report the development of a novel targeted NGS panel ("K9 lymphoma assay") and its use to obtain the genomic characterization of the largest dataset of clinically fully staged canine lymphomas.
In veterinary oncology, there is a growing demand for advanced genetic profiling to identify somatic mutations in canine cancers. Several diagnostic companies offer comprehensive pan-tumor genetic profiling for dogs, enabling more precise cancer care12,13. However, these platforms are designed more toward solid tumors and do not allow for the identification of some mutations that are more specific for lymphoid neoplasms. The availability of such an approach is expected to improve sensitivity in identifying known and actionable mutations, consequently boosting clinical effectiveness.
Moreover, in response to the evolving landscape of veterinary medicine, there's a critical need for research to move beyond the traditional "one-size-fits-all" approach. Notably, oncology clinical trials in dogs are gaining prominence as platforms to evaluate innovative anticancer strategies. These trials aim to advance treatment options for both canine and human patients, representing a significant shift in the field. Acknowledging this significant knowledge gap, the newly developed K9 lymphoma assay successfully identified protein-coding mutations in genes commonly altered in canine lymphomas. We initially tested it in a small dataset of DLBCL previously analyzed by WES, and the results demonstrated comparability and similarity, confirming the quality and coverage of the design.
In the present study, we included both B-cell and T-cell lymphomas, covering histotypes not previously genetically characterized in detail, specifically MZL, FL, BL, B-LL, and B-SLL. The genetic analysis largely captured the landscape of mutations previously reported by us and other groups8,9,10. Consistent with previous data, TRAF3 emerged as BCL's most frequently altered gene. The prevalence of truncating mutations within TRAF3 underscores the potential functional implications on the encoded protein.
Despite the apparent homogeneity of mutations across different BCL histotypes, our research revealed a distinct separation in the mutational landscape between leukemic lymphoid neoplasms (B-LL and B-SLL) and lymphomas primarily originating from the lymph nodes (BL, DLBCL, FL, and MZL) (Fig. 3). This new data presents a promising outlook and might serve as a foundation for upcoming research, potentially identifying new therapeutic strategies distinguishing these two subgroups.
Consistent with previous hypotheses generated from two gene expression profile studies6,7, the clustering of DLBCL and MZL implies a potential shared genetic origin between these two diseases. Certainly, the ongoing clinical challenge primarily lies in the histologic diagnosis. Nodal MZL has historically been interpreted as histologically indolent but biologically aggressive22. Despite being treated with similar protocols used for DLBCL, some studies focusing on this particular subtype have indicated a worse prognosis and varied prognostic cutoffs for peripheral and marrow blood infiltration23. The findings from the current research, however, suggest that despite differing histologic diagnoses (MZL vs. DLBCL), there is a shared molecular origin24. This also agrees with data from human lymphomas, in which at least a subset of DLBCL would arise from the transformation of MZL25,26. Notably, all MZL cases in the current series were categorized as late-stage MZL, indicating the presence of two types of cells: a minority characterized by medium to large-sized cells, exhibiting pale cytoplasm and multiple nucleoli peripheralized into the nucleus (centroblasts), and an intermediate-sized population with abundant pale cytoplasm, irregular nuclei containing peripheralized chromatin and a single central nucleolus (marginal cell morphology). Like immunoblastic DLBCL in human pathology, these features have not previously been examined in veterinary medicine and will require focused investigations.
Exploring co-mutation and mutual exclusivity patterns in aggressive BCL revealed intriguing relationships among mutated genes. The mutual exclusivity between TRAF3 mutations and MAP3K14 suggests potential functional overlap or epistatic interactions between these genes. Conversely, the co-occurrence of TP53 mutations with FAT4 suggests a potential synergistic effect in driving BCL development.
As dogs underwent comprehensive clinical staging and were continuously followed up during treatment, we were able to identify mutations with clinical relevance. The prominence of TP53 mutations was particularly striking as a pivotal clinical-genetic predictor of outcomes. This finding aligns with the TP53 established role as a tumor suppressor and its frequent mutation across various cancer types. While our previous experiments underscored its significance specifically in DLBCL10, our study reveals its clinical relevance across all BCLs, regardless of histotypes. This broadened insight into the prognostic importance of TP53 emphasizes its potential as a universal prognostic marker in canine BCLs. Importantly, this predictive power did not extend to T-cell lymphomas, indicating the phenotype-specific nature of this correlation.
In addition to TP53, we identified SETD2 mutations as contributors to a shorter TTP, underscoring the role of epigenetic regulation in BCL progression. The association between SETD2 mutations and unfavorable outcomes aligns with the growing recognition of epigenetic dysregulation as a pivotal factor in cancer pathogenesis and progression27. When stratifying the analysis by histotypes, we found that the co-occurrence of TP53 and SETD2 mutations led to the worst prognosis in aggressive BCLs, whereas this association was not observed in indolent lymphomas. Conversely, mutations in TBL1XR1 were linked to longer TTP and overall survival in aggressive BCLs.
In indolent BCLs, only TP53 mutations emerged as predictive of poorer outcomes in terms of both TTP and LSS. This distinction underscores the heterogeneity within BCLs, emphasizing the need for a tailored approach based also on the specific genetic characteristics of the tumor.
The detection of the tumor's mutational status also helps guiding therapeutic decisions. In fact, we have documented that chemo-immunotherapy provides better clinical benefit in the absence of TP53 mutations (LSS: 361 days vs. 254 days if dogs received chemotherapy only).
Chemotherapy alone can be considered in dogs with lymphoma without a TP53 mutation and with indolent histology: the LSS in this scenario was 345 days and only 169 days for aggressive histotypes.
Concerning TCL, the number of identified mutated genes was notably low. This result might be attributed to the relatively limited gene coverage of the K9 lymphoma assay, primarily derived from canine BCL databases. Notably, TCL genetic data were sourced from only two studies. One employed a WES approach, but exclusively focused on two pure breeds9. A more recent study utilized an amplicon-based targeted approach to analyze 16 canine PTCL-NOS28. However, the panel design relied on human T-cell lymphoma data, introducing marked limitations. While there was some overlap in the genes identified by these studies and our panel, the overall results were scarce. Consequently, it is conceivable that a more comprehensive sequencing approach, such as whole exome sequencing, is necessary to better understand TCL's genetic landscape, particularly when including various histotypes and multiple breeds. Nevertheless, we found SATB1 as the most significantly mutated gene in TCLs. SATB1 codes for a crucial, global transcription regulator and chromatin organizer. Its silencing leads to impaired T-cell development and function, and its dysregulation is associated with human cutaneous T cell lymphomas29. Our study indicates SATB1 as an intriguing target for further exploration in veterinary medicine. The identification of mutations in the tumor suppressor gene FBXW7 was also notable, due to its role in canine BCL and in multiple human cancers. FBXW7 encodes a conserved E3 ubiquitin-protein ligase involved in the degradation of critical oncoproteins within the SCF complex. In our case series, FBXW7 mutations were frequently found in a heterozygous state, potentially impacting the stability of the FBXW7 protein. This is noteworthy as the protein operates as a dimer, and alterations in one of the gene copies can affect its functionality.
Our study has several limitations that should be acknowledged. Firstly, there is an underrepresentation of TCL compared to BCL. However, the proportion of different histotypes is consistent with worldwide data on the distribution of canine lymphomas30. Secondly, the 31-gene panel, while enabling focused analysis of key drivers, may have been insufficient to comprehensively evaluate all genetic aberrations and copy number variations, potentially underestimating the full genetic complexity of the disease. Lastly, the lack of matched germline samples and the application of stringent bioinformatic filters might have reduced the detection of mutations, resulting in an incomplete depiction of the tumor mutational landscape.
Despite these limitations, the K9 lymphoma assay provides a comprehensive genomic characterization of clinically fully staged canine lymphomas. The design of genetic profiling platform tailored to canine lymphomas, rather than a broad pan-tumor approach, has proven valuable in identifying actionable mutations with potential clinical implications. The identification of shared molecular origins between DLBCL and MZL contributes to a nuanced understanding of canine lymphoma pathogenesis, drawing parallels with human lymphomas. The clinical significance of specific mutations, such as TP53, SETD2 SATB1 and FBXW7, across various lymphoma histotypes underscores the potential for these genetic markers to serve as prognostic indicators, guiding tailored therapeutic decisions in the management of canine lymphomas.
Methods
Animal recruitment and sample acquisition
Dogs with previously untreated B-cell and T-cell lymphoma of any WHO clinical stage and histological histotype were included in the study. The study and all methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org), where applicable.
The study did not fall within the application areas of Italian Legislative Decree 26/2014 which governs the protection of animals used for scientific or educational purposes; therefore, ethical approval was waived for this study by Department of Veterinary Sciences at University of Turin. Dogs were treated according to the current standards, and dog owners gave written informed consent. All methods were carried out in accordance with relevant guidelines and regulations.
All dogs underwent a complete staging work-up, consisting of history and physical examination, complete blood cell count with differential, serum biochemistry profile including LDH activity, thoracic radiographs and abdominal ultrasound, fine-needle aspiration (FNA) of liver and spleen regardless of their sonographic appearance, cytologic evaluation and immunophenotyping by flow cytometry (FC) of a FNA of an enlarged peripheral lymph node (LN), BM aspirate and PB. Before the initiation of therapy, all dogs underwent lymphadenectomy to confirm the histotype by routine histology and immunohistochemistry (CD3 and CD20)3 and to provide material for nucleic acid extraction. The excised LN was preserved in formalin for histopathologic evaluation; one small tissue fragment was preserved at -20 °C for nucleic acid extraction. The treatment protocol was in keeping with approved standards.
Dogs with aggressive and indolent B-cell lymphoma received a CHOP-based chemotherapeutic protocol31 with or without an autologous vaccine5, based on the clinician’s and owner’s preferences.
Dogs with aggressive T-cell lymphoma received VELCAP-TSC or CCNU-CHOP32,33, two alkylating-rich protocols with similar clinical activities, based on the clinician’s and owner’s preferences. Dogs with indolent T-cell lymphoma were treated with oral chlorambucil and prednisolone if indicated by the tumor load and/or the presence of clinical signs (substage b).
Based on the criteria of the Veterinary Cooperative Oncology Group consensus statement34, treatment response was classified as complete remission (CR), partial remission (PR), stable disease (SD) or progressive disease (PD) at each therapeutic session. The best response was required to last for at least 28 days.
At the end of treatment, all dogs underwent end-staging, including FC on a peripheral LN, PB and BM, and imaging. Follow-up evaluation consisted of monthly physical examination, peripheral LN size measurement, cytologic evaluation during the first year, and every other month thereafter. Relapse was defined as the clinical reappearance and cytologic evidence of lymphoma with or without FC confirmation in any anatomical site in dogs having experienced CR.
TTP was calculated as the interval between initiation of treatment and PD or relapse, whereas LSS was measured as the interval between initiation of treatment and lymphoma-related death. Dogs lost to follow-up or dead from lymphoma-unrelated causes before PD, as well as those still in CR at the end of the study, were censored for TTP analysis. Dogs alive at the end of the study, lost to follow-up or dead owing to causes other than lymphoma or its treatment were censored for LSS analysis.
Targeted gene panel design
The sequencing gene panel was designed using the Agilent SureDesing tool (https://earray.chem.agilent.com/suredesign/) to target the coding exons, 5’- and 3’- untranslated regions (UTRs) and 10 base pairs (bp) across intron–exon junctions of the genes of interest, using the CanFam3.1 as reference genome. The target spanned a total of 196 kbp covered by 14,436 unique probes. Design ID is available on request.
DNA extraction, library preparation, and sequencing
DNA was extracted from fresh-frozen samples using the DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. DNA quality and concentration were assessed using NanoDrop OneC (Thermo Fisher, Waltham, MA, USA) and Quantus Fluorometer (Promega, Madison, WI, USA), respectively. A total of 257 libraries for targeted sequencing were prepared using the SureSelect XT HS2 DNA Reagent Kit (Agilent Technologies, Santa Clara, CA, USA). Briefly, 100 ng input DNA of each sample underwent enzymatic fragmentation using the SureSelect Enzymatic Fragmentation Kit (Agilent Technologies) to obtain a 180–250 bp target fragment size. The end-repaired and adaptor-ligated libraries were PCR-amplified and then purified with the AMPure XP beads (Beckman Coulter, Brea, CA, USA). The concentration and fragment size of the pre-capture libraries were assessed using the Bioanalyzer 2100 instrument (Agilent Technologies). Then, 1,000 ng from each library were hybridized to the biotinylated RNA probes and captured with streptavidin-coated DynaBeads MyOne T1 (Thermo Fisher). Captured libraries were PCR-amplified and further purified with AMPure XP beads (Beckman Coulter), and finally eluted in Low TE (Thermo Fisher). The final libraries’ concentration was assessed using the Quantus Fluorometer (Promega) and Bioanalyzer 2100 (Agilent Technologies). Libraries were pooled together in equimolar amounts and sequenced on an Illumina NextSeq 550 instrument (Illumina, San Diego, CA, USA) using a Mid-output flow cell (Illumina) to generate 2 × 150 bp paired-end reads. Raw Illumina sequencing data are deposited in BioProject database with BioProject ID PRJNA1074236 .
Bioinformatic analysis
Sequence reads were aligned to the canine reference genome CanFam3.1 retrieved from UCSC (https://genome-euro.ucsc.edu/cgi-bin/hgGateway?db=canFam3) using Burrows-Wheeler Aligner (BWA, v0.7.17) 35. Data preprocessing was performed following the Genome Analysis Toolkit (GATK, v4.3.0.0) guidelines36. Briefly, the aligned reads were converted to BAM format, sorted with Samtools v1.13 and provided of the missing information fields (Instrument used, Sample ID, etc.). Duplicates were marked with Picard and BaseQualityRecalibration was applied sample-wise. Mutect2 variant caller37 from GATK was used in combination with Varscan2 v2.4.338 and Freebayes v1.3.639 to identify SNVs and indels. The variants detected by each caller were then filtered to have a minimum coverage of 30X and an Allele Frequency (AF) less than 90% and more than 10%. A majority voting filtering was applied and only the variants called by two out of the three callers were kept. As matched-normal samples were not sequenced in the present study, all variants included in the DoGSD database (https://ngdc.cncb.ac.cn/idog/index.jsp) and in the European Variant Archive (EVA) Release 5 database (https://www.ebi.ac.uk/eva/), excluding those reported among the “Lymphoma som SNPs9” from the Broad Improved Canine Annotation v1 track at UCSC Genome Browser, were filtered out as putative germline variants. Lastly, VCF files were annotated with ANNOVAR40. The predicted impact of missense mutations was assessed using SIFT41.
Statistical analysis
Statistical analyses were performed in R (R software v4.3.0). The Fisher Exact test was used to correlate recurrently mutated genes and clinicopathological features. TMB, defined as the number of protein-coding gene mutations (namely exonic variants excluding synonymous variants) per megabase sequenced, was calculated as previously described10 and was correlated with the recurrently mutated genes by means of Student’s t-test, if data were normally distributed and the variance were comparable, or alternatively Wilcoxon rank-sum test. Maftools R package was used to assess mutual exclusivity and co-occurrence patterns among the recurrently mutated genes and to generate oncoplots and lollipop plots. Survival analysis was carried out using survival and survminer packages.
The following clinicopathological variables were tested for their influence on both TTP and LSS by univariate Cox proportional hazard model: breed (pure vs. mixed), sex (female vs. male), age (< 7 years vs. ≥ 7 years; median used as cut-off), weight (< 26.6 kg vs. ≥ 26.6 kg; median used as cut-off), stage (III and IV vs. V), substage (a vs. b), presence of BM infiltration (< 3% vs. ≥ 3%, except for PTCLs: < 5% vs. ≥ 5%), presence of PB infiltration (< 3.8% vs. ≥ 3.8%, median used as cut-off), LDH activity (normal vs. increased), pretreatment with steroids (yes vs. no), treatment (chemotherapy vs. chemo-immunotherapy for BCL and chemotherapy vs. prednisolone only for TCL). The impact of the status of recurrently mutated genes (mut vs. wt) on TTP and LSS was also assessed.
Variables with a p ≤ 0.2 were included in the multivariate analysis. For the statistically significant features obtained by multivariate Cox proportional hazard analysis, Kaplan–Meier (KM) curves were drawn and compared by means of a log-rank test. Finally, a matrix including the percentage of mutated samples by genes and by histotypes was constructed. This matrix was then fed into a hierarchical clustering algorithm to access ensemble information.
To further investigate prognostic-related data structures, two distinct regression tree models were developed utilizing the rpart package, employing exponential method. Subsequently, these trees underwent pruning to set a maximum complexity factor and a maximum depth, thereby mitigating potential artifacts. Following pruning, each terminal leaf was assigned to one of three distinct classes: POOR, FAIR, and GOOD. Specifically, FAIR nodes comprise cases that exhibit performance closely aligned with the overall population, while GOOD nodes display longer times with respect to the population. Conversely, POOR nodes demonstrate times below the overall population. These three categories were employed to create a new categorical feature, subsequently subjected to validation within a Cox proportional hazards model.
Data availability
Raw Illumina reads of targeted sequencing are publicly available in BioProject database with BioProject ID PRJNA1074236. Any further request about data and materials can be addressed to corresponding authors (luca.aresu@unito.it; laura.marconato@unibo.it).
References
Zandvliet, M. Canine lymphoma: A review. Vet. Q. 36, 76–104. https://doi.org/10.1080/01652176.2016.1152633 (2016).
Valli, V. E. et al. Classification of canine malignant lymphomas according to the World Health Organization criteria. Vet. Pathol. 48, 198–211. https://doi.org/10.1177/0300985810379428 (2011).
Aresu, L. et al. Canine indolent and aggressive lymphoma: Clinical spectrum with histologic correlation. Vet. Comp. Oncol. 13, 348–362. https://doi.org/10.1111/vco.12048 (2015).
Modiano, J. F. et al. Distinct B-cell and T-cell lymphoproliferative disease prevalence among dog breeds indicates heritable risk. Cancer Res. 65, 5654–5661. https://doi.org/10.1158/0008-5472.CAN-04-4613 (2005).
Marconato, L. et al. Opportunities and challenges of active immunotherapy in dogs with B-cell lymphoma: A 5-year experience in two veterinary oncology centers. J. Immunother. Cancer 7, 146. https://doi.org/10.1186/s40425-019-0624-y (2019).
Richards, K. L. et al. Gene profiling of canine B-cell lymphoma reveals germinal center and postgerminal center subtypes with different survival times, modeling human DLBCL. Cancer Res. 73, 5029–5039. https://doi.org/10.1158/0008-5472.CAN-12-3546 (2013).
Aresu, L. et al. New molecular and therapeutic insights into canine diffuse large B-cell lymphoma elucidates the role of the dog as a model for human disease. Haematologica 104, e256–e259. https://doi.org/10.3324/haematol.2018.207027 (2019).
Bushell, K. R. et al. Genetic inactivation of TRAF3 in canine and human B-cell lymphoma. Blood 125, 999–1005. https://doi.org/10.1182/blood-2014-10-602714 (2015).
Elvers, I. et al. Exome sequencing of lymphomas from three dog breeds reveals somatic mutation patterns reflecting genetic background. Genome Res. 25, 1634–1645. https://doi.org/10.1101/gr.194449.115 (2015).
Giannuzzi, D. et al. The genomic landscape of canine diffuse large B-cell lymphoma identifies distinct subtypes with clinical and therapeutic implications. Lab Anim. 51, 191–202 (2022).
Kamps, R. et al. Next-generation sequencing in oncology: Genetic diagnosis, risk prediction and cancer classification. Int. J. Mol. Sci. https://doi.org/10.3390/ijms18020308 (2017).
Chon, E. et al. Genomic tumor analysis provides clinical guidance for the management of diagnostically challenging cancers in dogs. J. Am. Vet. Med. Assoc. 261, 668–677. https://doi.org/10.2460/javma.22.11.0489 (2023).
Wu, K. et al. Analyses of canine cancer mutations and treatment outcomes using real-world clinico-genomics data of 2119 dogs. NPJ Precis. Oncol. 7, 8. https://doi.org/10.1038/s41698-023-00346-3 (2023).
Wang, G. et al. Actionable mutations in canine hemangiosarcoma. PLoS One 12, e0188667. https://doi.org/10.1371/journal.pone.0188667 (2017).
Megquier, K. et al. Comparative genomics reveals shared mutational landscape in canine hemangiosarcoma and human angiosarcoma. Mol. Cancer Res. 17, 2410–2421. https://doi.org/10.1158/1541-7786.MCR-19-0221 (2019).
Wang, G. et al. Molecular subtypes in canine hemangiosarcoma reveal similarities with human angiosarcoma. PLoS One 15, e0229728. https://doi.org/10.1371/journal.pone.0229728 (2020).
Wong, K. et al. Comparison of the oncogenomic landscape of canine and feline hemangiosarcoma shows novel parallels with human angiosarcoma. Dis. Model. Mech. https://doi.org/10.1242/dmm.049044 (2021).
Estabrooks, T. et al. Identification of genomic alterations with clinical impact in canine splenic hemangiosarcoma. Vet. Comp. Oncol. 21, 623–633. https://doi.org/10.1111/vco.12925 (2023).
Gardner, H. L. et al. Canine osteosarcoma genome sequencing identifies recurrent mutations in DMD and the histone methyltransferase gene SETD2. Commun. Biol. 2, 266. https://doi.org/10.1038/s42003-019-0487-2 (2019).
Sakthikumar, S. et al. SETD2 is recurrently mutated in whole-exome sequenced canine osteosarcoma. Cancer Res. 78, 3421–3431. https://doi.org/10.1158/0008-5472.CAN-17-3558 (2018).
Hendricks, W. P. D. et al. Somatic inactivating PTPRJ mutations and dysregulated pathways identified in canine malignant melanoma by integrated comparative genomic analysis. PLoS Genet. 14, e1007589. https://doi.org/10.1371/journal.pgen.1007589 (2018).
Cozzi, M. et al. Canine nodal marginal zone lymphoma: Descriptive insight into the biological behaviour. Vet. Comp. Oncol. 16, 246–252. https://doi.org/10.1111/vco.12374 (2018).
Marconato, L. et al. Prognostic significance of peripheral blood and bone marrow infiltration in newly-diagnosed canine nodal marginal zone lymphoma. Vet. J. 246, 78–84. https://doi.org/10.1016/j.tvjl.2019.02.002 (2019).
Giannuzzi, D. et al. Integrated analysis of transcriptome, methylome and copy number aberrations data of marginal zone lymphoma and follicular lymphoma in dog. Vet. Comp. Oncol. 18, 645–655. https://doi.org/10.1111/vco.12588 (2020).
Chapuy, B. et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 24, 679–690. https://doi.org/10.1038/s41591-018-0016-8 (2018).
Schmitz, R. et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med. 378, 1396–1407. https://doi.org/10.1056/NEJMoa1801445 (2018).
Ilango, S., Paital, B., Jayachandran, P., Padma, P. R. & Nirmaladevi, R. Epigenetic alterations in cancer. Front. Biosci. (Landmark Ed) 25, 1058–1109. https://doi.org/10.2741/4847 (2020).
McDonald, J. T. et al. Comparative oncology DNA sequencing of canine T cell lymphoma via human hotspot panel. Oncotarget 9, 22693–22702. https://doi.org/10.18632/oncotarget.25209 (2018).
Fredholm, S. et al. SATB1 in malignant T cells. J. Invest. Dermatol. 138, 1805–1815. https://doi.org/10.1016/j.jid.2018.03.1526 (2018).
Ito, D., Frantz, A. M. & Modiano, J. F. Canine lymphoma as a comparative model for human non-Hodgkin lymphoma: Recent progress and applications. Vet. Immunol. Immunopathol. 159, 192–201. https://doi.org/10.1016/j.vetimm.2014.02.016 (2014).
Simon, D. et al. Treatment of dogs with lymphoma using a 12-week, maintenance-free combination chemotherapy protocol. J. Vet. Intern. Med. 20, 948–954. https://doi.org/10.1892/0891-6640(2006)20[948:todwlu]2.0.co;2 (2006).
Goodman, I. H., Moore, A. S. & Frimberger, A. E. Treatment of canine non-indolent T cell lymphoma using the VELCAP-TSC protocol: A retrospective evaluation of 70 dogs (2003–2013). Vet. J. 211, 39–44. https://doi.org/10.1016/j.tvjl.2016.03.003 (2016).
Marconato, L. et al. Prognostic value of peripheral blood and bone marrow infiltration assessed by flow cytometry in dogs with de novo nodal peripheral T-cell lymphoma receiving alkylating-rich chemotherapy. Vet. J. 303, 106057. https://doi.org/10.1016/j.tvjl.2023.106057 (2023).
Vail, D. M. et al. Response evaluation criteria for peripheral nodal lymphoma in dogs (v1.0)–a Veterinary Cooperative Oncology Group (VCOG) consensus document. Vet. Comp. Oncol. 8, 28–37. https://doi.org/10.1111/j.1476-5829.2009.00200.x (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the cloud: Using docker, GATK and WDL in Terra (O’Reilly Media Inc., 2020).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. https://doi.org/10.1038/nbt.2514 (2013).
Koboldt, D. C. et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. https://doi.org/10.1101/gr.129684.111 (2012).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907. https://doi.org/10.48550/arXiv.1207.3907 (2012).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. https://doi.org/10.1093/nar/gkq603 (2010).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. https://doi.org/10.1093/nar/gkg509 (2003).
Acknowledgements
This work was funded by MyLav Laboratory, Milan, Italy.
Author information
Authors and Affiliations
Contributions
L.A. and L.M. designed the study, interpreted the data, and wrote the manuscript; A.F. and F.B. contributed to study design, data interpretation and manuscript revision; L.M. and I.M. provided samples and clinical data; A.F., L.L., S.D., M.M. and A.R. carried out library preparation and sequencing experiments; E.M. conducted bioinformatic and statistical analyses and performed data visualization; all authors contributed to manuscript revision and approved the final draft. L.A. and L.M. equally contributed to this work and share senior authorship.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fanelli, A., Licenziato, L., Mazzone, E. et al. The K9 lymphoma assay allows a genetic subgrouping of canine lymphomas with improved risk classification. Sci Rep 14, 18687 (2024). https://doi.org/10.1038/s41598-024-69716-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69716-6
- Springer Nature Limited