
Abstract
Alterations in miRNA levels have been observed in various types of cancer, impacting numerous cellular processes and increasing their potential usefulness in combination therapies also in brain tumors. Recent advances in understanding the genetics and epigenetics of brain tumours point to new aberrations and associations, making it essential to continually update knowledge and classification. Here we conducted molecular analysis of 123 samples of childhood brain tumors (pilocytic astrocytoma, medulloblastoma, ependymoma), focusing on identification of genes that could potentially be regulated by crucial representatives of OncomiR-1: miR-17-5p and miR-20a-5p. On the basis of microarray gene expression analysis and qRTPCR profiling, we selected six (WEE1, CCND1, VEGFA, PTPRO, TP53INP1, BCL2L11) the most promising target genes for further experiments. The WEE1, CCND1, PTPRO, TP53INP1 genes showed increased expression levels in all tested entities with the lowest increase in the pilocytic astrocytoma compared to the ependymoma and medulloblastoma. The obtained results indicate a correlation between gene expression and the WHO grade and subtype. Furthermore, our analysis showed that the integration between genomic and epigenetic pathways should now point the way to further molecular research.
Similar content being viewed by others

Introduction
Accurate brain tumor histopathological and molecular integrative analysis is essential in the age of emerging precision medicine. The main goal of a well-defined diagnosis is the possibility of using tailored therapies. Currently, the modulation of microRNAs (miRNAs) activity in different types of cancers is also considered an option for future anticancer therapeutics. miRNAs are one of the most characterized non-coding RNAs regulating gene expression at the posttranscriptional level. Alterations of miRNA levels were observed in various types of cancer and could result in abnormalities of numerous cellular processes, which raises their potential usefulness in combination therapies. It has increasing value as there is proof that a multi-targeted approach in cancer therapies is still more effective also in brain tumors.
Recently published papers showed the presence of miRNA targets that modify various signaling pathways responsible for tumor formation and growth in the glioblastoma and medulloblastoma, which suggests their potential for use in such type of treatment1,2,3.
Due to the scientific research trends, we provide comprehensive molecular analysis of the most common childhood brain tumors (pilocytic astrocytoma, medulloblastoma, ependymoma) which focuses on the miRNA alterations and their target identification.
In our previous research4,5 quantitative real-time polymerase chain reaction (qPCR) was used to analyse the level of miR-17-5p and miR-20a, which are key members of OncomiR-1. The findings of our study indicated an upregulation of miR-17-5p and miR-20a-5p in the paediatric brain tumours, accompanied by a correlation between miRNA levels and tumour grade.
In this study, we specifically examined the expression patterns of the target genes regulated by the key members of the miR-17-5p and miR-20a-5p clusters and investigated their potential association with cellular activities. The bioinformatics research reveals that the sequences of miR-17-5p and miR-20a-5p are in concordance with numerous 3’UTR regions of mRNA molecules (Fig. 1). After a microarray analysis of gene expression, we conducted a thorough examination of the genes that may be controlled by miR-17-5p and miR-20a-5p. A larger cohort was subjected to real-time PCR profiling to evaluate the expression levels of 84 genes that may be controlled by the core members of OncomiR-1. Then we have chosen six candidates that show the most potential for a thorough evaluation: WEE1, CCND1, VEGFA, PTPRO, TP53INP, and BCL2L11.

Target genes for miR-17-5p and miR-20a-5p, generated from TargetScan 8.0 database.
Materials and methods
Patients and tissue samples
The analyses included 123 cases of childhood brain tumors (all patients were under 18 years of age) located infratentorially: 35 medulloblastomas, WHO grade 4, (MB), including 3 WNT-activated (WNT-MB), 2 SHH-activated (SHH-MB), 30 non-WNT/non-SHH; 52 ependymomas, WHO grade 2/3 (EP), including 25 posterior fossa group A (PFA) and 27 posterior fossa group B (PFB) 31 pilocytic astrocytomas,WHO grade 1, (PA).
Total RNA was extracted according to the manufacturer’s instructions, using miRNeasy Mini Kit (Qiagen, Hilden, Germany, cat. no. 217004). The quantity and purity of RNA were analysed quantitatively and qualitatively. The Human Brain Total RNA (Invitrogen, cat. no. AM7962) was used in all analyses as a control material.
Microarray profiling
Gene expression analysis was performed using the Human Genome U133 + PM Array Strip (901569, Affymetrix) on 16 brain tumors, comprising 11 cases of WHO grade 2/3 ependymoma (3 PFA, 8 PFB), 5 cases of medulloblastoma (non-WNT/non-SHH). Furthermore, a control sample of Human Brain Total RNA (Invitrogen, cat. no. AM7962) was incorporated.
The procedure entailed synthesizing cDNA via reverse transcriptase and an oligo-dT primer. The cDNA served as a template for an in vitro transcription, resulting in the production and amplification of biotin-labeled antisense mRNA (cRNA). Before hybridization, the cRNA was fragmented into parts varying in length from 50 to 200 bases. Following this, the GeneChip matrix was washed, stained, and scanned utilizing the GeneAtlas™ System from Affymetrix. The obtained outcomes were analysed with the use of dedicated software, specifically, the Transcriptome Analysis Console from Applied Biosystems.
Analysis of miR-17-5p and miR-20-5p targets
Initially, we used The Human miR-17 & miR-20a Targets RT2 profiler PCR array (Qiagen, Hilden, Germany, cat. no. 330231) to profile the expression of predicted target genes regulated by miR-17-5p and miR-20a-5p and also target genes regulated by other miRNAs that have the same seed sequence as miR-17-5p and miR-20a (miR-20b-5p, miR-93-5p, miR-106a-5p, miR-106b-5p, miR-519d). Reverse transcription for the RT2 Profiler PCR Array was prepared using the RT2 First Strand Kit (Qiagen, Hilden, Germany, cat. no. 330401). The analysis included 17 cases of WHO grade 2/3 ependymoma (11 PFA, 6 PFB), a control sample was Human Brain Total RNA. Normalized relative expression levels of each gene were calculated in the tested samples compared with the control based on the sample’s average Ct value, according to the formula in the Eq. (1):
Gene expression validation analysis
After examining the results of the microarray and RT2 Profiler PCR analyses, as well as conducting a literature review, we selected six genes for investigation using TaqMan probes (Life Technologies, Carlsbad, CA, USA): WEE1 (ID Hs00268721_m1), CCND1 (ID Hs00277039_m1), VEGFA (ID 00900054_m1), PTPRO (ID Hs00243097_m1), TP53INP (ID Hs01003820_m1), BCL2L11 (ID Hs01076940_m1). The study was carried out on 91 cases: 31 pilocytic astrocytomas, 30 WHO grade 2 ependymoma (15 PFA and 15 PFB) and 30 medulloblastoma (3 WNT-MB, 2 SHH-MB and 25 non-WNT/non-SHH), a control sample was Human Brain Total RNA (Invitrogen, cat. no. AM7962). cDNA dedicated for gene expression analysis was synthesized from 500 ng of total RNA of each sample (miScript II RT Kit, Qiagen, cat. no. 218161). The real-time quantitative PCR analysis was performed in duplicate using Fast Advanced Master Mix. The GAPDH (ID Hs99999905_m1) gene was used as the reference gene. Normalized relative expression levels of each gene were calculated according to the Eq. (1).
Data analysis
Statistical analysis of gene expression results was performed using Statistica software (version 13.1). Normal distribution of data was determined using the Shapiro–Wilk and Lilliefors-corrected Kolmogorov–Smirnov tests. To compare different levels of genes between groups, a non-parametric Kruskal–Wallis test was used. Correlation between genes and miRNA was determined using the Pearson test. Results were considered significant at p ≤ 0.05.
Ethics statement
All methods were carried out to the tenets of the Declaration of Helsinki and was approved by the Bioethical Committee at the Medical University of Lodz (permit no. RNN/122/17/KE). A informed consent was obtained from all a parent and/or legal guardian of patient.
Results
Microarray analysis
The microarray data was analysed to examine the expression of target genes for miR-17-5p and miR-20a-5p. Gene expression was compared between medulloblastoma and ependymoma (Table 1). All genes showed increased expression levels compared to the control. 23 of 30 genes showed statistically significant differences (p ≤ 0.05) between medulloblastoma and ependymoma included BCL2, BCL2L11, CCND1, E2F1, PPARG, PTPRO, VEGFA, WEE1.
Expression profiles of genes regulated by miR-17-5p and miR-20a-5p
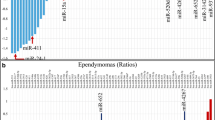
Expression profiles of predicted genes regulated by miR-17-p and miR-20a-5p were analysed for 17 cases of ependymoma (11 PFA, 6 PFB) (Fig. 2). Between the analysed groups, we noted statistically significant differences for 32 genes with p < 0.05 and all of these genes had increased expression in the PFA group. Increased expression levels in the PFA group were recorded for the following genes: WEE1 (ddCt = 4.12), CCND (ddCt = 3.75), TP53INP1 (ddCt = 2.89), BCL2L11 (ddCt = 2.70), TCF7L1 (ddCt = 2.66), VEGFA (ddCt = 2.60), E2F1 (ddCt = 2.54), in PFB group for: CCND1 (ddCt = 2.86), WEE1 (ddCt = 2.85), PPARG (ddCt = 2.72), VEGFA (ddCt = 2.38), KPNA2 (ddCt = 1.45), PBX3 (ddCt = 1.32), RUNX1 (ddCt = 1.18).

Heatmap showing expression profile of predicted target genes regulated by miR-17-5p and miR-20a-5p in ependymoma. The genes with increased normalized expression are marked in red; the genes with reduced levels of normalized expression are marked in green. The p-value is given for each gene. If the p-value is less than 0.05 (p < 0.05) we report the result as statistically significant (indicated in red).
Gene expression analysis
As a next step in our research, we conducted a detailed validation of the mRNA expression of selected genes in three different WHO groups of tumors: 30 medulloblastomas WHO 4 (25 non-WNT/non-SHH, 3 WNT-MB, 2 SHH-MB), 30 ependymomas WHO grade 2 (15 PFA, 15 PFB), and 31 pilocytic astrocytomas WHO 1 (Fig. 3).

Expression of WEE1, CCND1, VEGFA, PTPRO, TP53INP1 and BCL2L11 genes in three groups of pediatric brain tumors: medulloblastomas, ependymomas, and pilocytic astrocytomas. Relative expression given in ddCt values with an indication of the differences in expression between the groups studied; *p < 0.05, **p < 0.01, ***p < 0.001.
The WEE1 gene showed the highest expression in the ependymomas, ddCt = 4,21 (PFA ddCt = 4.51, PFB ddCt = 4.1), followed by medulloblastomas, ddCt = 3,69, (non-WNT/non-SHH ddCt = 3.49, WNT-MB ddCt = 3.92, SHH-MB ddCt = 4.72) and pilocytic astrocytomas, ddCt = 1,15. Statistically significant differences were found between MB and PA (p < 0.001), EP and PA (p < 0.001).
The CCND1 gene was characterized by down-expression in MBs, ddCt = −3.45(non-WNT/non-SHH ddCt = −3.74, WNT-MB ddCt = −2.83, SHH-MB ddCt = −0.07), while in EP and PA the expression was increased, in EP ddCt = 2.94 (PFA ddCt = 4.42, PFB ddCt = 3.75), in PAs ddCt = 1.49. Differences in expression were noted between MB and PA, as well as MB and EP groups (p < 0.001). Statistically significant differences in gene expression were also found between ependymomas subtypes (p = 0.04).
An inverse correlation was observed for PTPRO, the highest level was found in the MBs ddCt = 3.35 (non-WNT/non-SHH ddCt = 3.43, WNT-MB ddCt = 2.97, SHH-MB ddCt = 2.53), while in PAs ddCt = 0,70 and EPs ddCt = 0,72 (PFA ddCt = −0.33, PFB ddCt = 1.18) a very low level was noticed. Statistically significant differences in expression were found between MB and EP (p < 0.001), as well as between MB and PA (p < 0.001).
Increased gene expression was also reported for TP53INP1, with ddCt values of 1.84 for MBs (non-WNT/non-SHH ddCt = 1.7, WNT-MB ddCt = 2.55, SHH-MB ddCt = 2.88), 1.61 for EPs (PFA ddCt = 1.78, PFB ddCt = 1.5), and 0.76 for PAs. Statistically significant differences in expression were observed between the MB and EP groups, as well as between the EP and PA groups (p < 0.01).
The VEGFA gene showed high expression values in the ependymoma group ddCt = 2. 22 (PFA ddCt = 3.58, PFB ddCt = 1.82 where p = 0.02). In the medulloblastoma and pilocytic astrocytoma groups was below control, with ddCt = −0.25 for MBs (non-WNT/non-SHH ddCt = −0.26, WNT-MB ddCt = −0.24, SHH-MB ddCt = 0.21) and ddCt = −0.08,for PAs. ddCt values close to control were found for the BCL2L11 gene, MBs ddCt = −0.86 ( non-WNT/non-SHH ddCt = −1.0, WNT-MB ddCt = −0.57, SHH-MB ddCt = 0.87), EPs ddCt = 0.94 (PFA ddCt = 1.45, PFB ddCt = 0.68), PAs ddCt = 0.97. Differences between groups were observed between MB and EP, as well as MB and PA (p < 0.001).
Correlation between microRNA and genes
The analysis of Pearson correlation between genes and miRNA was conducted in three groups: pilocytic astrocytoma (Table 2a), ependymoma (Table 2b) and medulloblastoma (Table 2c). In the pilocytic astrocytoma group, the highest correlation coefficient (r < 0.7) was recorded for genes: WEE1-CCND1, WEE1-TP53INP1, WEE1-BCL2L11 and CCND1-BCL2L11. There was no strong correlation between genes and miRNA expression. Only between miR 17-5p-WEE1 and miR 17-5p-CCND1 it reached the level of statistical significance with r values of 0.42. In the ependymoma group, three pairs of genes reached statistical significance with r value above 0.7: BCL2L11-CCND1, TP53INP1-CCND1 and TP53INP1-BCL2L11. There was no strong correlation between genes and miRNA expression (the correlation coefficient ranged from −0.2 to 0.3). Statistically significant Pearson correlation results (r < 0.7) were obtained for several gene pairs in medulloblastoma, including WEE1-VEGFA, WEE1-TP53INP1, WEE1-BCL2L11, CCND1-BCL2L11, and VEGFA-BCL2L11. Additionally, the expression of PTPRO was found to negatively correlate with the expression of miR-17-5p (r = −0.51) and miR-20a (r = −0.49).
Discussion
Here, we comprehensively examined the expression levels of the target genes for miR-17-5p and miR-20a-5p, which are the primary members of the OncomiR-1 cluster. The results of our study suggest a positive association between elevated WEE1 expression and both the grade and subtype of tumor. Medulloblastomas and ependymomas, which are more aggressive tumors, exhibited the highest levels of WEE1 expression followed by pilocytic astrocytomas. The findings suggest a correlation between the activity of WEE1 and disturbances in the cell cycle regulation mechanisms that are intensified before entering mitosis in fast-growing lesions. The WEE1 checkpoint protein is essential in the regulation of the G2/M phase transition, by phosphorylating the cyclin 1 dependent kinase (CDK1) on Tyr15 residue, which inactivates the CDK1/cyclin B1 complex and arrests the cell cycle6. Empirical evidence has established that WEE1 is excessively expressed in tumors exhibiting genomic instability, where cell cycle checkpoints play a critical role in preventing cell death (apoptosis)7. Thus far, altered expression of WEE1 in brain tumors, such as medulloblastoma, diffuse intrinsic pontine glioma, and glioblastoma have been reported so it could be considered an important part of the plausible targeted therapy approach in brain tumors7,8,9,10,11.
As the last two World Health Organization (WHO) classification systems have evolved, information about the molecular background of the lesions has gradually been added to help make the correct diagnosis but despite many advances in this intensively researched field, traditional treatment, including resection and adjuvant therapy, remains the gold standard for treating pediatric brain tumours. This is why the search for new targets and the discovery of molecular relationships in brain cancer cells is so important.
Some results showed an association of WEE1 expression with poor survival, however increased WEE1 activity was also observed in low-grade gliomas, suggesting that it could be a marker of tumorigenesis rather than disease progression12,13. The relevance of such observations has been confirmed in recently published results describing the possibility of using a selective small-molecule inhibitors of WEE1 in sonic hedgehog (SHH) medulloblastoma and relapsed ependymoma14,15. The ongoing clinical trials demonstrated its antitumor activity in monotherapy and/or in combination therapy15,16. The most promising synergistic effect has been observed with using multi-targeted therapy focusing on replication stress response pathways with a PARP inhibitors 17. Furthermore, when combined with a PARP inhibitor, the treatment exhibited established effects and suggests that multi-targeted therapy is particularly beneficial in reducing the risk of drug resistance in tumor cells9,14. Our results are an indication that the beneficial effects of this type of therapy are also possible in other types of pediatric tumors.
Next, we observed a unobvious results of the expression of the essential cell cycle regulator CCND1 which has been decreased in medulloblastomas and showed higher levels in ependymomas, especially in posterior fossa type A, and pilocytic astrocytomas. Studies in mice have shown that cyclin D1 plays a role in the development of medulloblastoma and the absence of cyclin D1 reduces the likelihood of disease development18. In an analysis of clinical samples of brain tumors the expression of cyclin D1 was generally increased but its relation to outcome is still unclear19. On the other hand an elevated levesl of CCND1 were noted as a positive factor in breast and renal cancer what suggests plausible gene's different cellular roles20,21,22. Cyclin D1 is a known regulator of cell cycle progression, but there are evidences that it binds to an miRNAs promoter sequences and regulates a specific miRNA signature23. So vague expression and unclear predictive value of CCND1 in various type of tumors could be the consequences of alterations in epigenetic regulatory systemss including miRNA regulatory feedback loop26,27. Therefore, information on such relationships would seem to be very important for the development of the most appropriate therapy. The statistically significant difference in this oncogene expression observed here in PFA and PFB ependymomas may suggest the potential use of this gene in optimizing therapy of the more aggressive ependymal tumors in the future.
Thus, the next gene analysed by us, BCL2L11, coding for the proapoptotic protein (BIM), could be one of the factors related to the treatment success. There are pieces of evidence showing that cancer cells have developed mechanisms that suppress the protein expression, which is necessary for tumor progression. Such a phenomenon is associated with the immune system and apoptosis via various cellular pathways. It was shown that activation of the MEK/ERK pathway reduced BCL2L11 expression at the protein level especially due to regulation of phosphorylation24,25.
It has been shown that in several solid tumors and hematological malignancies, the effectiveness of MAPK inhibitors is influenced by the levels of BIM. When BIM levels are increased, the sensitivity to these inhibitors is enhanced. Conversely, when the expression of this pro-apoptotic protein is reduced, the effectiveness of the inhibitors is diminished26,27,28. Disrupted apoptotic pathways are significant contributors to the development and advancement of cancer, as well as how well cancer responds to treatment. Apoptosis can be initiated using either the extrinsic or intrinsic pathways. BIM plays a crucial role in regulating the intrinsic pathway by interacting with the BCL2 family, which comprises both anti-apoptotic and pro-apoptotic proteins29,30. BCL2L11 mRNA can be regulated by several microRNAs (e.g. miR-9, −181a, −17∼92, −25, −32, −301a) as shown in our Oncomir-1 prediction target analysis24. In the gene analysis, we have demonstrated a statistically significant decrease in gene expression levels in medulloblastomas, whereas ependymomas and pilocytic astrocytomas exhibited increased gene expression. The significance of MAPK inhibitors in the treatment of MB is underscored by the compelling evidence from both in vitro and in vivo studies demonstrating their potential usefulness31,32. Considering that BCL2L11 functions as a mediator of miRNAs that regulate cellular processes and play a crucial role in the MAPK pathway, the number of molecular variables that need to be considered for predicting the prognosis of pediatric brain tumors increases33. This suggests that only the combined analysis of genetic information and epigenetic modifications, including the regulation of ncRNAs, can improve treatment, especially when miRNA can have different mRNA targets and each miRNA can have opposite roles according to the tumor type.
Furthermore, the TP53INP1 and PTPRO genes, which are directly associated with apoptosis, exhibited a consistent pattern of expression. Notably, the expression levels of these genes were significantly elevated in all tested samples compared to the control samples. TP53INP1 gene encodes a protein that can induce programmed cell death and inhibit cell proliferation. The function of this protein is influenced by multiple factors through distinct signaling pathways, which hampers the assessment of its impact on tumor development. The expression of TP53INP1 can be suppressed by many miRNAs, including miR-155-5p34,35, miR-106a36, miR-17-5p37, and miR-124-3p38. Opposite expression levels with a distinct influence on tumor growth and progression were noted for that gene in different types of cancer39,40,41. In the current investigation, we observed low expression of TP53INP1 in brain cancers of various origins, which is consistent with previous findings42. PTPRO is expressed mainly in the renal and brain epithelium, especially in the cells of the glomerular epithelium43,44. It can serve as a tumor suppressor and a predictor for diagnosis and a worse prognosis in various cancers45,46,47,48,49. Our research indicates that medulloblastoma has the most elevated level of PTPRO expression. However, there is a lack of corroborating evidence in the existing literature, with a limited number of documents on the gene’s role in the nervous system50.
Likewise, VEGFA, which as an undisputed member of the VEGF family is responsible for angiogenesis, can also function in an antiapoptotic manner51,52. In the current study, the highest levels of VEGFA expression were found for ependymoma with higher expression related to more aggressive posterior fossa type A. The results obtained are consistent with previously suggested observations that highlight the role of hypoxia-related genes, including VEGFA, in the mesenchymal stem cells in the tumor microenvironment as a key factor of tumor progression which could be cured53 Moreover, the differences in expression between the analysed three groups showed a statistical significance. Interestingly VEGFA, especially in the light of their role in the activation of the PI3K/AKT pathway, is an important regulator of angiogenesis, where miR-17-5p plays a key role in the regulation of oncogenes from that signaling pathway54,55.
Conclusions
Our investigation revealed that pediatric brain tumors exhibit a wide range of variations in both coding and non-coding RNA expression. As an understanding of the molecular background of cancer deepens, the complexity of this scenario increases, necessitating the exploration of driver factors. In addition, our results and more recent studies underlined numerous cross-talks between genes activity and various epigenetic processes together with tumor’s metabolic patterns56,57. As these findings continue to emerge, their integration with the WHO classification and therapeutic implications of paediatric brain tumours offers the prospect of improved treatment outcomes.
The example is clinical trial with Adavosertib, inhibitor of WEE1 in monotherapy and in combination therapy also in pediatric patients with aggressive brain tumors15. Our research has revealed the existence of molecules exhibiting altered levels of expression and their complex regulatory system. We identified genes and non-coding molecules that show great potential for being utilized in the detection and treatment of pediatric brain tumors in the future. Furthermore, the successful regulation of PI3K/AKT and RAS/MAPK/ERK, which are functionally associated with cell growth and survival, makes them potential targets for anti-cancer therapies, including those for brain tumors.
Such a trend is of particular importance in cancers in which continuous molecular subgrouping has not had the expected clinical implications, such as medulloblastomas and ependymomas where resection and adjuvant therapy, remains still the gold standard.
Data availability
Microarray data has been deposited in the ArrayExpress database under the number E-MTAB-14259 (www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-14259?key=880a5bbb-ed20-4d2c-8f10-8a9811f89dce). The other datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Otmani, K., Rouas, R. & Lewalle, P. OncomiRs as noncoding RNAs having functions in cancer: Their role in immune suppression and clinical implications. Front. Immunol. 13, 913951 (2022).
Manea, A. J. & Ray, S. K. Advanced bioinformatics analysis and genetic technologies for targeting autophagy in glioblastoma multiforme. Cells. 12(6), 897 (2023).
Murphy, B. L. et al. Silencing of the miR-17~92 cluster family inhibits medulloblastoma progression. Cancer Res. 73, 7068–7078 (2013).
Gruszka, R., Zakrzewski, K., Liberski, P. P. & Zakrzewska, M. mRNA and miRNA expression analyses of the MYC/E2F/miR-17–92 network in the most common pediatric brain tumors. Int. J. Mol. Sci. 22(2), 543 (2021).
Gruszka, R., Zakrzewski, K., Liberski, P. P. & Zakrzewska, M. microRNA interaction with MAPK and AKT pathways in paediatric brain tumours-preliminary results and review of the literature. Folia Neuropathol. 58, 123–132 (2020).
Matheson, C. J., Backos, D. S. & Reigan, P. Targeting WEE1 kinase in cancer. Trends Pharmacol. Sci. 37, 872–881 (2016).
de Jong, M. R. W. et al. WEE1 inhibition enhances anti-apoptotic dependency as a result of premature mitotic entry and DNA damage. Cancers 11(11), 1743 (2019).
Wu, S. et al. Activation of WEE1 confers resistance to PI3K inhibition in glioblastoma. Neuro Oncol. 20, 78–91 (2018).
Harris, P. S. et al. Integrated genomic analysis identifies the mitotic checkpoint kinase WEE1 as a novel therapeutic target in medulloblastoma. Mol. Cancer 13, 72 (2014).
Tashnizi, A. H., Jaberipour, M., Razmkhah, M., Rahnama, S. & Habibagahi, M. Tumour suppressive effects of WEE1 gene silencing in neuroblastomas. J. Cancer Res. Ther. 12, 221–227 (2016).
Music, D. et al. Expression and prognostic value of the WEE1 kinase in gliomas. J. Neurooncol. 127, 381–389 (2016).
Magnussen, G. I. et al. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: Potential for targeted therapy. PLoS ONE 7, e38254 (2012).
Mueller, S. et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 16, 352–360 (2014).
Lukoseviciute, M., Theodosopoulou, A., Holzhauser, S., Dalianis, T. & Kostopoulou, O. N. Combination of PARP and WEE1 inhibitors in vitro: Potential for use in the treatment of SHH medulloblastoma. Oncol. Rep. https://doi.org/10.3892/or.2023.8562 (2023).
Cole, K. A. et al. Phase I clinical trial of the WEE1 inhibitor adavosertib (AZD1775) with irinotecan in children with relapsed solid tumors: A COG phase I consortium report (ADVL1312). Clin. Cancer Res. 26, 1213–1219 (2020).
Gatz, S. A. & Harttrampf, A. C. Phase I/II study of the WEE1 inhibitor adavosertib (AZD1775) in combination with carboplatin in children with advanced malignancies: Arm C of the AcSé-ESMART trial. Clin. Cancer Res. 30, 741–753 (2024).
Keller, K. M. et al. Target actionability review: A systematic evaluation of replication stress as a therapeutic target for paediatric solid malignancies. Eur. J. Cancer 162, 107–117 (2022).
Pogoriler, J., Millen, K., Utset, M. & Du, W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development 133, 3929–3937 (2006).
Zhao, X., Song, T., He, Z., Tang, L. & Zhu, Y. A novel role of cyclinD1 and p16 in clinical pathology and prognosis of childhood medulloblastoma. Med. Oncol. 27, 985–991 (2010).
Wang, Q. S. et al. Low level of cyclin-D1 correlates with worse prognosis of clear cell renal cell carcinoma patients. Cancer Med. 8, 4100–4109 (2019).
Siraj, A. K. et al. High expression of cyclin D1 is an independent marker for favorable prognosis in middle eastern breast cancer. Onco Targ. Ther. 14, 3309–3318 (2021).
Mylona, E. et al. Cyclin D1 in invasive breast carcinoma: Favourable prognostic significance in unselected patients and within subgroups with an aggressive phenotype. Histopathology 62, 472–480 (2013).
Yu, Z. et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J. Cell Biol. 182, 509–517 (2008).
Sionov, R. V., Vlahopoulos, S. A. & Granot, Z. Regulation of bim in health and disease. Oncotarget 6, 23058–23134 (2015).
Balmanno, K. & Cook, S. J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 16, 368–377 (2009).
Rose, M., Sharma, V., Espinoza, V., Hoffman, M. C. & Schweppe, R. Regulation of BIM by cooperative Src and MAPK signaling. Eur. J. Cancer 174, S19–S20 (2022).
Chakraborty, A. R. et al. MAPK pathway activation leads to bim loss and histone deacetylase inhibitor resistance: Rationale to combine romidepsin with an MEK inhibitor. Blood 121, 4115–4125 (2013).
Rose, M. M. et al. BCL2L11 induction mediates sensitivity to src and MEK1/2 inhibition in thyroid cancer. Cancers 15(2), 378 (2023).
Barillé-Nion, S., Bah, N., Véquaud, E. & Juin, P. Regulation of cancer cell survival by BCL2 family members upon prolonged mitotic arrest: Opportunities for anticancer therapy. Anticancer Res. 32, 4225–4233 (2012).
Kalkavan, H. & Green, D. R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 25, 46–55 (2018).
Schönholzer, M. T. et al. Real-time sensing of MAPK signaling in medulloblastoma cells reveals cellular evasion mechanism counteracting dasatinib blockade of ERK activation during invasion. Neoplasia 22, 470–483 (2020).
Zagozewski, J. et al. Combined MEK and JAK/STAT3 pathway inhibition effectively decreases SHH medulloblastoma tumor progression. Commun. Biol. 5, 697 (2022).
Jiang, Z., Zheng, X., Lytle, R. A., Higashikubo, R. & Rich, K. M. Lovastatin-induced up-regulation of the BH3-only protein, bim, and cell death in glioblastoma cells. J. Neurochem. 89, 168–178 (2004).
Li, N., Cui, T., Guo, W., Wang, D. & Mao, L. MiR-155-5p accelerates the metastasis of cervical cancer cell via targeting TP53INP1. Onco Targ. Ther. 12, 3181–3196 (2019).
Liu, F., Kong, X., Lv, L. & Gao, J. MiR-155 targets TP53INP1 to regulate liver cancer stem cell acquisition and self-renewal. FEBS Lett. 589, 500–506 (2015).
Han, L. et al. MicroRNA-106a regulates autophagy-related cell death and EMT by targeting TP53INP1 in lung cancer with bone metastasis. Cell Death Dis. 12, 1037 (2021).
Wei, Q., Li, Y. X., Liu, M., Li, X. & Tang, H. MiR-17-5p targets TP53INP1 and regulates cell proliferation and apoptosis of cervical cancer cells. IUBMB Life 64, 697–704 (2012).
Margolin-Miller, Y. et al. Prognostic relevance of miR-124-3p and its target TP53INP1 in pediatric ependymoma. Genes Chromosomes Cancer 56, 639–650 (2017).
Nishimoto, M. et al. Prognostic impact of TP53INP1 gene expression in estrogen receptor α-positive breast cancer patients. Jpn. J. Clin. Oncol. 49, 567–575 (2019).
Seux, M. et al. TP53INP1 decreases pancreatic cancer cell migration by regulating SPARC expression. Oncogene 30, 3049–3061 (2011).
Giusiano, S. et al. TP53INP1 overexpression in prostate cancer correlates with poor prognostic factors and is predictive of biological cancer relapse. Prostate 72, 117–128 (2012).
Wang, Y. & Lin, G. TP53INP1 3’-UTR functions as a ceRNA in repressing the metastasis of glioma cells by regulating miRNA activity. Biotechnol. Lett. 38, 1699–1707 (2016).
Bodden, K. & Bixby, J. L. CRYP-2: A receptor-type tyrosine phosphatase selectively expressed by developing vertebrate neurons. J. Neurobiol. 31, 309–324 (1996).
Wiggins, R. C., Wiggins, J. E., Goyal, M., Wharram, B. L. & Thomas, P. E. Molecular cloning of cDNAs encoding human GLEPP1, a membrane protein tyrosine phosphatase: characterization of the GLEPP1 protein distribution in human kidney and assignment of the GLEPP1 gene to human chromosome 12p12-p13. Genomics 27, 174–181 (1995).
Liu, Z. et al. Protein tyrosine phosphatase receptor type O expression in the tumor niche correlates with reduced tumor growth, angiogenesis, circulating tumor cells and metastasis of breast cancer. Oncol. Rep. 33, 1908–1914 (2015).
Gan, J. & Zhang, H. PTPRO predicts patient prognosis and correlates with immune infiltrates in human clear cell renal cell carcinoma. Transl. Cancer Res. 9, 4800–4810 (2020).
Xie, F., Dong, H. & Zhang, H. Regulatory functions of protein tyrosine phosphatase receptor type O in immune cells. Front. Immunol. 12, 783370 (2021).
Huang, Y. T. et al. PTPRO promoter methylation is predictive of poorer outcome for HER2-positive breast cancer: Indication for personalized therapy. J. Transl. Med. 11, 245 (2013).
Ming, F. & Sun, Q. Epigenetically silenced PTPRO functions as a prognostic marker and tumor suppressor in human lung squamous cell carcinoma. Mol. Med. Rep. 16, 746–754 (2017).
Beltran, P. J., Bixby, J. L. & Masters, B. A. Expression of PTPRO during mouse development suggests involvement in axonogenesis and differentiation of NT-3 and NGF-dependent neurons. J. Comp. Neurol. 456, 384–395 (2003).
Lee, S. H., Jeong, D., Han, Y. S. & Baek, M. J. Pivotal role of vascular endothelial growth factor pathway in tumor angiogenesis. Ann. Surg. Treat. Res. 89, 1–8 (2015).
Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2, 795–803 (2002).
Fu, R. et al. Spatial transcriptomic analysis delineates epithelial and mesenchymal subpopulations and transition stages in childhood ependymoma. Neuro Oncol. 25, 786–798 (2023).
Cuzziol, C. I. et al. Regulation of VEGFA, KRAS, and NFE2L2 oncogenes by microRNAs in head and neck cancer. Int. J. Mol. Sci. 23(13), 7483 (2022).
Claesson-Welsh, L. & Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 273, 114–127 (2013).
Su, J., Xie, Q. & Xie, L. Identification and validation of a metabolism-related gene signature for predicting the prognosis of paediatric medulloblastoma. Sci. Rep. 14, 7540 (2024).
Graziani, V. et al. Metabolic rewiring in MYC-driven medulloblastoma by BET-bromodomain inhibition. Sci. Rep. 13, 1273 (2023).
Author information
Authors and Affiliations
Contributions
RG: conceptualization, methodology, investigation, statistical analysis, writing—original draft, visualization; JZ: visualization, writing—review and editing; EN: sampling, writing—review and editing; WG: sampling, writing—review and editing; MZ: conceptualization, methodology, investigation, writing—review and editing. All authors reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gruszka, R., Zakrzewski, J., Nowosławska, E. et al. Identification and validation of miRNA-target genes network in pediatric brain tumors. Sci Rep 14, 17922 (2024). https://doi.org/10.1038/s41598-024-68945-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68945-z
- Springer Nature Limited